


Abstract
The activity of human liver microsomal cytochrome P450 1A2 (CYP1A2) is readily estimated by following the O-deethylation of [O-ethyl 14C]phenacetin (PODase). The basis of the assay is the quantitative measurement of [14C]acetaldehyde, remaining in the supernatant of assay incubates, after extraction of unmetabolized [O-ethyl 14C]phenacetin with charcoal. In the presence of native human liver microsomes (Km = 54 ± 27 μM;Vmax = 14 ± 2.3 nmol/hr/mg; mean ± SD; N = 3 different livers) and human B-lymphoblastoid cell microsomes containing cDNA-expressed CYP1A2 (Km = 46 μM;Vmax = 55 nmol/hr/nmol CYP), PODase activity conformed to monophasic Michaelis-Menten kinetics. Furthermore, PODase activity in a panel of microsomes prepared from a series of human livers was significantly correlated (r= 0.91; p < 0.001; N = 11) with CYP1A2-selective 7-ethoxyresorufin O-deethylase activity, and was markedly inhibited (≥ 92%) by furafylline (FURA, IC50 = 0.4 μM) and 7,8-benzoflavone (ANF, IC50 = 0.1 μM), two well known CYP1A2 inhibitors. Inhibitors selective for other forms of CYP (e.g. CYP3A, CYP2C, CYP2D6, CYP2E1) elicited a marginal effect (≤ 17% inhibition) at relatively high concentrations (≥ 10·Ki ). It is concluded that the inhibition of human liver microsomal CYP1A2 activity can be readily determined by using a charcoal-based radiometric method employing [O-ethyl 14C]phenacetin as substrate.
Members of the human liver microsomal CYP1 pool (EC1.14.14.1) are now known to play a central role in the oxidative metabolism of a large number of drugs (1). Therefore, given that the treatment of most diseases warrants the use of more than one therapeutic agent, the potential for a deleterious drug-drug interaction exists. This is especially evident when multiple drugs are metabolized by the same form(s) of CYP. One such example is CYP1A2 (P450PA, P450HLd), the most abundant CYP1A subfamily member in the human liver (1, 2), which has been shown to metabolize a large number of drugs (1, 3-6). At the same time, the enzyme is also known to be induced in individuals consuming charbroiled foods, in smokers, and in subjects receiving omeprazole (1, 7, 8). In addition, compounds such as fluvoxamine, ANF, and FURA have been shown to be particularly potent inhibitors of CYP1A2 (1, 9, 10).
From a drug development standpoint, it is now acknowledged that one can attempt to predict metabolism-based drug-drug interactions using human liver microsomes (11, 12). The data can often be used in the selection of the most appropriate “lead compound,” when inhibition of drug-metabolizing enzymes is considered a liability. Toward this end, a number of assays have been developed to selectively monitor CYP1A2 activity in human liver microsomes (13-15). However, many of these warrant the use of HPLC or are prone to fluorescence quenching (e.g. 7-ethoxyresorufin O-deethylase). One or both factors can negate the usefulness of these assays, particularly during the high-throughput screening of large numbers of structurally diverse compounds. In the present study, we briefly describe a relatively simple and rapid assay procedure for measuring CYP1A2-dependent monooxygenase activity in human liver microsomes using [O-ethyl 14C]phenacetin. The assay involves the radiometric measurement of [14C]acetaldehyde, after a single-step extraction procedure, which obviates the need for HPLC and is considered particularly amenable to automation.
Materials and Methods
Reagents
Activated charcoal (untreated powder, 100–400 mesh; cat. no. C-5260; lot no. 54H0279) was obtained from the Sigma Chemical Co. (St. Louis, MO). The charcoal was freshly prepared as a suspension (0.8% w/v) in 0.1 M potassium phosphate buffer (pH 7.4) and was stirred continuously while in use. [1-14C]Ethyl iodide (50 mCi/mmol) and [1, 2-14C]acetaldehyde (110 mCi/mmol) were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). All other reagents were purchased commercially at the best obtainable grade. Human B-lymphoblastoid microsomes containing cDNA-expressed CYP protein were purchased from Gentest Corp. (Woburn, MA). Human liver microsomes (N = 11 subjects) were prepared from organ donor tissue and were characterized as previously described (3, 4, 16, 17).
