


Abstract
The 2012 drug-drug interaction (DDI) guidance from the European Medicines Agency (EMA) and the draft DDI guidance from the Food and Drug Administration (FDA) have proposed that metabolites present at >25% of the parent area under the time-concentration curve (AUC) (EMA and FDA) and >10% of the total drug-related exposure (EMA) should be investigated in vitro for their DDI potential. This commentary attempts to rationalize the clinically relevant levels of metabolite(s) that contribute to DDI by considering not only the abundance but also inhibition potency, physicochemical properties, and structural alerts of the metabolite. A decision tree is proposed for levels of metabolites that could trigger in vitro DDI assessment. When the parent is an inhibitor of cytochrome P450s (P450s), clinical DDI studies will assess the in vivo DDI effect of the combination of parent and metabolite(s). When the parent is not a P450 inhibitor, it is important to assess the inhibition potential of abundant metabolites in vitro. The proposal is to apply a default cutoff value of metabolite level which is 100% of the parent AUC. It is important to note that exceptions can occur, and different metabolite levels may be considered depending on the physiochemical properties of metabolites (e.g., increased lipophilicity) and whether the metabolite contains structural alerts for DDI (e.g., mechanism-based inhibition). A key objective of this commentary is to stimulate discussions among the scientific community on this important topic, so that appropriate in vitro metabolism studies are conducted on metabolites, to ensure the safety of drugs in development balanced with the desire to avoid creating unnecessary studies that will add little to no value in ensuring patient safety.
Introduction
The recent European Medicines Agency (EMA) Guideline on Investigation of Drug Interactions (www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf) and the draft guidance on drug interaction studies from the Food and Drug Administration (FDA) (www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf) have both proposed that metabolites that are present at >25% of the parent area under time-concentration curve (AUC) (EMA and FDA) and >10% of the total drug-related exposure (EMA) should trigger further in vitro characterization of the metabolite as a possible contributor to drug-drug interactions (DDIs), as a consequence of inhibition or induction. There is already an appreciation that metabolites can contribute to the main component of the pharmacology of a parent molecule [e.g., morphine 6-glucuronide (Hanna et al., 1990)] or be partial contributors [e.g., N-desmethylsertraline (Rudorfer and Potter, 1997)]. Metabolites have also been implicated in adverse effects [e.g., the quinone-imine metabolite of acetaminophen (Manyike et al., 2000) and trifluoroacetyl chloride of halothane (Satoh et al., 1989)].
In cases where the parent compound is an inhibitor or an inducer of drug-metabolizing enzyme(s), based on in vitro data, in vivo DDI studies are generally conducted to confirm DDI potential of parent their DDI in vivo. Metabolites that contribute to inhibition or induction, in combination with the parent molecule, will, by default, be accounted for in clinical studies at relevant levels. In those situations, the question to ask is whether there are populations in which the metabolite could disproportionately contribute to the overall DDI effect by being present at higher levels. On the other hand, when the parent compound is not an inhibitor or inducer, based on in vitro data, in vivo DDI studies are generally not conducted. In those cases, how do we identify metabolites that are inhibitors or inducers of drug-metabolizing enzyme(s)? There are a few examples of metabolites being the main contributor to clinically relevant DDI reported in the literature. For example, threohydrobupropion and erythrohydrobupropion have 4- and 12-fold lower inhibition potency (Ki) values for CYP2D6, respectively, compared with the parent bupropion, and circulate at higher concentrations than bupropion (Reese et al., 2008). Gemfibrozil glucuronide was identified as an unusual example of a conjugated metabolite that was considerably more potent an inhibitor for CYP2C8 compared with the parent molecule (Ogilvie et al., 2006). This commentary attempts to rationalize the clinically relevant levels of a metabolite that could contribute to DDI by considering abundance, inhibition potency, physicochemical properties, and structural alerts of metabolites in addition to the available in vitro/in vivo DDI data for the parent drug.
