


Abstract
The enzyme kinetics of the metabolism of ezlopitant in liver microsomes from various species have been determined. The rank order of the species with regard to the in vitro intrinsic clearance of ezlopitant was monkey ≫ guinea pig > rat ≫ dog > human. CJ-12,764, a benzyl alcohol analog, was observed as a major metabolite, and a dehydrogenated metabolite (CJ-12,458) was equally important in human liver microsomes. Scale-up of the liver microsomal intrinsic clearance data and correcting for both serum protein binding and nonspecific microsomal binding yielded predicted hepatic clearance values that showed a good correlation with in vivo systemic blood clearance values. Including microsomal binding was necessary to achieve agreement between hepatic clearance values predicted from in vitro data and systemic clearance values measured in vivo. Cytochrome P450 (CYP) 3A4, 3A5, and 2D6 demonstrated the ability to metabolize ezlopitant to CJ-12,458 and CJ-12,764. However, in liver microsomes, the CYP3A isoforms appear to play a substantially more important role in the metabolism of ezlopitant than CYP2D6, as assessed through the use of CYP-specific inhibitors, correlation to isoform-specific marker substrate activities, and appropriate scale-up of enzyme kinetic data generated in microsomes containing individual heterologously expressed recombinant CYP isoforms. The apparent predominance of CYP3A over CYP2D6 is consistent with observations of the pharmacokinetics of ezlopitant in humans in vivo.
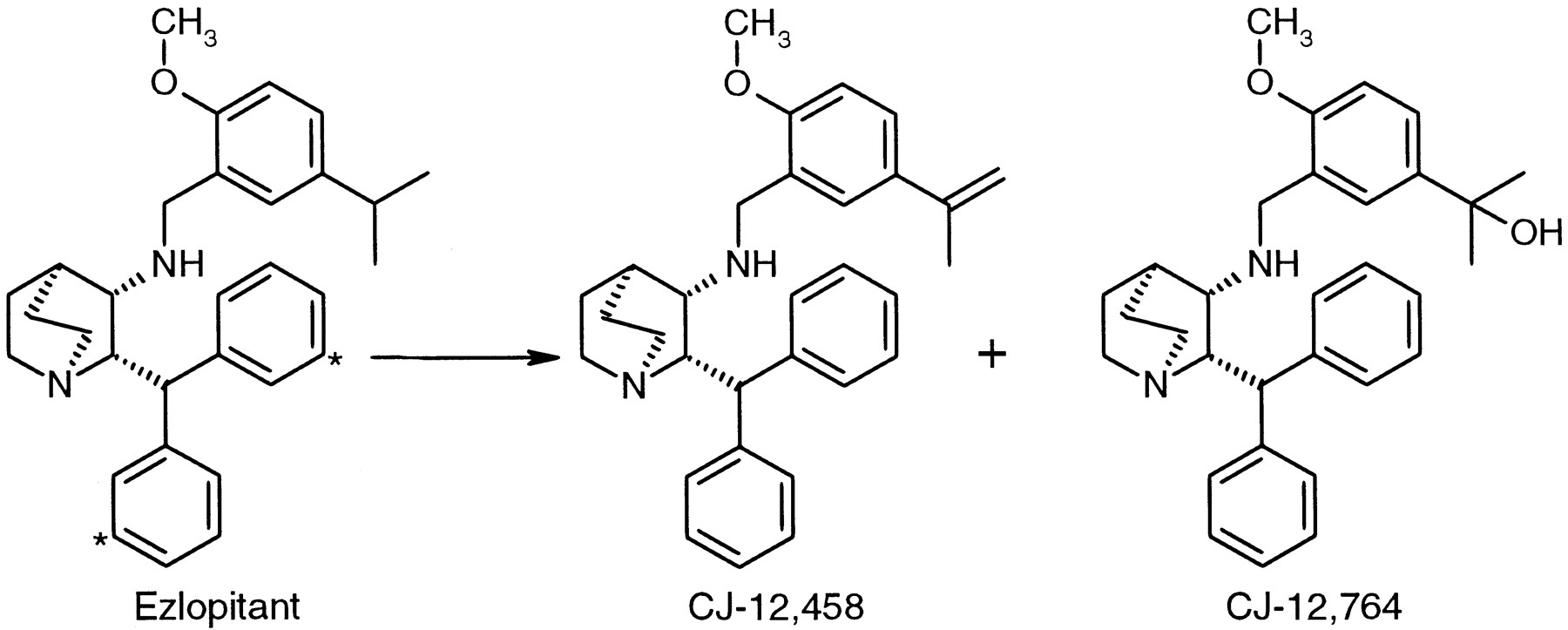
Ezlopitant, (2S,3S,4S)-2-diphenylmethyl-3-[(5-isopropyl-2-methoxybenzylamino]-1-azabicyclo[2.2.2]octane (Fig. 1), represents a novel, potent, nonpeptidic antagonist of the Substance P receptor similar in structure to the well characterized analog CP-96,345 (Snider et al., 1991). It is believed that an antagonist of the human Substance P receptor will be of therapeutic utility in disorders in which Substance P is believed to play a role, such as inflammatory diseases, depression, pain, and emesis (Kramer et al., 1998; Hesketh et al., 1999). The pharmacokinetics of ezlopitant and two pharmacologically active metabolites have been measured in several preclinical species of pharmacological and toxicological relevance (Reed-Hagen et al., 2000). In rat, guinea pig, dog, and monkey, ezlopitant is characterized by a high systemic clearance and low oral bioavailability, presumably due to high first-pass hepatic extraction. However, initial data suggest that the clearance of this compound in humans is substantially lower than in preclinical species (unpublished observations). Thus, despite the poor pharmacokinetic characteristics in animals, ezlopitant has pharmacokinetics in humans amenable to commercial utility (i.e., good oral exposure, half-life suitable for once-per-day administration).