Synthesis of [O-Ethyl 14C]Phenacetin
[1-14C]Ethyl iodide (5.3 mCi, 0.1 mmol) was transferred to methyl ethyl ketone (0.2 mL) under vacuum. Acetaminophen (23 mg, 0.15 mmol) and powdered anhydrous potassium carbonate (28 mg, 0.2 mmol) was added in a mini reflux apparatus. [1-14C]Ethyl iodide in methyl ethyl ketone (0.2 mL) was then added to the mini reflux apparatus and rinsed with more methyl ethyl ketone (0.3 mL). The suspension was heated to reflux for 8 hours. The reaction mixture was then diluted with water (2 mL) and extracted three times with methylene chloride (1 mL). The organic layers were combined and washed with 1 N sodium hydroxide (2 mL), brine (2 mL), and water (2 mL) to remove any unreacted starting materials. The clear methylene chloride solution was dried over magnesium sulfate, filtered, and concentrated to dryness in vacuo. The residue (4.17 mCi, 79%) was dissolved in ethanol (2.5 mL), chilled, and filtered once again, and the radiochemical purity of the final product (> 99.5%) was determined by radio-thin layer chromatography on silica gel (Silica Gel 60, 5 × 20 cm, 250 μm thick; EM Separations Technology, Gibbstown, NJ), by using a Radiomatic Model RA radio-thin layer chromatography scanner (Packard Instrument Co., Meriden, CT). Methylene chloride: ethyl acetate (20:80, v/v) was employed as the solvent system, and the final radiolabeled product and authentic phenacetin standard were similarly eluted (RF = 0.54). Mass spectrometry data confirmed this material to be [O-ethyl 14C]phenacetin, since the spectra and pseudo molecular ion (m/z at 180 amu for [M + H]+) were identical to that of authentic phenacetin standard. The m/z 180 amu: m/z 182 amu abundance ratio indicated that approximately 88% of the sample was labeled with a single carbon-14 atom. This corresponded to a specific activity of about 55 mCi/mmol, in reasonable agreement with the manufacturers specifications for the [1-14C]ethyl iodide. The final working solution of [O-ethyl 14C]phenacetin was 33.4 mM (1.67 mCi/mL ethanol; 50 mCi/mmol) and was used for all subsequent experiments. In some cases, unlabeled phenacetin was added to the solution to obtain a lower specific activity.
PODase Activity
Incubations were carried out in 2.0-mL polypropylene microcentrifuge tubes (ClickSealTM microtubes, Research Products International Corp., Mount Prospect, IL) in a Dubnoff shaking water bath at 37 °C (under air). Briefly, the final assay volume was 0.5 mL and consisted of the following at the indicated final concentration: 0.1 M potassium phosphate buffer (pH 7.4), EDTA (0.1 mM), MgCl2 (3.0 mM), microsomal protein (0.25–1.0 mg/mL), and [O-ethyl14C]phenacetin (1–100 μM, 50 mCi/mmol; 200 μM, 17.2 mCi/mmol; added in 2 μL of ethanol, 0.4%, v/v). After a 3-min preincubation period, the reaction was initiated by addition of a NADPH-generating system containing NADP+ (4.0 mM), D-glucose 6-phosphate (10 mM) and D-glucose 6-phosphate dehydrogenase (Sigma Type VII, from baker’s yeast, 2.0 U/mL). Incubations with human B-lymphoblastoid microsomes containing cDNA-expressed CYP1A2 (133 pmol CYP1A2/mg protein) were carried out as described for native human liver microsomes, except that the reactions were started by addition of rapidly thawed microsomal protein (final assay concentration was 0.25 mg/mL; 33 pmol CYP1A2/mL) (3). No metabolism was detected in microsomes prepared from B-lymphoblastoid cells devoid of CYP.
At the required time, the reaction was terminated with 50 μL of 1 M HCl, and the samples were placed on ice for at least 5 min. After thorough vortexing, the charcoal suspension (0.45 mL) was added and the sample was vortexed once again. All samples were immediately centrifuged at 4°C (16,000 g, 10 min). Thereafter, 0.5 mL of supernatant was mixed with 15 mL of Ultima Gold-XR scintillation cocktail (Packard Instrument Co., Meriden, CT), and the samples were analyzed for [14C]acetaldehyde by radioassay. The majority of the added [14C]phenacetin (> 99%) was bound to charcoal.
Although the further oxidation of acetaldehyde, to acetic acid and/or CO2, was not addressed, the recovery of total radioactivity in the supernatant was 90 ± 4% (mean ± SD of over 30 experiments performed during a 4-month period) after incubation of [1, 2-14C]acetaldehyde (0.05 μM to 50 μM) with human liver microsomes and a NADPH-generating system (≤ 40 min, 37 °C). In addition, no metabolism of [O-ethyl14C]phenacetin was detected in the absence of a NADPH-generating system.
