


Abstract
An in vitro model is proposed to account for the clinically observed inhibition of cytochrome P450 (CYP) 3A that results from administration of clarithromycin, fluoxetine, or diltiazem. Rates for loss of CYP3A4 enzymatic activity resulting from metabolic intermediate complex formation and the concentration dependencies thereof were determined in vitro for clarithromycin, fluoxetine, andN-desmethyl diltiazem, which is the primary metabolite of diltiazem. Using the in vitro concentration-dependent rates for loss of activity, in vivo rates of CYP3A4 inactivation were predicted for these compounds at a clinically relevant unbound plasma concentration of 0.1 μM. Based on the predicted rates combined with published rates for in vivo CYP3A degradation, our model predicts that fluoxetine, clarithromycin, and the primary metabolite of diltiazem reduce the steady-state concentration of liver CYP3A4 to approximately 72, 39, or 21% of initial levels, respectively. These reductions correspond to 1.4-, 2.6-, or 4.7-fold increases, respectively, in the area under the plasma concentration-time curve of a coadministered drug that is eliminated exclusively by hepatic CYP3A4 metabolism. These predicted results are in good agreement with reported clinical data. The major implication of this work is that fluoxetine, clarithromycin, and the primary metabolite of diltiazem, at clinically relevant concentrations, inactivate CYP3A4 enzymatic activity at rates sufficient to affect in vivo concentrations of CYP3A4 and thereby affect the clearance of compounds eliminated by this pathway. We speculate that mechanisms involving substrate-mediated mechanistic inactivation of CYPs play a major role in many clinically observed drug-drug interactions.
The cytochromes P450 (CYPs)1 are a superfamily of heme-containing enzymes that catalyze the oxidation of a wide variety of compounds. An important role of these enzymes is to aid in the elimination of lipophilic xenobiotics, including environmental toxins and drugs. It is well known that many drugs can modulate the activity of the CYPs and thereby alter the pharmacokinetic profile of coadministered drugs. Such an effect is referred to as a metabolic drug-drug interaction. The consequences of these interactions can range from loss of therapeutic efficacy to the introduction of potentially lethal toxic effects (Honig et al., 1993; Olkkola et al., 1993; Azie et al., 1998).
Considerable effort has been expended in an attempt to predict the magnitude of in vivo metabolic drug-drug interactions using in vitro data (Rodrigues and Wong, 1997). Metabolic drug-drug interactions can be loosely classified into two groups: those in which the capacity of a metabolic pathway is enhanced and those in which it is diminished. Most examples from the former group have been attributed to elevated steady-state levels of CYP. Increased rates of synthesis (Gillum et al., 1993, 1996) and attenuated rates of degradation (Chien et al., 1997) have both been implicated as mechanisms underlying these elevations. In contrast, attempts to explain how the presence of one drug can diminish the elimination of another have largely ignored rates of CYP synthesis and degradation. Instead, despite the fact that most drugs never reach plasma concentrations that approach their in vitro determined Ki values, competitive and noncompetitive interactions have been most often proposed to explain interactions between drugs sharing a common route of elimination.
This work demonstrates that enzyme inactivation, namely mechanistic inactivation through metabolic intermediate complex (MIC) formation, can occur at rates that are sufficient to significantly diminish steady-state levels of CYP in vivo. A mechanism-based inactivator first binds to and then becomes catalytically activated by the enzyme (Silverman, 1998). The activated species irreversibly alters the enzyme to remove it permanently from the pool of active enzyme. In the case of mechanistic inactivation through MIC formation, compounds are catalytically oxidized to intermediates or products that coordinate tightly to the prosthetic heme of the CYP (Franklin, 1977). This coordination can only be displaced under nonphysiological experimental conditions (e.g., potassium ferricyanide). Primary amines are required for the MIC formation, although secondary and tertiary amines are also appropriate precursors. The primary amines are hydroxylated then further oxidized to a nitroso group that appears to chelate to the heme, resulting in a more stable, ferrous state of iron (Bensoussan et al., 1995). This ferrous state exhibits a spectrum with an absorbance maximum of 445 to 455 nm (Franklin, 1977).