Structures of ezlopitant and metabolites CJ-12,458 and CJ-12,764.
The asterisks denote the sites of tritium incorporation.
Ezlopitant is converted primarily to two pharmacologically active metabolites, both of which have been observed in the systemic circulation of preclinical species (Reed-Hagen et al., 2000) as well as in humans (unpublished observations). CJ-12,458, an alkene metabolite, and CJ-12,764, a benzyl alcohol metabolite (Fig. 1), are compounds with in vitro Substance P receptor antagonist activities in the same range as the parent compound. CJ-12,764 is the major metabolite in preclinical species, with less CJ-12,458 observed, although the two metabolites are observed in nearly equal abundance in human circulation. These metabolites can potentially contribute to the pharmacological activity observed after administration of the parent compound.
Hepatic microsomes contain many drug-metabolizing enzymes, the most notable being the heme-thiolate-containing cytochrome P450s (CYP)1 (Ortiz de Montellano, 1996). This large family of enzymes catalyze a wide variety of reactions of xenobiotic metabolism as well as metabolism of endogenous compounds such as lipids and steroids. Because CJ-12,458 and CJ-12,764 represent metabolites of ezlopitant that arise from oxidative processes, the metabolism of ezlopitant to these two compounds was investigated in hepatic microsomes in vitro.
The use of enzyme kinetic data of drug metabolism reactions measured in hepatic microsomes in attempting to quantitatively describe in vivo pharmacokinetics has been an area of exploration for approximately 20 years, with the first demonstration of the potential utility of this approach done using rat liver microsomes (Rane et al., 1977; reviewed in Houston, 1994; Iwatsubo et al., 1997). The use of such data can be a powerful tool in the drug discovery and development processes because human and human-derived materials can be used to predict the clearance and hepatic extraction of drugs in humans before undertaking the large costs associated with administering new chemical entities in the clinic. An early demonstration of a correlation between in vitro enzyme kinetic data and pharmacokinetic data, as applied to a human system, was described for felodipine (Baarnhielm et al., 1986).
In addition to using human hepatic microsomes in the quantitative prediction of in vivo human pharmacokinetic data, this in vitro approach can be used in examining the roles of individual drug-metabolizing enzymes, most importantly CYP enzymes, in the overall metabolism of a given compound as well as specific biotransformation reactions. Measurement of the contribution of individual CYP isoforms involved in the metabolism of drugs can have important implications for interindividual variability because some CYP isoforms are expressed in widely variable amounts due to environmental or genetic factors (reviewed in Wrighton and Stevens, 1992). Furthermore, such knowledge can provide a better understanding of the potential for pharmacokinetic drug-drug interactions. Therefore, experimental results are also included that address the roles of human CYP isoforms in the metabolism of ezlopitant to the major metabolites, CJ-12,458 and CJ-12,764. The impact of the findings as they relate to expected pharmacokinetic behavior in humans and the potential impact of CYP2D6 metabolizer phenotype are discussed.
Thus, the primary objectives of these experiments are to: 1) attempt to make a cross-species comparison of the enzyme kinetics of liver microsomal metabolism of ezlopitant and to compare the enzyme kinetics of formation of the active metabolites CJ-12,458 and CJ-12,764 in the development of an in vitro-in vivo correlation, and 2) attempt to determine the contribution of various isoforms of human CYP to both the overall metabolism of ezlopitant and the specific biotransformation pathways giving rise to CJ-12,458 and CJ-12,764.
Experimental Procedures
Materials.