The quantity of acetaldehyde formed was calculated from the net dpm observed (dpm in supernatant at time t minus dpm in supernatant at time zero), corrected to the total volume of the supernatant (0.5 mL aliquot/1.0 mL supernatant), and corrected for daily acetaldehyde recovery. The total net dpm’s were then converted to nanomoles of product from the specific activity of the substrate (typically 111,000 dpm/nmol) (3, 16). Estimates of apparentKm and Vmaxwere obtained by using PCNONLIN (3). The kinetic parameters were determined under linear reaction conditions (≤ 40 min; 0.25–1.0 mg protein/mL) over a substrate concentration range of 1–200 μM.
Inhibition Studies.
The inhibition of PODase activity was investigated using a number of purported CYP form selective inhibitors. Except for 4-methylpyrazole and diethyldithiocarbamate (dissolved in water), all inhibitors were dissolved in ethanol. The final volume of ethanol in the assay did not exceed 1.0% (v/v). In all cases, the concentrations chosen exceeded (≥ 10-fold) apparent Ki and/orKm (3, 4, 9, 11, 14).
Results and Discussion
The O-deethylation of phenacetin has long been established as a method for measuring CYP1A2 (P450PA, P450HLd) activity in native human liver microsomes (9, 14, 15, 18). However, in an attempt to circumvent the need for HPLC, a method was devised for the synthesis and purification of [O-ethyl14C]phenacetin. The availability of [O-ethyl 14C]phenacetin, and measurement of [14C]acetaldehyde formed upon incubation, allows one to determine CYP1A2-dependent PODase activity by using a rapid and relatively simple single-step extraction procedure.
In our hands, at relatively low concentrations of [O-ethyl14C]phenacetin (≤ 0.1 mM), native human liver microsomal PODase activity conformed to monophasic kinetics (data not shown) and was characterized by a mean (± SD, N = 3 livers) apparent Km ,Vmax, andVmax/Km ratio of 54 ± 27 μM, 14 ± 2.3 nmol/hr/mg, and 0.30 ± 0.16 mL/hr/mg, respectively. When employing native human liver microsomes, low concentrations of phenacetin ensure that only “lowKm PODase activity” is studied, which has been shown to be largely mediated by CYP1A2 (14, 15, 18-21). However, the apparent Km reported by various investigators has varied considerably (6.0–60 μM) (14, 15,18-22). Given the biphasicity of PODase activity in native human liver microsomes, variability in Km may partly result from differences in the concentration range of phenacetin used (e.g. 2–200 μM vs. 5–2000 μM). Likewise, the apparent Vmax characterizing lowKm (Km1 ) PODase activity has been reported to range from 9–32 nmol/hr/mg (10,18, 21, 22).
Additional kinetic studies were performed with human B-lymphoblastoid cell microsomes containing cDNA-expressed CYP1A2.2 In this instance, PODase activity also conformed to monophasic kinetics and was characterized by an apparent Km (± SE, PCNONLIN) andVmax (± SE, PCNONLIN) of 46 (± 5.6) μM and 55 (± 3.2) nmol/hr/nmol CYP, respectively (data not shown). The apparent Km was similar to that obtained with CYP1A2 purified from native human liver tissue or CYP1A2 expressed heterologously in COS-7 and V79 cells (18-20).
The CYP selectivity of the charcoal-based PODase assay was further evaluated with native human liver microsomes, by employing correlation analysis and CYP form selective chemical inhibitors. As expected from the work of others (2), PODase activity was significantly correlated (r = 0.91; p < 0.001;N = 11)3 with CYP1A2-selective 7-ethoxyresorufin O-deethylase activity (data not shown). The correlation with activities selective for other CYP forms (e.g. erythromycin N-demethylase, coumarin hydroxylase, (S)-(+)-mephenytoin 4′-hydroxylase, andN,N-dimethyl-nitrosamine N-demethylase) was relatively weak (≤ 0.38). On the other hand, the correlation with CYP2C9-selective tolbutamide methyl hydroxylase (r = 0.64; p < 0.05; N = 10) and CYP2D6-selective [O-methyl14C]dextromethorphan O-demethylase (r = 0.74; p < 0.01; N= 11) activity is considered fortuitous, because the appropriate CYP form selective inhibitors were relatively ineffective at inhibiting PODase activity (fig. 1).