Proposed Strategy for Assessing the Contribution of Metabolites to DDI
Pharmaceutical companies frequently conduct in vitro studies to assess DDI potential (e.g., P450 inhibition and induction) for new chemical entities in preclinical and early-stage clinical development. An early readout of the clinical relevance of DDI potential can be achieved through targeted studies (e.g., midazolam clearance for CYP3A or cocktail studies). A common strategy is to rank order the likelihood of in vivo DDI and conduct a probe substrate interaction in the clinic targeting the most potent interaction, based on the in vitro DDI data (Obach et al., 2005). Alternatively, a clinical study, using a cocktail of probe substrates, has been used to assess DDI potential (Streetman et al., 2000; Chainuvati et al., 2003). Both approaches have their merits (Bjornsson et al., 2003; U.S. FDA, 2006). By this point in the development of the drug, many companies have evaluated circulating metabolite profiles in humans, and can assess the relative plasma levels of parent to metabolites. Thus, data from in vitro and early clinical studies, including DDI characterizations of the parent drug with a consideration of metabolites, can, and should be, used in determining whether separate inhibition or induction studies for the metabolites are warranted. An important consideration in these early analyses is whether metabolites (or even the parent molecule) will accumulate with multiple dosing, and so conducting the DDI assessment at steady state becomes even more important. If inhibition or induction is not observed in these clinical studies, further in vitro characterization of the metabolites for DDI potential is generally not warranted, regardless of their abundance.
A more interesting challenge to this strategy is the situation in which the parent drug does not inhibit or induce based on in vitro data, and so in vivo DDI studies for the parent are generally not conducted. Under what circumstances should efforts be extended to evaluate metabolites for their potential to inhibit or induce? There can be significant challenges in trying to synthesize metabolites. The efforts required to synthesize metabolites should not be minimized or dismissed when debating whether such studies should be conducted. The problem is often exacerbated by not having early definitive identification of metabolites (e.g., mass spectrometry fragmentation patterns suggesting structures rather than definitive NMR). Enantiomers and other structural analogs can also complicate identification and synthesis. Separation of these isomers is important, as