Ezlopitant dihydrochloride and CJ-12,764 dibenzenesulfonate were obtained from the Process Research and Development Department, Pfizer Central Research, Groton, CT. CJ-12,458 was obtained from the Medicinal Chemistry Department, Pfizer, Nagoya, Japan. [3H]Ezlopitant was prepared by catalytic reductive dehalogenation of a dibromo analog by Tokai Research Laboratories, Ibaraki, Japan. The specific activity of this material was 20.0 Ci/mmol (radiochemical purity > 99%), and the positions of tritium incorporation were the meta positions of the two phenyl rings of the diphenylmethyl moiety. [3H]CJ-12,764 was biosynthesized as described by Reed-Hagen et al. (2000). Hepatic microsomes were prepared from Sprague-Dawley rat (Charles River Labs, Wilmington, MA), Hartley guinea pig (Charles River Labs), beagle dog (Marshall Farms, North Rose, NY), cynomolgus monkey (BRF/Charles River Labs, Houston, TX), and human livers (IIAM, Exton, PA) using standard procedures. Protein concentrations were determined using the BCA assay method (Pierce Chemical Co., Rockland, IL), total CYP was measured by the method ofOmura and Sato (1964), and microsomes were characterized using CYP-specific marker activities by the Drug Metabolism Department, Pfizer Central Research, Groton, CT. Microsomes used in the following experiments represent combinations of preparations from the following numbers of individual animals: 4 (rats), 4 (guinea pigs), 4 (dogs), and 3 (monkeys). Pooled human liver microsomes represent an equal mixture from 10 individual donors. Sf9 cell microsomes containing recombinant heterologously expressed CYP isoforms and reductase were obtained from the Molecular Sciences Department, Pfizer Central Research, and microsomes from human β-lymphoblastoid cells were obtained from Gentest Corp. (Woburn, MA).
Incubation Conditions.
Ezlopitant, mixed with [3H]ezlopitant (1.2 μCi) as a radiotracer, was incubated in the presence of liver microsomes at various protein concentrations (0.05–4.0 mg/ml), NADP (1.3 mM), glucose 6-phosphate (3.3 mM), MgCl2(3.3 mM), and glucose 6-phosphate dehydrogenase (5 U) in a total volume of 0.2 ml of 25 mM potassium phosphate, pH 7.5. In preliminary experiments, time courses of product formation were measured to establish conditions of reaction linearity. The following microsomal protein concentrations were used in substrate saturation experiments: rat, 0.5 mg/ml; guinea pig, 0.2 mg/ml; dog, 1.0 mg/ml; monkey, 0.05 mg/ml; and human, 4.0 mg/ml. Incubations were commenced by addition of the cofactors, incubated for 10 min at 37°C in a shaking water bath open to the atmosphere, followed by termination of the reactions by addition of two volumes of CH3CN. The precipitated materials were removed by centrifugation, and the supernatant was analyzed by radiometric HPLC as described below.
Inhibition Studies.
Inhibition of ezlopitant metabolism by triacetyloleandomycin (TAO) (a CYP3A inactivator) was assessed by comparison of the activity of an individual lot of human liver microsomes versus the same lot that had been preincubated 30 min with TAO (50 μM) and an NADPH generating system, followed by washing the microsomes of excess inhibitor. Quinidine inhibition (CYP2D6) was assessed by examining the metabolism of ezlopitant by pooled human liver microsomes in incubations containing 0, 1.0, 3.0, 10, 30, and 100 μM quinidine. Ketoconazole inhibition (CYP3A inhibitor) was assessed using inhibitor concentrations ranging from 0.05 to 10 μM and an ezlopitant concentration of 10 μM. Sulfaphenazole inhibition (CYP2C9) was assessed by examining the metabolism of ezlopitant by an individual lot of human liver microsomes containing a high CYP2C9-tolbutamide hydroxylase activity in incubations containing 0, 1.0, 3.0, 10, 30, and 100 μM sulfaphenazole. Incubations (total volume = 0.2 ml) were conducted at an ezlopitant concentration of 10 μM (containing 1.2 μCi [3H]ezlopitant) and cofactors at concentrations as described above. Reactions were incubated and terminated as above, and analyzed by HPLC as described below.
Correlation Studies.
Ezlopitant (mixed with 1.2 μCi [3H]ezlopitant as a radiotracer) at substrate concentrations of 0.3, 10, and 100 μM (hence, specific activities of 20, 0.6, and 0.06 μCi/nmol) was incubated with cofactors as described above and human liver microsomal protein (2.5–4.6 mg/ml) from 11 individual donors in duplicate determinations. Reactions were incubated and terminated as above, and analyzed by HPLC as described below.
Metabolism of Ezlopitant by Heterologously Expressed CYP Isoforms.
Ezlopitant, at a substrate concentration of 50 μM, was incubated in the presence of microsomes from cells containing heterologously expressed CYP isoforms coexpressed with CYP/NADPH reductase. CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5 were expressed in a baculovirus system, and CYP1A1, CYP1A2, CYP2A6, and CYP2E1 were expressed in human β-lymphoblastoid cells (Gentest). Microsomes were used at protein concentrations ranging from 0.3 to 6.2 mg/ml, commensurate with the relative activities reported for marker substrate activities for each of the preparations. Reactions were incubated and terminated as above, and analyzed by HPLC as described below. Substrate saturation experiments using CYP2D6, CYP3A4, and CYP3A5 were conducted at substrate concentrations ranging from 0.1 to 50 μM using unlabeled ezlopitant, cofactor, and microsomal protein concentrations as above. Analysis of the CJ-12,458 and CJ-12,764 products was accomplished using HPLC-mass spectrometry (MS) as described below.
Analysis of [3H]Ezlopitant Incubation Mixtures by Radiometric HPLC.