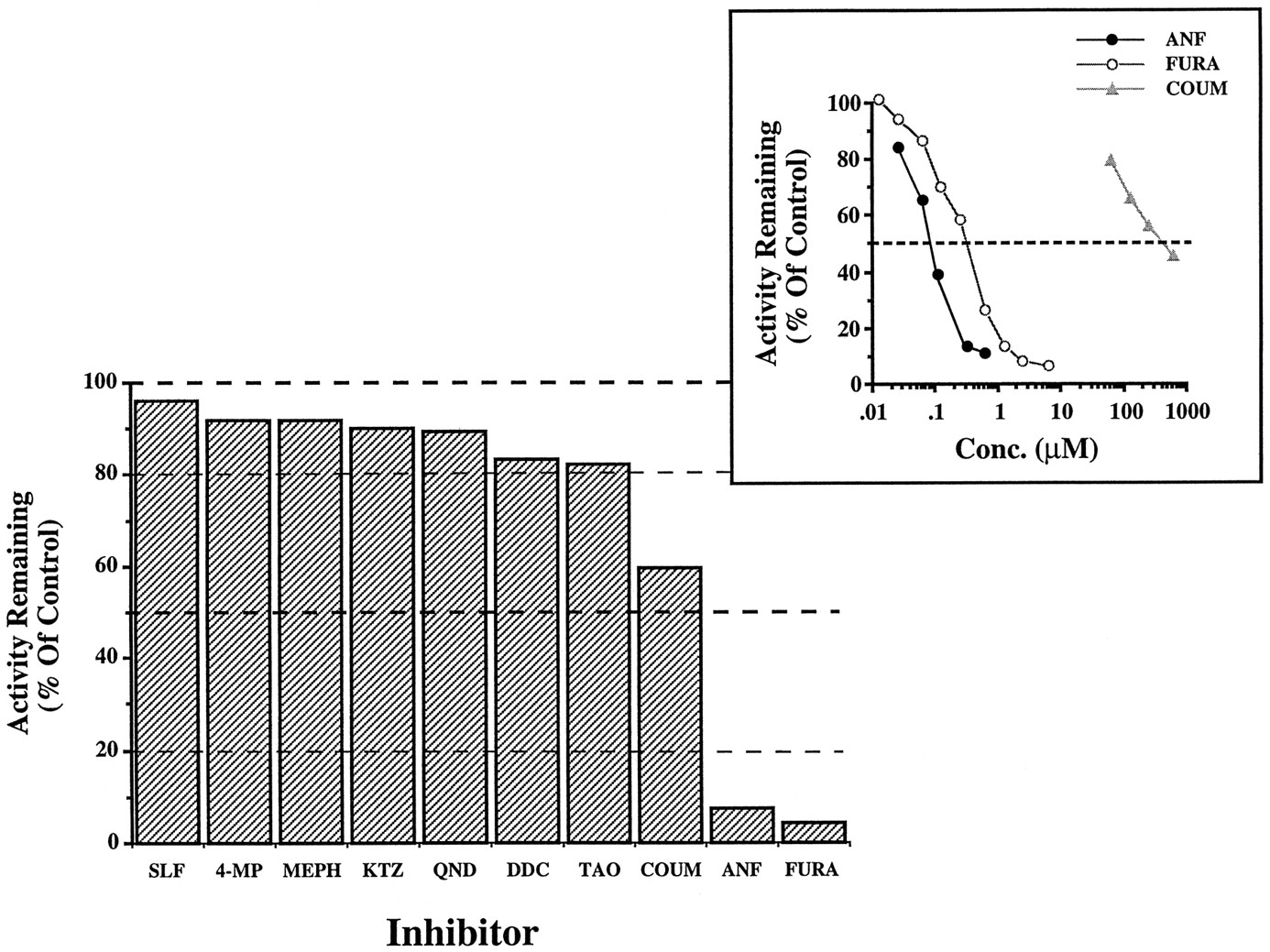
Effect of various CYP inhibitors on the O-deethylation of [O-ethyl 14C]phenacetin in native human liver microsomes.
Data are presented as the mean of two different livers (subjects H.G.D. and G.F.E.), which differed by ≤10%, and are expressed as percentage of control (ethanol or water alone, ≤1.0% v/v). Incubations were performed as described in Materials and Methods. MEPH (0.5 mM), COUM (0.2 mM), QND (10 μM), SLF (10 μM), KTZ (2.0 μM), 4-MP (50 μM), and ANF (1.0 μM) are known inhibitors of CYP2C19, CYP2A6, CYP2D6, CYP2C9, CYP3A, CYP2E1, and CYP1A2, respectively. Similarly, TAO (0.2 mM), FURA (10 μM), and diethyldithiocarbamate (30 μM) are mechanism-based inhibitors of CYP3A, CYP1A2, and CYP2E1/2A6, respectively. The final concentration of [O-ethyl14C]phenacetin was 40 μM (∼Km), and the final concentration of each inhibitor (≥ 10·Ki) was based on published Ki values. Inset: Inhibition plot for ANF (closed circles; IC50 = 0.1 μM) and FURA (open circles; IC50 = 0.4 μM) with native human liver microsomes. Data showing the effect of COUM on PODase activity (phenacetin concentration ≈ apparent Km), catalyzed by B-lymphoblastoid cell microsomes containing cDNA-expressed CYP1A2 (closed triangles; IC50 = 0.4 mM), are also presented. SLF, sulfaphenazole; 4-MP, 4-methylpyrazole; MEPH, (S)-(+)-mephenytoin; KTZ, ketoconazole; QND, quinidine; DDC, diethyldithiocarbamate; TAO, troleandomycin.