We have chosen three widely prescribed drugs, diltiazem, clarithromycin, and fluoxetine, as clinically relevant examples of irreversible inactivators of CYP3A enzymes. Diltiazem (Backman et al., 1994; Ahonen et al., 1996; Azie et al., 1998) and clarithromycin (Albani et al., 1993; Sketris et al., 1996; Gorski et al., 1998) have been confirmed to reduce elimination of drugs by CYP3A, and reports have suggested that fluoxetine also affects this pathway, albeit to a lesser extent (Greenblatt et al., 1992). Furthermore, each of these drugs or metabolites thereof has been shown to form MICs (Tinel et al., 1989; Bensoussan et al., 1995; Franklin, 1995; Jones et al., 1999). In this report, we will attempt not only to rank-order the effects of these compounds from in vitro data but also to predict the expected changes in area under the plasma concentration-time curve (AUC) that their long-term administration will impart on a coadministered substrate of CYP3A.
Experimental Procedures
Theoretical Background.
Several approaches for modeling mechanism-based enzyme inactivation have been described in the literature (Walsh et al., 1978; Waley 1980,1985; Tatsunami et al., 1981; Tudela et al., 1987; Silverman, 1998). The critical features of mechanism-based enzyme inactivation are portrayed in Scheme FS1.

Scheme FS1 illustrates that, on binding to an enzyme, an inactivator (I) is destined to one of three fates: it can be released from the enzyme through reversible binding, it can be converted to a product (P) through a productive catalytic cycle, or it can inactivate the enzyme by forming with it an irreversible complex (E·MI).
For the model depicted in Scheme FS1, the rate of enzyme inactivation is defined by
In the case where two simplifying assumptions are applied to the model of Scheme FS1, the parameters kinact andKI of eq. 1 have physical meanings that are intuitively clear. The first assumption is that binding of inactivator to enzyme is in a rapid equilibrium. The second assumption is that catalytic steps common to both productive catalysis and irreversible inactivation are rapid and nonrate-limiting and therefore can be ignored. Where these assumptions are valid,kinact represents the rate constant for mechanistic inactivation to form the inactive complex, andKI represents the equilibrium disassociation constant for the inactivator. It is to be noted that, where the above simplifying assumptions are not appropriate, the parameters kinact andKI of eq. 1 represent complex functions, dependent on several rate constants, that have no simplistic interpretation. However, irrespective of the mechanistic interpretation, KI can be operationally defined as the inactivator concentration that supports half the maximal rate for mechanistic inactivation. In turn, the maximal rate of inactivation is defined by the product ofkinact and the active enzyme concentration.
Equation 1 can be integrated to provide a relationship that has been widely used to estimate the desired parameters when the concentration of active enzyme can be measured:

The steady-state concentration of enzyme is defined by the following equation:

If we assume that ksynth andkdegrad remain constant in the presence of the mechanistic inactivator, the new steady-state enzyme concentration for the situation depicted in Scheme FS3 is defined by
The apparent rate constant λ of eq. 6 is the same apparent rate constant λ for enzyme degradation as defined in eq. 4. Substituting eq. 4 into eq. 6, yields the following:
Materials.
Fluoxetine was purchased from Sigma Chemical Company (St. Louis, MO). Clarithromycin was a gift from Abbott Laboratories (North Chicago, IL).N-Desmethyl diltiazem (MA) was a gift from Tanabe Seiyaku Co. (Osaka, Japan). Midazolam, 1′-hydroxy midazolam, and 1′-hydroxy-15N3-midazolam were gifts from Hoffmann-LaRoche (Nutley, NJ). All gas chromatography (GC), mass spectroscopy (MS), and microsomal preparation supplies were of the highest grade available from standard commercial sources. NADPH was purchased from Boehringer Mannheim (Indianapolis, IN). Insect cell microsomes containing baculovirus cDNA-expressed CYP3A4 [+cytochromeb5 (b5)] (Supersomes) were purchased from Gentest Corp. (Woburn, MA).
Specimens.
A human adult liver specimen, designated IUL-10, was obtained at surgery in accordance with protocols approved by the Committee for the Conduct of Human Research of Indiana University, Indianapolis, IN. The handling, preparation, and storage of microsomes along with characteristics of the microsomal sample and relative CYP3A level have been previously described (Gorski et al., 1994a,b). The CYP concentration for human liver microsomal sample IUL-10 was determined to be 0.61 nmol of CYP/mg of protein by the method of Omura and Sato (1964).
Metabolic Intermediate Complex Formation in Expressed Enzyme.