Incubation mixtures were analyzed on an HPLC system consisting of an LDC Analytical (Riviera Beach, FL) 4100 ConstaMetric gradient pump, 3200 SpectroMonitor variable wavelength ultraviolet detector, membrane degasser, Perkin-Elmer (Norwalk, CT) ISS 200 injector, and Inus Systems (Tampa, FL) β-RAM radioactivity detector with a 500-μl mixing cell. The supernatants from terminated incubation mixtures were injected (120 μl) onto a Waters Novapak C-18 column (3.6 × 150 mm) pre-equilibrated in 20 mM CH3COOH/CH3CN (87.5:12.5) containing 0.1% triethylamine (TEA) at a flow rate of 1.0 ml/min. Initial conditions were maintained for 5 min, followed by a linear gradient to 20 mM CH3COOH/CH3CN (20:80) with 0.1% TEA at 30 min. The final conditions were held for 10 min, after which the column was re-equilibrated for 12 min. The eluent was monitored for radioactivity using β-Blend scintillation fluid at 4.0 ml/min. Ezlopitant, CJ-12,458, and CJ-12,764 eluted at 35.8, 34.5, and 25.5 min, respectively.
Analysis of CJ-12,764 and CJ-12,458 by HPLC-MS.
In some cases, determination of concentrations of CJ-12,458 and CJ-12,764 was accomplished by liquid extraction followed by HPLC-MS analysis. Incubation mixtures were terminated with vortex mixing with 3 ml of methyl tertiary butyl ether and 100 ng of internal standard CJ-11,957, in 0.1 ml of water was added. (CJ-11,957 is identical with ezlopitant, with the exception that the isopropyl group in the latter is replaced with an ethyl group.) The mixtures were vortex mixed for 1 min, followed by separation of the layers by spinning at 3000 rpm at ambient temperature in a Jouan model CT422 swinging bucket tabletop centrifuge. The samples were placed in a dry-ice acetone bath to effect freezing of the aqueous layer, and the organic layer was decanted into a fresh silylated glass test tube. The solvent was removed under N2 at 30°C in a Zymark TurboVap, and the residue was reconstituted in 0.1 ml of HPLC mobile phase.
The HPLC-MS system consisted of a Hewlett-Packard 1100 HPLC system coupled to a PE Sciex API 100 single quadrupole mass spectrometer containing an atmospheric pressure chemical ionization interface. The column was a Waters Symmetry C18 (3.9 × 150 mm; 5-μm particle size packing), and the initial mobile phase consisted of 45.5% CH3CN in 20 mM acetic acid, adjusted to pH 4 with NH4OH at a flow rate of 0.8 ml/min. Samples (75 μl) were injected, and the initial mobile phase composition was maintained for 2 min, after which a linear gradient was applied, resulting in 95% CH3CN at 6 min.
The entire flow was introduced into the atmospheric pressure chemical ionization source operated in the positive ion mode. The orifice voltage was 45 V, and the nebulizer temperature was set at 500°C. For each analyte, protonated molecular ions were followed (m/z 471.2 for CJ-12,764;m/z 453.2 for CJ-12,458;m/z 455.2 for CJ-11,974; andm/z 441.0 for CJ-11,957 internal standard) with a dwell time for each ion of 150 ms. The retention times were 1.8 min for CJ-12,764, 5.2 min for CJ-12,458, 5.6 min for CJ-11,974, and 5.0 min for CJ-11,957 internal standard. Quantitation was accomplished by extrapolation from a standard curve with linear dynamic range from 0.1 to 100 ng/ml (1/x weighting).
Metabolite Profile of [3H]CJ-12,764 in Liver Microsomes.
CJ-12,764 (0.3 μM) with biosynthesized [3H]CJ-12,764 as a radiotracer (1.2 μCi; final specific activity = 20 μCi/nmol) was incubated with liver microsomes from rat, guinea pig, dog, monkey, and human at a protein concentration of 4.0 mg/ml and cofactors under conditions as described above. Aliquots (0.2 ml) were removed at time points of 5 and 30 min and processed as described above.
Results
Comparison of Liver Microsomal Metabolite Profiles Across Species.
HPLC radiochromatograms of liver microsomal incubations of [3H]ezlopitant are shown in Fig.2, A–E. Both CJ-12,764 and CJ-12,458 were formed in liver microsomes from all five species. CJ-12,764 was the predominant metabolite in rat, guinea pig, dog, and monkey, whereas CJ-12,764 and CJ-12,458 were formed to similar extents in human liver microsomes. In addition to these two metabolites, two additional metabolites of unknown structure were observed: one in rat (retention time = 32 min; referred to as RRT = 0.900), and the other in guinea pig, monkey, and human (retention time = 22 min; referred to as RRT = 0.616). Examination of the metabolic profile of CJ-12,764 in liver microsomes was done with the primary intention of determining whether the dehydrogenated metabolite, CJ-12,458, arose via a simple dehydration of the benzylic alcohol under the incubation conditions used in the examination of the metabolic profile of the parent compound ezlopitant. In no case was CJ-12,458 observed in any of the incubations of CJ-12,764 (Fig. 3). Several other metabolites of CJ-12,764 of unknown structure were observed.