differences in inhibition potency for isomers have been observed (Reese et al., 2008). On the basis of internal experience, we have come across a number of clear examples of extensive time and resource commitments in synthesizing metabolites. Approaches to biologic generation of metabolites (through recombinant enzymes, tissue fractions, etc.) are often restricted to making small amounts of material.
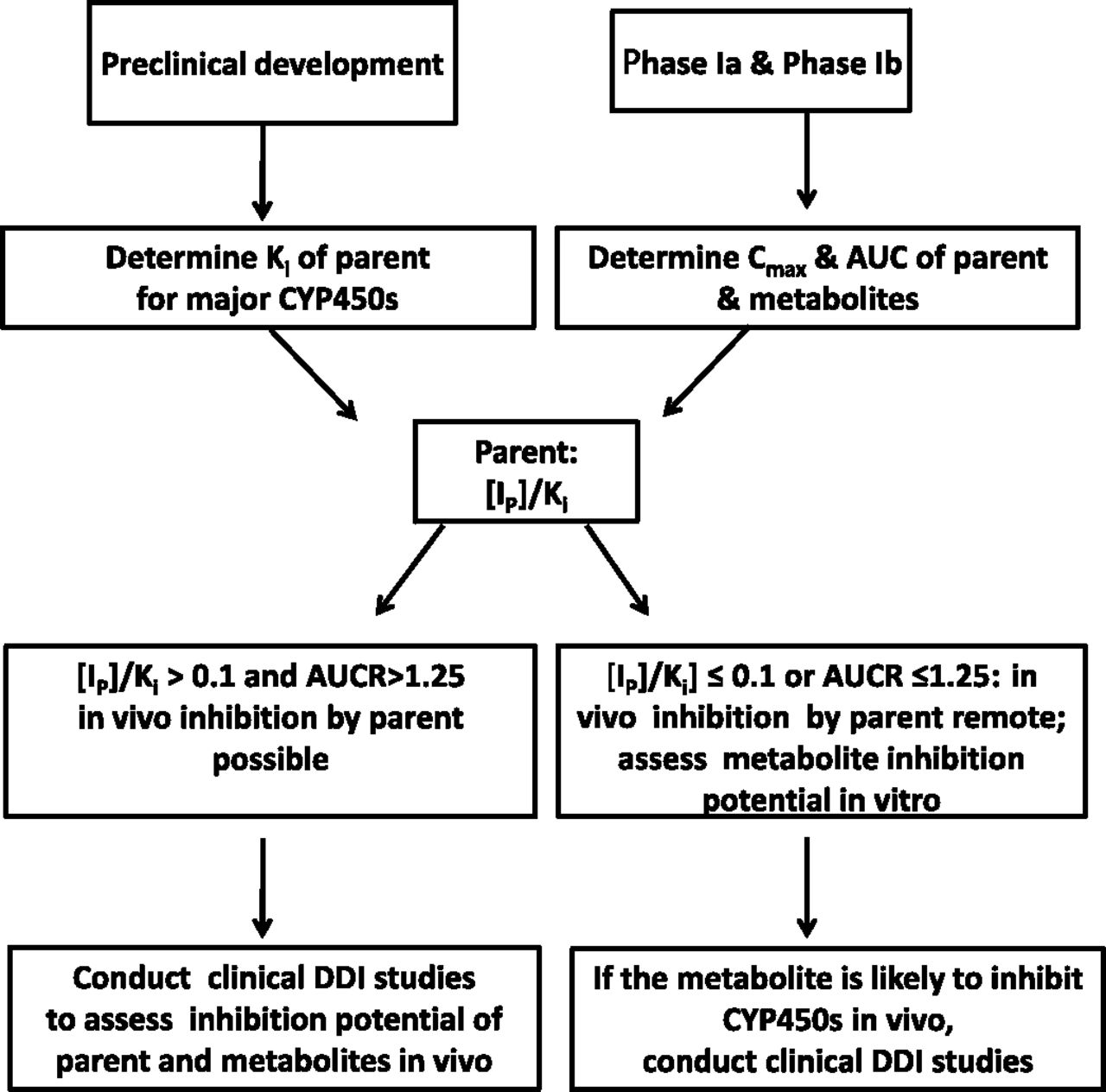
Currently, we are not aware of any reported cases where a metabolite is a P450 inducer while the parent drug is not. As such, assessment of the parent induction potential (in vitro and/or in vivo) should cover both the parent and its metabolites, and therefore separate evaluation of the induction potential of metabolites is generally not warranted. The strategy to cover metabolites, when the parent molecule has no potential DDI liabilities, should focus on inhibition. A proposed strategy for assessing the possible contribution of metabolites to P450 inhibition is outlined in Fig. 1. The IC50 and/or Ki values of the parent drug are generally determined in preclinical development. The plasma concentrations and exposure (total and/or free) of the parent drug and possibly the major metabolites are obtained in early clinical development (phases Ia and Ib). The ratio of total plasma concentration of the parent as an inhibitor ([Ip]) to Ki and/or the AUC ratio (AUCR) value for the parent can be calculated following the equations outlined in Fig. 4 of the 2012 FDA draft DDI guidance (U.S. FDA, 2012). If [Ip]/Ki is >0.1 and AUCR is >1.25 for one or more P450s, the parent is determined to be a possible inhibitor of these P450s in vivo. In this case, a dedicated clinical DDI study can be conducted to assess the in vivo DDI potential of the parent and its metabolites. If [Ip]/Ki is ≤0.1, the parent drug is not likely to cause inhibition in vivo. In this case, a dedicated clinical DDI study is generally not conducted, with the caveat that the metabolite(s) could be a more potent P450 inhibitor than the parent drug. Therefore, it is important to assess the DDI potential of the abundant circulating metabolites in vitro to obtain a more complete assessment of the DDI potential of drug-related material. If the metabolite(s) is likely to inhibit P450s, based on the [I]/Ki ratio, there will now be a need to conduct clinical DDI studies of the parent at steady state to confirm the in vitro results.

A proposed strategy to assess the inhibition potential of metabolites by taking into account the available in vitro and in vivo DDI data of the parent compound.