Supersomes [CYP3A4 (+b5)] were used to characterize MIC formation associated with the metabolism of clarithromycin, fluoxetine, diltiazem, and MA. MIC formation was measured at various times in samples containing clarithromycin, fluoxetine, diltiazem, or MA at 100 μM in microsomal buffer (100 mM sodium phosphate buffer, 5 mM magnesium chloride, pH = 7.4). MIC formation was observed with dual-beam spectroscopy (Uvikon 933 double-beam UV/VIS spectrophotometer; Research Instruments International, San Diego, CA) by scanning from 380 to 500 nm to monitor the formation of an absorbance maximum at ∼455 nm. In each case, the sample cuvette contained 200 pmol of CYP3A4 (+b5), inactivator, and 1 mM NADPH, whereas the reference cuvette contained 200 pmol of CYP3A4 (+b5), inactivator vehicle, and 1 mM NADPH. All MIC formation experiments were maintained at 37°C and initiated by the addition of NADPH. The final incubation volume for each cuvette was 1 ml.
Simultaneous MIC Formation and Loss of Enzyme Activity.
Clarithromycin, fluoxetine, MA, or alprazolam was incubated in human liver microsomes with NADPH to quantify time- and concentration-dependent MIC formation simultaneously with time- and concentration-dependent loss of midazolam hydroxylase activity. Alprazolam was selected as the control compound because it is metabolized by CYP3A but does not form an MIC based on preliminary experiments, and thus it served to assess whether all CYP3A substrates cause loss of midazolam hydroxylation activity. Large reaction tubes were prepared containing IUL-10 at 250 μg of protein/ml of reaction buffer and clarithromycin, fluoxetine, MA, or alprazolam at various concentrations, and the tubes were maintained at 37°C. Spectroscopic measurement of MIC formation was accomplished as follows: from the large reaction tubes, 0.8-ml samples were transferred to cuvettes. Buffer containing NADPH was added to each cuvette to start the reactions. The final NADPH and protein concentrations were 1 mM and 200 μg/ml, respectively, and the final cuvette volume was 1 ml. The time dependence of MIC formation was measured at each concentration by simultaneously sampling absorbance at 490 nm and at 455 nm at 30-s intervals. The absorbance at 490 nm measured at a given time was subtracted from the absorbance at 455 nm measured at the same time. In each case, the sample cuvette contained microsomal protein, inactivator at a given concentration, and 1 mM NADPH, whereas the reference cuvette contained microsomal protein, inactivator vehicle, and 1 mM NADPH. All MIC formation experiments were maintained at 37°C.
Measurement of loss of enzyme activity was accomplished as follows: preincubation reactions were conducted at 37°C. Buffer containing NADPH was added to the remaining sample in the above-described large sample tubes to start the preincubation reactions. The final NADPH and protein concentrations in each preincubation reaction tube were 1 mM and 200 μg/ml, respectively. Samples (100 μL) were removed from the preincubation reaction tubes at various times and were transferred to small tubes already containing midazolam and NADPH. After transfer, the total incubation volume in each small tube was 1 ml, and the final midazolam and NADPH concentrations in each small tube were 200 μM and 1 mM, respectively. After a 10-min incubation at 37°C, the midazolam metabolism was terminated by the addition of acetonitrile, then the tubes were partially submerged into an acetone/dry ice bath to instantly freeze the aqueous matrix. Samples were dried and derivatized withN-methyl-N(trimethylsilyl)-trifluoracetamide, and the amounts of 1′-hydroxy midazolam formed during the 10 min incubations were quantified by GC/MS. All samples were analyzed by GC/MS using 1′-hydroxy-15N3-midazolam as internal standard.
Data Analysis and Statistics.
The microsomal activity and spectroscopic data represent the mean of duplicate or triplicate assays for every experiment. Untransformed kinetic data were analyzed by nonlinear regression without weighting (Scientist v 2.1; MicroMath Software, Salt Lake City, UT). Appropriateness of fit was determined by visual inspection of residual patterns and lines of best-fit, residual sums of squares, and precision of the parameter estimates.
The time- and inactivator concentration-dependent loss of midazolam 1′-hydroxylase activity is described by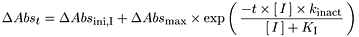 Equation 10where ΔAbsmax is a parameter for estimating maximal absorption, and ΔAbsini,I represents a separate parameter for each inactivator concentration, each for estimating the initial absorbances for its respective concentration-dependent time course.