HPLC radiochromatograms of microsomal incubations of [3H]ezlopitant.
Individual chromatograms are from rat liver microsomes (panel A; 0.5 mg/ml; 0.33 μM CYP), guinea pig liver (panel B; 0.2 mg/ml; 0.14 μM CYP), dog liver (panel C; 1.0 mg/ml; 0.42 μM CYP), monkey liver (panel D; 0.05 mg/ml, 0.05 μM CYP), human liver (panel E; 4.0 mg/ml; 1.0 μM CYP), heterologously expressed CYP2D6 (panel F; 0.05 mg/ml; 0.6 nM CYP), and heterologously expressed CYP3A4 (panel G; 0.6 mg/ml; 0.028 μM CYP).

HPLC radiochomatogram of human liver microsomal incubation of [3H]CJ-12,764.
Substrate Saturation Experiments.
An example of a substrate saturation plot for the metabolism of ezlopitant in human liver microsomes is presented in Fig.4. Apparent enzyme kinetic constants for the overall metabolism of ezlopitant, formation of CJ-12,764, CJ-12,458, and other selected major metabolites are presented in Table1 for five species. Intrinsic clearance was determined by dividing Vmax byKMapp. The rank order of the species with respect to overall ezlopitant intrinsic clearance is monkey > guinea pig > rat > dog > human. Apparent Michaelis constants (KMapp) for all reactions in rat and guinea pig (except one) were below 10 μM. In both of these species, conversion of ezlopitant to CJ-12,764 occurred to a greater extent than conversion to CJ-12,458, by virtue of both a lowerKMapp value and higherVmax value. The intrinsic clearance (CL′int) of the metabolism of ezlopitant to CJ-12,764 accounted for approximately three-fourths of the CL′int of ezlopitant, whereas the conversion to CJ-12,458 was only about 10% of the total.

Substrate saturation plot of ezlopitant metabolism by human liver microsomes.
Data represent individually plotted triplicate determinations. ▪, CJ-12,458 formation; ●, CJ-12,764 formation.
Enzyme kinetic parameters of ezlopitant metabolism by rat, guinea pig, dog, monkey, and human liver microsomes
In dog microsomes, the data suggested the presence of two enzymes involved in the metabolism of ezlopitant, one lowKMapp-low Vmaxenzyme and one high KMapp-highVmax enzyme. However, limited data were obtained at substrate concentrations near the lowKMapp value (due to assay sensitivity limitations); thus the presence of the lowKmapp activity must be interpreted with caution. In dog, CJ-12,764 accounted for the majority of ezlopitant metabolism; both of the observed enzyme activities generated this metabolite. CJ-12,458 was formed to a much lesser extent, and a lowKMapp activity could not be observed under these assay conditions.
Ezlopitant had a high CL′int value in monkey liver microsomes, and a large portion of this value consisted of conversion to CJ-12,764 (65%). The portion of total ezlopitant intrinsic clearance consisting of conversion to CJ-12,458 was comparable with that observed in rat, guinea pig, and dog microsomes (10%).
The enzyme kinetics of ezlopitant metabolism in pooled human liver microsomes were distinctly different from those observed in the animal species. The KMapp values for formation of CJ-12,458 and CJ-12,764 were highest in this species, with values determined to be 80 and 89 μM, respectively (Table 1; Fig. 4). Conversion to CJ-12,458 and CJ-12,764 was approximately equal (CL′int values of 0.0041 and 0.0029 ml/min/mg of microsomal protein, respectively).
Inhibition Studies.
Pretreatment of human liver microsomes with TAO resulted in an 75% loss of activity toward metabolism of ezlopitant at a substrate concentration of 10 μM. Additionally, the specific conversion to CJ-12,458, CJ-12,764, and the unidentified metabolite RRT = 0.616 was inhibited by 79, 42, and 87%, respectively. In these microsomes, TAO inhibited testosterone 6β-hydroxylase, a marker activity for human CYP3A4, by 72% (D. J. Tweedie and R. Whalen, unpublished data). Ketoconazole inhibited the formation of CJ-12,458 and CJ-12,764 with IC50 values of 0.32 and 0.28 μM, respectively (Fig. 5).