Consideration of Clinically Relevant Levels and Inhibition Potency of Metabolites That Contribute to Enzyme Inhibition
Based on literature data, the risk of DDI, as a result of inhibition by metabolites alone (the parent is not an inhibitor), appears to be low. Two recent publications indicated that the majority (90%) of drugs (including their metabolites) are not P450 inhibitors in vivo (with a survey of 1323 drugs from the U.S. market) (Isoherranen et al., 2009; Yeung, et al., 2011). Of the remaining 10% of drugs (129) that were P450 inhibitors, inhibition data for the individual metabolites were only available for 21 drugs (1.6% of the total or 16% of drugs showing DDI). The lack of data certainly calls into question whether the 16% of drugs truly represent the overall potential. In only three out of 21 drugs were the metabolites P450 inhibitors, when the parent was not. These three drugs (amiodarone, bupropion, and sertraline) represent 0.2% of all drugs, 2.3% of drugs exhibiting DDI, and 14% of drugs in which metabolites were evaluated. Therefore, for the vast majority of drugs, assessing the DDI potential of the parent drug is adequate to assess the DDI potential of both the parent drug and its metabolites. There is certainly a valid argument that a false negative should be viewed with much greater concern than a false positive, particularly as we strive to ensure patient safety. To minimize further the low risk of DDI caused by metabolites, there are additional considerations. Some structural alerts can be used as a trigger to conduct in vitro DDI assessments for inhibition and inactivation. For example, metabolites derived from N-dealkylation (VandenBrink and Isoherranen, 2010) or containing an epoxide moiety (Brown and Ford-Hutchinson, 1982) have the potential to inactivate P450s and should be further evaluated.
In assessing the contribution of metabolites to clinically relevant enzyme inhibition, it is important to consider both the levels of a metabolite(s) (individually or summed) and the inhibition potency of these metabolites. Several literature reports have demonstrated that hydrophobicity is one of the most important factors in determining the inhibition potency of P450 inhibitors. For example, QSAR studies by Lewis et al. (2006) showed that a linear relationship existed between lipophilicity and potency for 15 inhibitors/substrates of CYP2C9, including nonsteroidal anti-inflammatory drugs. Modeling by Didziapetris et al. (2010) demonstrated that an increase in the size of the molecule with the incorporation of hydrophobic aliphatic or aromatic residues resulted in a higher probability for the compound to inhibit CYP3A4. Roy and Pratim Roy (2009) also demonstrated that logP appeared to be the most important factor affecting the inhibition potency of CYP3A4 inhibitors. From an evolutionary perspective, biotransformation of xenobiotics by host enzymes is a protective mechanism, resulting in the generation of (generally) more polar compounds, which will then be more readily excreted compared with the parent. The decreased hydrophobicity of metabolites generally leads to a lower affinity for the drug-metabolizing enzymes. As such, metabolites are expected to be less potent inhibitors than the parent drug. Clearly there are some exceptions. However, we believe the generalization that metabolites are less potent inhibitors than the parent is applicable, and is appropriate in defining the relative contribution to inhibition by the parent and metabolite(s). Thus, calculations of relevant concentrations of metabolites, when considering their contribution to inhibition, cannot be simple arithmetic summations of metabolite and parent molecule levels. Adjustments for expected lower inhibition potency of metabolites should be considered.
In rationalizing clinically relevant levels of metabolites, an important consideration is the change in AUC of the victim drug that warrants a concern for DDI. The FDA draft guidance on drug interaction studies proposes a 25% increase or 20% decrease in the AUC of the victim drug, which, although in line with defining bioequivalence [80%–125% (U.S. FDA, 2012)], should not merit full consideration for clinically relevant DDI. Additional safety concerns are warranted for drugs with narrow therapeutic ranges, such as theophylline, warfarin, and paclitaxel, and these can be, and should be, treated as a special case. This conservative value (80%–125%) should be addressed through scholarly discussion, but is not the focus of this commentary. As such, we will use the conservative value of a 25% increase in victim drug AUC as the basis for discussion.