Equation 10where ΔAbsmax is a parameter for estimating maximal absorption, and ΔAbsini,I represents a separate parameter for each inactivator concentration, each for estimating the initial absorbances for its respective concentration-dependent time course.
Where only midazolam 1′-hydroxylase activity data were available for a given inactivator, the data were fit to eq. 9 only. Where both loss of midazolam 1′-hydroxylase activity data and MIC formation data were available for a given inactivator, the data were fit simultaneously to eqs. 9 and 10, respectively. Equation 9 is analogous to eq. 2 but with two important differences. In the first, the equation includes an additional term, kbase, to account for the baseline loss of enzyme activity measured in the presence of alprazolam at 400 μM (0.003687 min−1). This baseline loss does not differ significantly from the loss of enzyme activity measured without inactivator in the preincubations. In the second difference, separate parameters for the initial activities (Actini,I) at each inactivator concentration replace [E]o, the known enzyme concentration. Equation 10 is analogous to eq. 3 but also differs in two important ways. In the first, a parameter for maximal absorption (ΔAbsmax) replaces [E]o. In the second difference, separate parameters for the initial absorbances (ΔAbsini,I) at each inactivator concentration are added. The parametersActini,I and ΔAbsini,I are included to allow the curve-fitting algorithm to estimate a true initial value for concentration-dependent time course rather than depend on a single point.
Simulated Effects on CYP3A Concentrations.
Substituting the calculated parameters forKI and kinactinto eq. 4, apparent in vivo rates of CYP3A4 inactivation (λ) were predicted for fluoxetine, clarithromycin, and MA at a clinically relevant unbound plasma concentration of 0.1 μM. The rationale for using unbound concentrations is that bound inactivator would not be available to participate in enzyme inactivation. To predict how these compounds affect the steady-state levels of active CYP3A4 ([E]ss), the calculated λ values were substituted into eq. 7, where kdegradwas assigned the value of 0.000825 (t1/2 = 14 h) as reported in the literature (Watkins et al., 1986).
The time courses for decay from [E]ss to [E]′ss resulting from exposure to fluoxetine, clarithromycin, or MA at unbound plasma concentration of 0.1 μM were simulated using Equation 11where [E]t is enzyme content at a given time t. Becauseksynth values are not available for CYP3A, we set ksynth =kdegrade so that the function has the convenient value of unity at time 0. It should be noted that the rate constant for decay is λ, and thus the value ofksynth has no effect on the rate of decay calculated using eq. 11.
Equation 11where [E]t is enzyme content at a given time t. Becauseksynth values are not available for CYP3A, we set ksynth =kdegrade so that the function has the convenient value of unity at time 0. It should be noted that the rate constant for decay is λ, and thus the value ofksynth has no effect on the rate of decay calculated using eq. 11.
The time courses for post-treatment return from [E]′ss to [E]ss were simulated using Equation 12where [E]t has an initial value equal to [E]ss. As with the decay phase defined by eq. 11, the simulations were conducted usingksynth =kdegrade. However, unlike the decay phase, the rate of recovery defined by eq. 12 depends entirely byksynth, and thus recovery phase was simulated for illustrative purposes only.
Equation 12where [E]t has an initial value equal to [E]ss. As with the decay phase defined by eq. 11, the simulations were conducted usingksynth =kdegrade. However, unlike the decay phase, the rate of recovery defined by eq. 12 depends entirely byksynth, and thus recovery phase was simulated for illustrative purposes only.
Results
Spectroscopic Determination of MIC Formation.
On incubation of clarithromycin, fluoxetine, diltiazem, or MA with baculovirus-derived microsomes containing expressed CYP3A4(+b5) (Supersomes) and NADPH, a peak absorbance difference was observed at ∼455 nm (Figs. 1, a–d). For all compounds, the magnitude of the MIC increased with time. Both fluoxetine and MA exhibited maximum absorbance differences of ∼0.0050 at 455 nm (Fig. 1, b and d). Clarithromycin and diltiazem exhibited maximum absorbance differences of ∼0.0043 and ∼0.0038, respectively, at 455 nm (Fig. 1, a and b). None of the compounds produced the characteristic peak at 455 nm in the absence of NADPH (baselines of Fig. 1, a–d). These results confirm that clarithromycin, fluoxetine, diltiazem, and MA form MIC in Supersomes. Analogous experiments confirmed that these compounds also form MIC in human liver microsomes (data not shown).