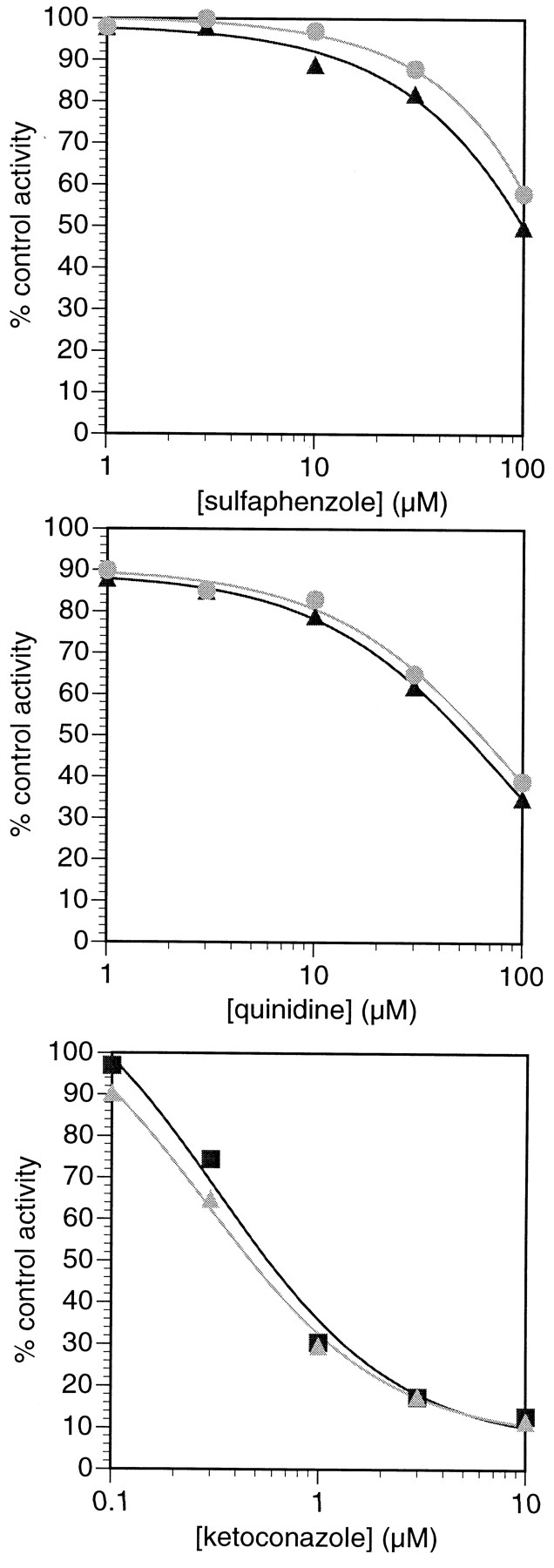
Inhibition of ezlopitant metabolism in human liver microsomes by CYP isoform specific inhibitors.
Each point represents the average of duplicate determinations. (▪), CJ-12,458 formation; (▴), CJ-12,764 formation. Average control activities in the sulfaphenazole inhibition experiments were 15 and 16 pmol/min/mg for CJ-12,458 and CJ-12,764 formation, respectively. Average control activities in the ketoconazole and quinidine experiments were 25 and 29 pmol/min/mg for CJ-12,458 and CJ-12,764 formation, respectively.
The effects of quinidine and sulfaphenazole on the metabolism of ezlopitant in human liver microsomes are shown in Fig. 5. At 1.0 μM quinidine, the metabolism of ezlopitant was inhibited by approximately 10%. Formation of CJ-12,458 and CJ-12,764 was inhibited by 10% as well, but no effect was observed on the formation of RRT = 0.616. Inhibition increased with increasing quinidine concentration, and IC50 values for inhibition of overall ezlopitant metabolism, as well as inhibition of formation of CJ-12,458, CJ-12,764, and RRT = 0.616, were in excess of 50 μM. Sulfaphenazole at concentrations ≤3 μM had no effect on ezlopitant metabolism. Inhibition increased as concentrations were raised and IC50 values for inhibition of ezlopitant metabolism and formation of CJ-12,458 and CJ-12,764 were ≥70 μM.
Correlation Studies.
Rates of ezlopitant metabolism in liver microsomes from 11 individual humans were measured at substrate concentrations of 0.3, 10, and 100 μM and plotted versus rates of metabolism of CYP-specific marker substrates (graphs not shown). No correlation was found between overall ezlopitant metabolism and activities specific to CYP1A2, CYP2C9, CYP2C19, or CYP2D6. Some correlations were found between ezlopitant metabolism and CYP3A-catalyzed testosterone 6β-hydroxylase activity. At a low ezlopitant substrate concentration, a correlation was only found to the formation of CJ-12,458, whereas at the high concentration (100 μM) correlations were found between CYP3A activity and the formation of both CJ-12,458 and CJ-12,764. A listing of correlation coefficients is in Table 2.
Summary of correlation data between ezlopitant metabolism and cytochrome P450 isoform specific marker substrate activities2-a
Metabolism of Ezlopitant by Heterologously Expressed P450s.
At a substrate concentration of 10 μM, ezlopitant was metabolized by microsomes containing CYP2D6, CYP3A4, and CYP3A5, but not by microsomes containing CYP1A1, CYP1A2, CYP2A6, CYP2C9, CYP2C19, or CYP2E1. The only metabolites observed in these incubations were CJ-12,458 and CJ-12,764. CYP2D6 and CYP3A4 formed CJ-12,458 and CJ-12,764 at similar intrinsic clearance values (Vm/KMapp), whereas CYP3A5 favored formation of CJ-12,764 over CJ-12,458 by 3-fold.