The magnitude of DDI caused by a metabolite depends on its concentration and inhibition. As stated, it is important to consider both the maximum concentration in human plasma (Cmax) and inhibition potency (Ki) values of the parent and metabolites in rationalizing a cutoff value to investigate the inhibition potential of metabolites. For the purpose of illustrating this principle, competitive inhibition and a static model (often most conservative) are used, and the focus is on cases where the [I]/Ki ratio for the parent drug ([Ip]/KiP) is ≤0.1 (i.e., parent is not an inhibitor; see Fig. 1). The following set of equations (eqs. 1–6) outline the rationale for setting 100% of the parent AUC as a reasonable level of metabolite that would trigger additional in vitro evaluation of DDI potential. [Im] and [Ip] are the total systemic Cmax of the metabolite and the parent, respectively. Although using total Cmax may generally lead to overprediction of in vivo DDI, it was demonstrated to predict in vivo DDI almost as well as using unbound hepatic inlet Cmax and superior than the unbound systemic Cmax (Obach et al., 2006). Kim and Kip are the inhibition potencies of the metabolite and the parent, respectively. [Im]/[Ip] is the cutoff value of metabolite (a surrogate for metabolite as a percentage of the parent AUC).
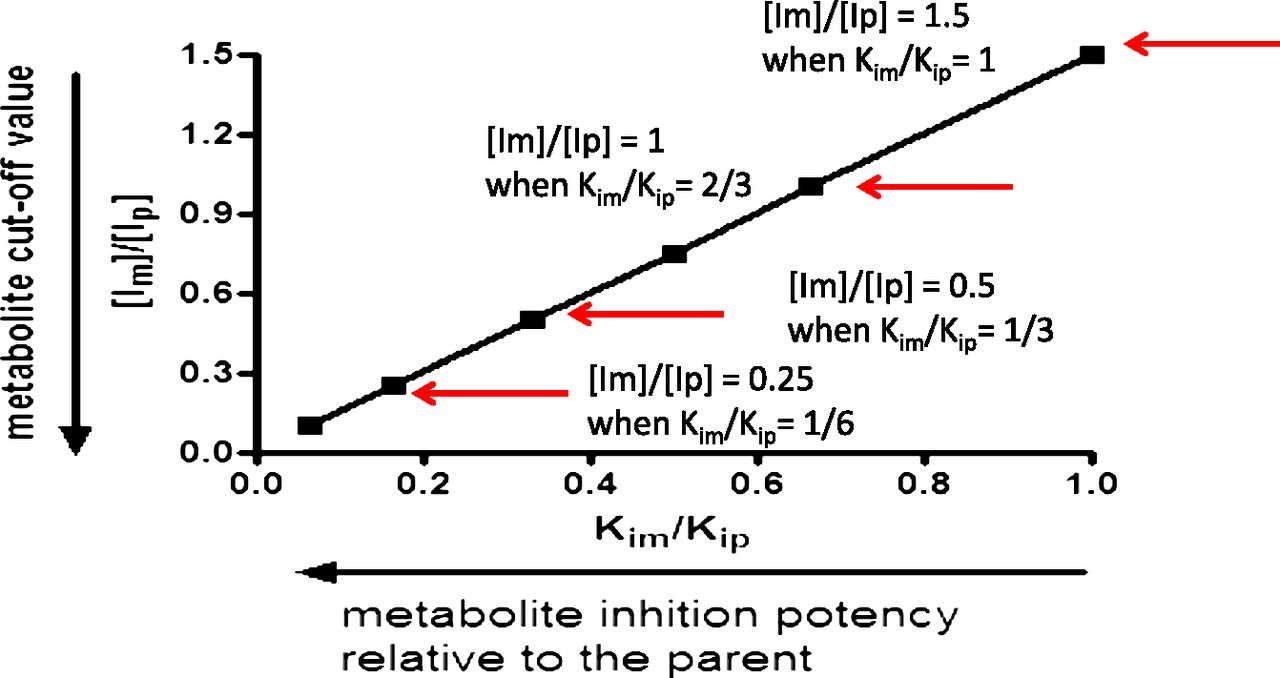
By use of the conservative value of a 25% increase in the AUC of a victim drug (AUCi/AUC = 1.25), eqs. 1 and 2 are used to calculate clinically relevant metabolite levels. Rearranging eq. 2, with eqs. 3 and 4 as the intermediate steps, yields eq. 5, which provides a ratio of inhibitor concentrations of metabolite to the parent ([Im]/[Ip]). Applying [Ip]/Kip of 0.1 (no DDI is anticipated for the parent) to eq. 5 results in eq. 6. Equation 6 clearly illustrates the importance of considering the inhibition potency ratio between the metabolite and parent (Kim/Kip) in proposing a cutoff value for metabolites ([Im]/[Ip]). The relationship between [Im]/[Ip] and Kim/Kip is illustrated in Fig. 2.