Metabolic intermediate complex formation by clarithromycin (a), fluoxetine (b), diltiazem (c), or MA (d) at 100 μM in baculovirus-derived microsomes containing expressed CYP3A4 + b5.
The sample cuvette contained 200 pmol of CYP3A4 + b5, inactivator, and 1 mM NADPH, whereas the reference cuvette contained 200 pmol of CYP3A4 + b5, inactivator vehicle, and 1 mM NADPH. The ribbons represent the change in absorbance difference for scans from 5 to 85 min (n = 1).
Concentration Dependence of MIC Formation and Loss of Enzyme Activity.
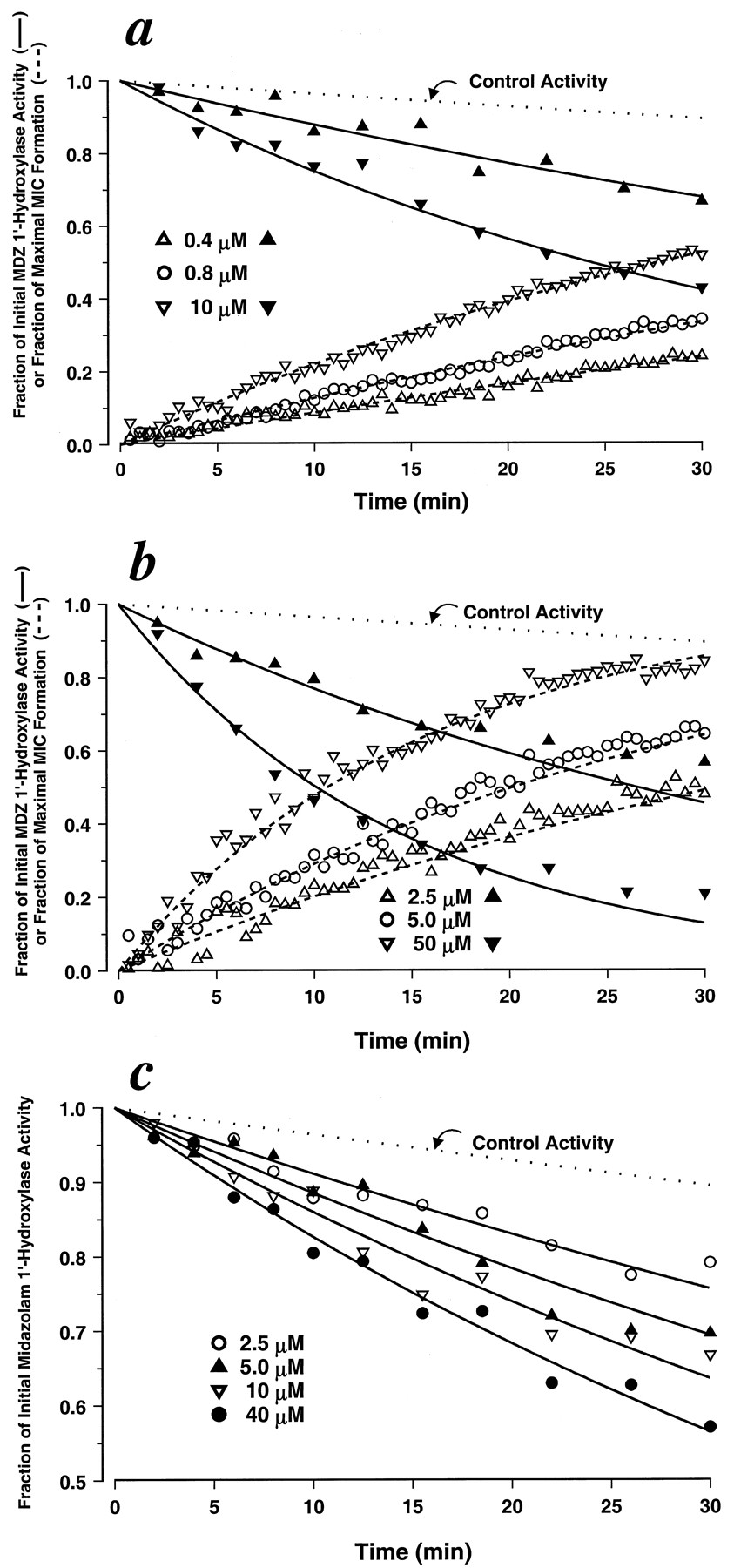
To investigate the relationship between MIC formation and inactivation of CYP3A activity and to assess the concentration dependencies of each, simultaneous preincubation and MIC formation experiments were performed at various concentrations of clarithromycin, fluoxetine, or MA using human liver microsomes. The concentrations ranged from 0.4 to 10 μM for MA, 2.5 to 50 μM for clarithromycin, and 2.5 to 40 μM for fluoxetine (Fig. 2, a–c). The rates of MIC formation, as measured by 455-490 nm difference spectra, showed clear dependencies on inactivator concentration for MA and clarithromycin (Fig. 2, a and b). Significant noise in the data and the modest extent of formation prevented assessing the inactivator concentration dependencies of MIC formation rates for fluoxetine (data not shown). For all cases, higher concentrations of inactivator resulted in higher rates of CYP3A4 inactivation as measured by loss of midazolam 1′-hydroxylase activity.