At a phenacetin concentration approaching apparentKm , PODase activity was markedly inhibited (≥ 92%) by FURA (IC50 = 0.4 μM) or ANF (IC50 = 0.1 μM) (fig. 1), two well-known inhibitors of CYP1A2 (9, 18, 22-24). In fact, similar IC50 values have been reported by other investigators (7, 9, 14, 15, 18, 23, 24). By comparison to ANF and FURA, reversible/competitive and/or mechanism-based chemical inhibitors of other CYP forms elicited a marginal effect on PODase activity (≤ 17% inhibition) (fig. 1), and it should be pointed out that the concentrations employed were far in excess (≥ 10·Ki ) of their reportedKi values. Nevertheless, inhibition (∼41%) in the presence of COUM (0.2 mM), a selective CYP2A6 substrate (11, 14, 16, 25), may indicate that CYP2A6 also contributes to PODase activity in human liver microsomes (fig. 1). In light of the fact that the correlation of PODase with coumarin hydroxylase activity was relatively weak (r = 0.24; N = 11) in our bank of microsomes (data not shown) and since both ANF and FURA (≤ 10 μM) have been reported to be relatively weak inhibitors of CYP2A6 (9, 14, 24), CYP2A6-mediated PODase activity appears unlikely.4 A second possibility is that COUM may inhibit CYP1A2, albeit with a relatively high IC50 (≥ 0.2 mM), without being a substrate for the enzyme. In concert, COUM was shown to inhibit PODase activity (IC50 = 0.4 mM; phenacetin concentration ≈ apparent Km ) catalyzed by B-lymphoblastoid cell microsomes containing cDNA-expressed CYP1A2 (fig. 1), while cDNA-expressed or native CYP1A2 has been shown not to metabolize COUM (0.1–1.0 mM) (25).
Based on the results of this study, it is concluded that human liver microsomal CYP1A2 activity, which represents a “lowKm ” PODase (Km ≤ 60 μM), can be readily measured by using a charcoal-based radiometric method employing [O-ethyl 14C]phenacetin as substrate. The method, which obviates the need for chromatography, an elaborate extraction procedure, and an internal standard, is considered amenable to automation and will allow potential CYP1A2 inhibitors and/or cosubstrates to be rapidly evaluated in a high-throughput screening format. In addition, the method permits rapid-prospective- evaluation of kinetic parameters such as apparentKm , which is essential when performing inhibition screens with large numbers of compounds based on IC50 values.
Footnotes
-
Send reprint requests to: A. David Rodrigues, Ph.D., Drug Metabolism I, Merck Research Laboratories, Sumneytown Pike, P. O. Box 4, WP26-A 2044, West Point, PA 19486-0004.
-
↵2 Nominal molar ratio of NADPH-CYP reductase (∼15 pmol/mg) to CYP1A2 (133 pmol CYP/mg) is 1:9.
-
↵3 At a final phenacetin concentration of 100 μM, PODase activity varied from 2.2-21.2 nmol/hr/mg (8.4 ± 5.6 nmol/hr/mg; mean ± SD; N = 11 livers).
-
↵4 No PODase activity was detected when [O-ethyl 14C]phenacetin (40 μM; ∼ Km) was incubated with human B-lymphoblastoid microsomes (Gentest Corp.) containing cDNA-expressed CYP2A6 (A. D. Rodrigues, unpublished results).
- Abbreviations used are::
- CYP
- cytochrome P450
- PODase
- [O-ethyl 14C]phenacetinO-deethylase
- ANF
- 7, 8-benzoflavone
- FURA
- furafylline
- COUM
- coumarin
- IC50
- concentration of drug required to inhibit activity by 50%
- Ki
- apparent inhibition constant (dissociation constant of the enzyme-inhibitor, or EI, complex)
- Km
- apparent Michaelis constant
- Vmax
- apparent maximal initial reaction velocity
- Received February 24, 1997.
- Accepted May 1, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}