Substrate saturation curves for formation of CJ-12,458 and CJ-12,764 by CYP2D6, CYP3A4, and CYP3A5 microsomes are shown in Fig.6, and apparent enzyme kinetic constants are listed in Table 3. The CYP2D6 microsomes had lower KMapp values than CYP3A isoforms (0.4–0.6 μM versus 5.6–11 μM). Expression levels of CYP2D6 and CYP3A4 in this system are not reflective of expression levels in human microsomes. Correction ofVmax values to reflect the range of actual activities of CYP2D6 and CYP3A in the individual preparations of human liver microsomes used in these experiments yielded estimated ranges of 0.1 to 8.0 pmol/min/mg of protein for CYP2D6 and 450 to 2500 pmol/min/mg of protein for CYP3A. Thus, for an estimated 20-fold difference in KM values for CYP2D6 versus CYP3A4-catalyzed metabolism of ezlopitant (0.5 μM versus 10 μM), the “highest” estimate of partial intrinsic clearance for CYP2D6 would still be only about one-third that of the “lowest” estimate of partial intrinsic clearance for CYP3A4.

Substrate saturation plots for the metabolism of ezlopitant by heterologously expressed CYP2D6 (A), CYP3A4 (B), and CYP3A5 (C).
(■), CJ-12,458 formation; (○), CJ-12,764 formation.
Summary of enzyme kinetic parameters of ezlopitant metabolism by heterologously expressed CYP2D6 and CYP3A4
Correlation Between In Vitro Enzyme Kinetic Data and In Vivo Pharmacokinetic Data.
A list of estimations of hepatic clearance values made from in vitro intrinsic clearance data is in Table 4along with actual blood clearance values in preclinical species (Reed-Hagen et al., 2000). Of the five species examined, only the guinea pig appeared to yield estimations of clearance that were inaccurate. When the values for nonspecific binding to microsomes in the incubation matrix were included, estimations of in vivo hepatic clearance were closer to in vivo blood clearance values than estimations that were made disregarding this factor, irrespective of the model used (well stirred or parallel tube, Pang and Rowland, 1977). In human, scale-up of the in vitro data yielded a prediction of moderate clearance (6.3–7.4 ml/min/kg), which is close to the in vivo value, albeit the in vivo value is calculated after oral administration and should be interpreted with caution. In animals, predictions of clearance were high (relative to hepatic blood flow values for each species), consistent with high clearance values measured after i.v. administration. Corresponding predictions of high hepatic extraction (Eh = 1 − Fh) were made for preclinical species and low hepatic extraction in human. Comparisons between predictions of Fh values and actual oral bioavailability values (F) are listed in Table 4.
Comparison of in vivo pharmacokinetic data with values predicted from in vitro liver microsomal intrinsic clearance data
Discussion
The experiments described above were designed to obtain a description of the in vitro metabolism of ezlopitant in five species for which pharmacokinetic data are available. These data can aid in the understanding of interspecies differences in the pharmacokinetics of ezlopitant as well as be used in the development of a cross-species in vitro-in vivo correlation.
To quantitatively correlate in vivo pharmacokinetic data to in vitro metabolism data obtained in hepatic microsomes, several criteria must be met: a) clearance must be primarily through metabolism; b) the structure of the compound is not readily amenable to conjugative or other non-CYP/non-FMO metabolism; c) the liver is the primary organ of metabolic clearance; and d) the compound does not possess physicochemical properties that are associated with absorption problems (i.e., limited solubility, low GI permeability). Ezlopitant possesses these properties and has been shown to be exclusively cleared by oxidative metabolic pathways in preclinical species and humans (data on file, Pfizer, Inc.). Furthermore, it must be assumed that metabolic rates measured in vitro are truly reflective of those that occur in vivo. Ezlopitant is a good example to examine in vitro because pharmacokinetic data have been gathered in several species, and pharmacokinetic data have also been obtained for the major oxidative metabolites (CJ-12,458 and CJ-12,764).
The overall correlation between in vivo clearance data and clearance values estimated from in vitro intrinsic clearance data for ezlopitant was good. Clearance in preclinical species was generally moderate to high, as was predicted from the in vitro intrinsic clearance data, whereas the oral clearance in human was low, which was also predicted from the human liver microsomal intrinsic clearance data (Table 4). Human was the outlier species, especially in regard to the highKMapp values observed in this species. Interestingly, predictions of clearance in which the values for nonspecific binding to liver microsomes were not included in the relationship between hepatic clearance and intrinsic clearance yielded very poor predictions of clearance. In all species, ezlopitant is highly bound to plasma proteins (fu < 0.03), and inclusion of these low free fraction values yielded low estimates of clearance. However, it was found that ezlopitant is highly bound to microsomes, and inclusion of the microsomal binding values improved the projections of clearance from in vitro data. The nonspecific binding to microsomes can be a significant factor in the estimation of clearance from in vitro intrinsic clearance data, especially for lipophilic amines (Obach, 1999). Both the well stirred and parallel-tube models of hepatic extraction (Pang and Rowland, 1977) were used to relate intrinsic clearance and clearance data. Neither model appeared to be generally more accurate than the other with regard to predictions of clearance of ezlopitant.