Simultaneous measurement of MIC formation and associated loss of CYP3A4 enzymatic activity by MA (a), clarithromycin (b), or fluoxetine (c) at various concentrations in human liver microsomes.
MIC formation was measured by monitoring the change in 455-490 difference spectra (n = 1; dashed line; fluoxetine data not shown). Loss of CYP3A4 enzymatic activity was assessed by measuring the effect of preincubation with the inactivators on midazolam 1′-hydroxylation (n = 2; solid line). Each line is the line of best fit obtained by fitting the data to eq. 9 (fluoxetine) or simultaneously fitting the data to eqs. 9 and 10 (MA and clarithromycin). Estimated parameter values forkinact and KIare provided in Table 1.
To further characterize the inactivator concentration dependencies for CYP3A activity, the loss of midazolam 1′-hydroxylase activity data shown in Fig. 2, a to c, were fit to eq. 9. We used the apparent rate constant (λ) for loss of midazolam 1′-hydroxylase determined as measured in the presence of alprazolam at 400 μM (0.003687 min−1) as our control for loss of activity. Alprazolam (400 μM) was chosen because it is a CYP3A4 substrate (Gorski et al., 1999) and because preincubation with it neither forms an MIC nor significantly inhibits midazolam 1′-hydroxylase activity compared with incubations in its absence (data not shown). In the cases of MA and clarithromycin, the loss of midazolam 1′-hydroxylase activity data