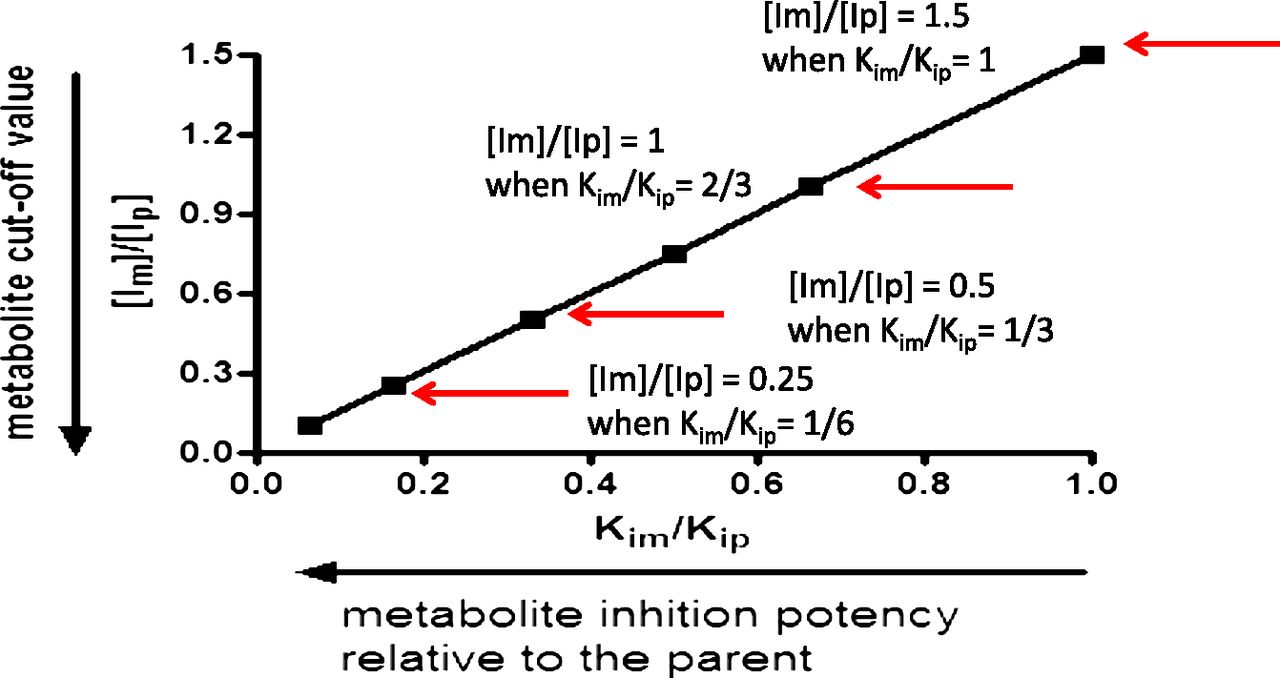
The metabolite cutoff value ([Im]/[Ip]) decreases with the increase in the inhibition potency ratio between the metabolite and parent (Kim/Kip).