Ezlopitant is converted primarily to two major metabolites in liver microsomes, CJ-12,458 (an alkene) and CJ-12,764 (a benzylic alcohol), both of which possess Substance P receptor binding activity similar to the parent compound. Human liver microsomes generated nearly equivalent amounts of these two metabolites, whereas the animal liver microsomes favored formation of the benzylic alcohol. This pattern appears to be consistent with in vivo data because the benzyl alcohol metabolite predominates in animals, whereas the two metabolites appear to be present in similar amounts in humans in vivo. Such an observation remains qualitative, because intrinsic clearance data on the metabolites themselves, as well as in vivo clearance of the metabolites, would be necessary to make quantitative predictions of metabolite exposures from in vitro metabolism data. The formation of an alkene represents an unusual reaction for P450. Some examples exist of CYP-catalyzed dehydrogenations of alkanes to alkenes including valproic acid (Rettie et al., 1987), testosterone (Nagata et al., 1986), lovastatin and simvastatin (Vickers et al., 1990; Vyas et al., 1990), ethyl hexanoic acid (Pennanen et al., 1996), and warfarin (Fasco et al., 1978). Many involve dehydrogenation adjacent to an sp2 hybridized center, which represents an electronically “activated” position for this reaction (Testa and Mihailova, 1978). The dehydrogenation of ezlopitant represents a dehydrogenation reaction adjacent to an sp2hybridized center (benzene ring). Experiments demonstrated that CJ-12,458 does not merely arise via chemical dehydration of the benzyl alcohol CJ-12,764, but truly represents a CYP-mediated metabolic product.
Data were also obtained to address the identity of human CYP isoforms involved in the metabolism of ezlopitant to CJ-12,458 and CJ-12,764. Heterologously expressed recombinant CYP3A4, CYP3A5, and CYP2D6 catalyzed these metabolic transformations, whereas other recombinant CYP isoforms did not. In human liver microsomes, the use of isoform selective inhibitors suggested that CYP3A predominates in the metabolism of ezlopitant, whereas CYP2D6 contributes a minor role. Ketoconazole, a CYP3A specific inhibitor, potently inhibited ezlopitant metabolism, whereas quinidine, a CYP2D6-specific inhibitor only yielded about 10% inhibition at concentrations in the range of its potency toward this enzyme (<0.1 μM). Quinidine did inhibit ezlopitant metabolism at higher concentrations, but this is likely due to inhibition of CYP3A as a competitive substrate (KM ≈ 10 μM; Guengerich et al., 1986). Correlation of CYP isoform-specific marker substrate activities with ezlopitant metabolic rates using human liver microsomes from individual donors suggested that CYP3A is involved in the metabolism of ezlopitant. However, the correlation data is less conclusive.
Substrate saturation data for ezlopitant metabolism by the recombinant CYP isoforms showed that the KMapp value for CYP2D6 was lower (≈0.5 μM) than that of CYP3A4 (≈10 μM). Scaling the intrinsic clearance data from the recombinant CYP enzyme kinetic data (Vmax/KMapp) yields a higher value for CYP2D6 (7.8 versus 1.0 μl/min/nmol of CYP). However, the amount of CYP3A4 in human liver is far greater than the amount of CYP2D6, which reaffirms the notion that CYP3A4, and not CYP2D6, is the major CYP isoform responsible for ezlopitant metabolism. In preliminary clinical studies, no differences in the pharmacokinetics of ezlopitant have been observed in CYP2D6 extensive and poor metabolizer phenotype subjects. However, the potential does exist for ezlopitant to inhibit, as a competitive substrate, the CYP2D6-mediated metabolism of other substrates.
In conclusion, the data presented demonstrate that the pharmacokinetics of ezlopitant observed in preclinical species and humans are correlated to estimates of pharmacokinetics made from in vitro intrinsic clearance data gathered in hepatic microsomes. They affirm that human is an outlier species with regard to the pharmacokinetics of ezlopitant. Qualitatively, the observations of major versus minor metabolites in vitro were also correlated to in vivo data on the relative systemic exposures to the metabolites. Furthermore, the contribution of CYP3A as the major CYP isoform in the metabolism of ezlopitant in humans was demonstrated.
Acknowledgments
I thank Drs. Larry M. Tremaine and Robert Ronfeld for helpful advice given during these studies. The preparation and characterization of liver microsomes and heterologously expressed CYP in the Pfizer Drug Metabolism Department Microsome Bank by Dr. Donald Tweedie, Dayna Mankowski, and Robert Whalen is greatly appreciated.
Footnotes
-
Send reprint requests to: Dr. R. Scott Obach, Drug Metabolism Department, Central Research, Pfizer, Inc., Groton, CT, 06340. E-mail: ronald_s_obach{at}groton.pfizer.com
- Abbreviations used are::
- CYP
- cytochrome 450
- TAO
- triacetyloleandomycin
- TEA
- triethylamine
- KMapp
- Michaelis constant
- CL′int
- intrinsic clearance
- fu
- unbound fraction
- MS
- mass spectrometry
- Received March 29, 2000.
- Accepted May 22, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}