were fit to eq. 9 and simultaneously fit to eq. 10. As determined by the visual inspection of fit, residual patterns, residual sums of squares, and precision of the parameter estimates, models assuming a hyperbolic dependence of inactivator concentration on loss of activity and MIC formation fit the data well. KI andkinact estimates for clarithromycin, fluoxetine, and MA are provided in Table1, and all lines of best fit in Fig. 2, a to c, were drawn using those parameters.
KI and kinact estimated in vitro using human liver microsomes
Time Dependent Changes in CYP3A Concentrations.
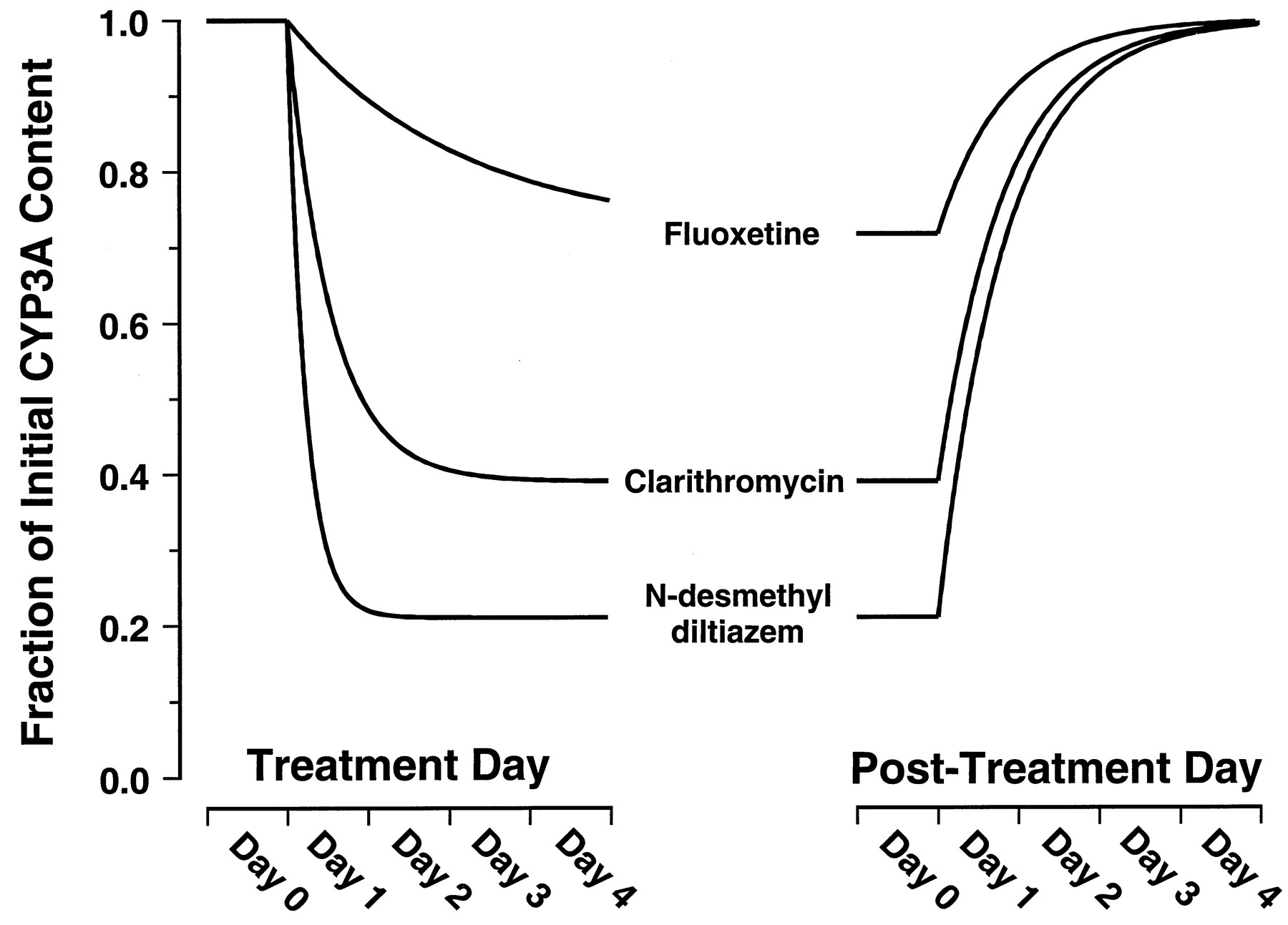
Figure 3 shows the simulated effects on steady-state concentration of active CYP3A4 resulting from prolonged exposure to fluoxetine, clarithromycin, or MA at an unbound plasma concentration of 0.1 μM. The simulation predicts that MA, followed closely by clarithromycin, would have the greatest effects on steady-state concentration of active enzyme. The simulation predicts only marginal effects would be seen on prolonged exposure to fluoxetine. Fluoxetine, clarithromycin, and MA are predicted to reduce the steady-state concentration of liver CYP3A4 to approximately 72, 39, and 21% of initial levels, respectively. According to eq. 8, these reductions correspond to 1.4-, 2.6-, or 4.7-fold increases, respectively, in the AUC of a coadministered drug that is eliminated exclusively by hepatic CYP3A4 metabolism.