 (1)
(1) (2)
(2)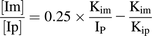 (3)
(3)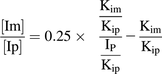 (4)
(4) (5)
(5)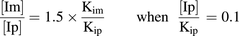 (6)
(6)As demonstrated in eq. 6 and in Fig. 2, the more potent the metabolite is as an inhibitor, relative to the parent, the lower the level of metabolite is that would trigger in vitro DDI studies. If the metabolite and the parent have the same inhibition potency (Kim/Kip = 1), the metabolite level to trigger in vitro inhibition studies would be 150% of the parent AUC ([Im]/[Ip] = 1.5; see Fig. 2 and Table 1). A metabolite present at 25% of the parent AUC, as suggested by the draft FDA guidance (U.S. FDA, 2012), would need to be 6-fold more potent in inhibition than the parent to cause an increase of 25% in AUC of the victim drug ([Im]/[Ip] = 0.25 and Kim/Kip = 1/6; see Fig. 2 and Table 1). Because metabolites are generally expected to be more polar and have a lower affinity for P450 and thus be less potent inhibitors than the parent drug (Kim/Kip ≥ 1) (Johnston et al., 1991; Backes et al., 1993), we propose that metabolite levels approaching 100% of the parent AUC is a more reasonable default value to trigger in vitro inhibition studies when the parent drug is not an in vitro inhibitor of P450s (i.e., [Ip]/Kip ≤ 0.1).
Metabolite cut-off value versus the metabolite/parent inhibition potency ratio
As stated in the previous section, the risk of a metabolite being the sole contributor to P450 inhibition is very small (three out of 1323 drugs) (Isoherranen et al., 2009; Yeung et al., 2011). The three drugs are bupropion, amiodarone, and sertraline. Bupropion metabolites (threohydrobupropion and erythrohydrobupropion) are present at 3.5- to 6-fold higher levels compared with the parent (Reese et al., 2008), and thus warrant further analysis by the criteria proposed herein (100% of the parent AUC). N-Desethylamiodarone would also be covered because the AUC ratio between N-desethylamiodarone and amiodarone is 1.5 (Marchiset et al., 1985). The same is true for the N-desmethyl metabolite of sertraline, which is∼3-fold higher in AUC than sertraline (Patel et al., 2009). Gemfibrozil glucuronide is often quoted as an example of a metabolite that causes significant DDI through inhibition of CYP2C8. This is certainly an exceptional case, and the presence of a mechanism-based inhibitor that is formed through further metabolism of a glucuronide is quite unique (Ogilvie et al., 2006; Jenkins et al., 2011). However, gemfibrozil is itself an inhibitor of CYP2C8 in vitro. Additionally, the glucuronide metabolite is present at ∼100% of the AUC of gemfibrozil (Tornio et al., 2008). As such, it would also be covered by the criteria proposed herein (i.e., metabolites = 100% of the parent AUC as the cutoff value to trigger in vitro DDI studies for metabolites).
We propose metabolite levels approaching 100% of the parent AUC as a default value to trigger in vitro inhibition studies of metabolites (see the proposed decision tree in Fig. 3). There are two notable exceptions when this default value needs to be adjusted: 1) metabolites are less polar than the parent drug, and 2) metabolites contain a structural alert(s) for mechanism-based inactivation (MBI) regardless of their change in polarity. Formation of less-polar metabolites can occur through biochemical or chemical reactions [e.g., converting carboxylic acid to lactone as in the case of atorvastatin (Jacobsen et al., 2000)]. These metabolites should be readily recognized from their structures, clogP calculations, and most practically, from the relative elution order on reverse phase high-performance liquid chromatography columns [e.g., threohydrobupropion and erythrohydrobupropion eluted later than bupropion (Petsalo et al., 2007)]. Examples of less polar metabolites that are associated with increased inhibition potency include the lactone of atorvastatin (Jacobsen et al., 2000) and threohydrobupropion and erythrohydrobupropion (Reese et al., 2008). It is worth noting that not all metabolites with decreased polarity would lead to more potent inhibition of P450s. We have compared the P450 inhibition potential between an internal compound and one of its less polar lactam metabolites. In this case, the lactam metabolite was a less potent inhibitor for major P450s than the parent (data not shown). Nonetheless, a lower cutoff value should be considered to trigger in vitro DDI studies for the less polar metabolites as we strive to predict the DDI potential of drug-related material (parent and metabolites) more accurately using in vitro studies. On the basis of the Kim/Kip ratios between metabolites and parents of 21 P450 inhibitors (Yeung et al., 2011), 80% of the Kim/Kip ratios are <1/6. As shown in Fig. 2 and Table 1, when Kim/Kip = 1/6, the cutoff value of [Im]/[Ip] is equal to 0.25. Therefore, it appears to be appropriate to propose a cutoff value of 25% of the parent AUC for metabolites that are less polar and can be more potent in inhibition than the parent drug. As mentioned previously, attention should be paid to metabolites containing structural alerts for MBI, which are absent in the parent molecule. Currently, a well characterized structural alert for MBI is the formation of a primary amine through N-dealkylation of the secondary and tertiary amines. The primary amine metabolites can cause MBI through the formation of the nitroso intermediate by metabolic activation (Orr et al., 2012). For these N-dealkylated primary amine metabolites, because their inactivation parameters (the dissociation constant for the enzyme-inactivator complex and the maximum rate for the inactivation) cannot be related to the parent, it would be prudent to conduct in vitro DDI studies of these metabolites, even if they are present at low but appreciable concentrations. For currently known drugs, it is interesting to note that these N-dealkylated metabolites tended to circulate at a concentration that was close to or higher than that of the parent (VandenBrink and Isoherranen, 2010).

Decision tree to trigger in vitro DDI studies for metabolites.
As shown in Fig. 3, polarity and structural alerts of metabolites are two key considerations in this decision tree. There will be exceptions. Because it is challenging to include all possible scenarios and exceptions, we intend to use this decision tree as a basis to stimulate discussions in the scientific community. The default cutoff value to trigger in vitro studies for metabolites is 100% of the parent AUC based on the premise that metabolites are generally more polar and less potent inhibitors of P450s than the parent. A lower cutoff value of 25% of the parent AUC is proposed for metabolites that are less polar than the parent drug. These less polar metabolites can be more potent inhibitors than the parent drug. A lower cutoff value should also be applied to metabolites carrying structural alerts for MBI that are absent from the parent drug on a case-by-case basis by considering multiple factors. These factors include 1) difficulty predicting the inactivation parameters of metabolites from those of the parent drug, 2) more severe safety consequences due to enzyme inactivation, and 3) the extent of formation of the metabolite with structural alerts in the inactivation experiment of the parent drug.
In conclusion, the DDI risk caused by metabolites alone is considered to be low based on the current literature. When the parent drug is an inhibitor of one or more P450 isoforms, clinical DDI studies are conducted to assess the in vivo inhibition potential for both the parent and metabolites. When the parent drug is not an inhibitor of one or more P450s, the proposed default cutoff value to trigger in vitro DDI studies for metabolites is ≥100% of the parent AUC. There can be important exceptions to this default cutoff value (e.g., metabolites are less polar or contain structural alerts for MBI). For metabolites that are less polar than the parent molecule, a lower cutoff value should be considered (25% of the parent AUC). Metabolites that contain structural alerts for MBI should be considered on a case-by-case basis, as it is not possible to ascribe a level of expected inhibition based simply on structure (or inhibition by the parent molecule). Pharmaceutical companies and regulatory authorities are concerned with patient safety. It is in this spirit that we make this proposal that rationally addresses the concern of DDI by metabolites. We welcome comments from the scientific community and hope to move the science on metabolite-related DDI forward through open dialogue.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Yu, Tweedie.
Footnotes
The views outlined in this article are those of the authors and not necessarily those of Boehringer Ingelheim.
Abbreviations
- AUC
- area under time-concentration curve
- clogP
- calculated logP
- Cmax
- maximum concentration in human plasma
- DDI
- drug-drug interaction
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- [Im]
- total plasma concentration of metabolite as an inhibitor
- [Im]/[Ip]
- metabolite to parent ratio
- [Im]/Kim
- [I]/Ki ratio for the metabolite
- [Ip]
- total plasma concentration of parent as an inhibitor
- [Ip]/Kip
- [I]/Ki ratio for the parent drug
- Ki
- inhibition potency
- Kim
- inhibition potency of the metabolite
- Kip
- inhibition potency of the parent drug
- MBI
- mechanism-based inhibition
- P450
- cytochrome P450
- Received September 7, 2012.
- Accepted November 9, 2012.
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}