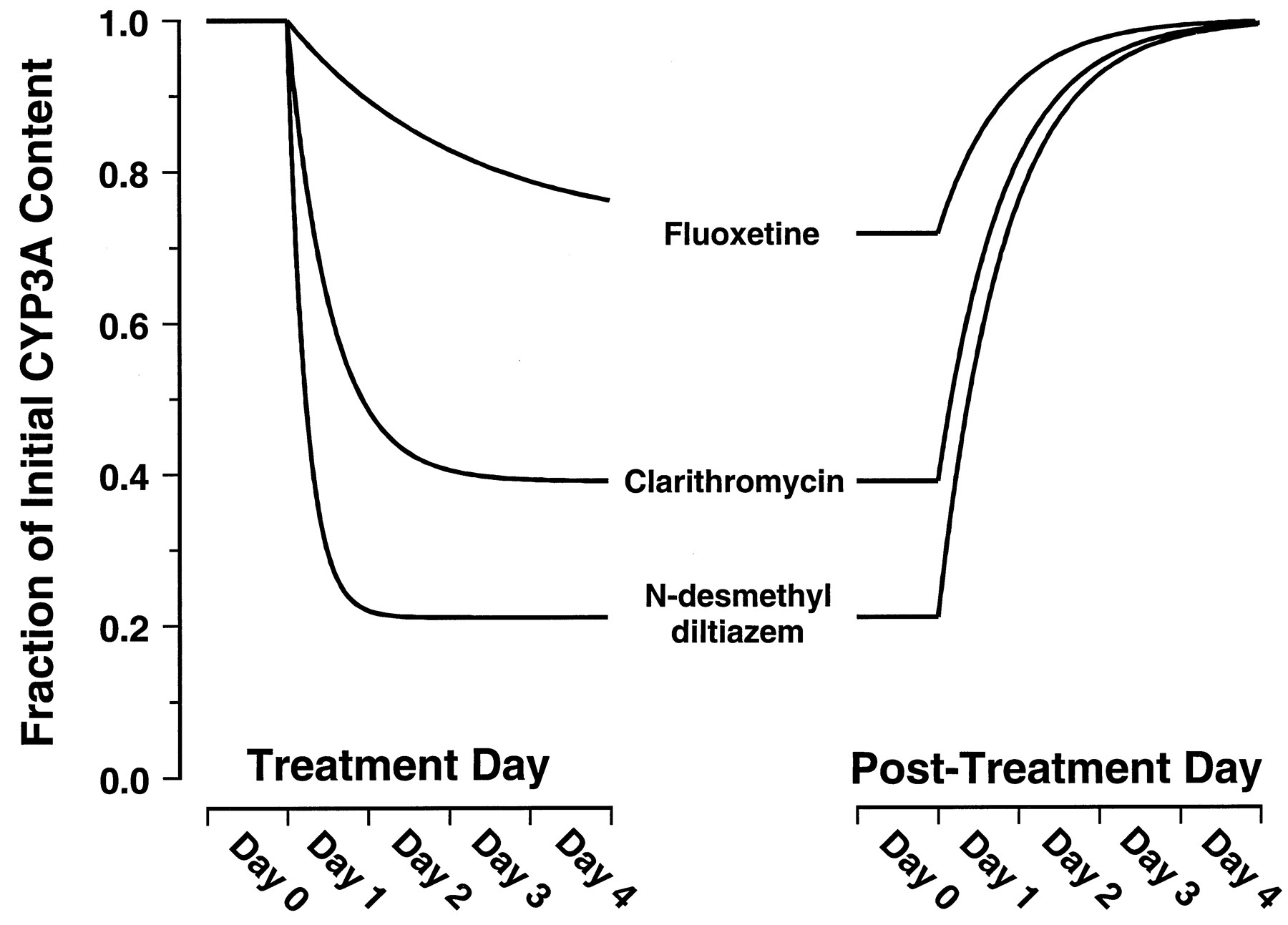
Simulated time courses for decay from [E]ss to [E]′ss resulting from exposure to fluoxetine, clarithromycin, or MA at unbound plasma concentrations of 0.1 μM.
The curves were constructed based on thekinact and KIvalues provided in Table 1 assuming akdegrad value of 0.000825 min−1(t1/2 = 14 h). Equation 11dictates that the rate constant for the decay phase is λ. Equation 12dictates that the rate constant for the recovery phase isksynth. Becauseksynth values are not available for CYP3A, the recovery phase was simulated for illustrative purposes usingksynth =kdegrad.
Discussion
CYP3A4 is responsible for the oxidative metabolism of more than half the drugs in current therapeutic use and therefore is often the locus of clinically important drug-drug interactions (Rodrigues and Wong, 1997). Diltiazem is a widely used calcium-channel blocker that has been shown to potently inhibit the metabolism of a variety of coadministered CYP3A substrates, including cyclosporin A (Brockmöller et al., 1990), quinidine (Laganière et al., 1996), midazolam (Backman et al., 1994), alfentanil (Ahonen et al., 1996), and lovastatin (Azie et al., 1998). For example, pretreatment with oral diltiazem significantly increased the oral AUCs of quinidine, midazolam, and lovastatin by approximately 2- to 4-fold (Backman et al., 1994; Laganière et al., 1996; Azie et al., 1998). Clarithromycin is a macrolide antibiotic for which several clinical interactions have been reported with CYP3A substrates (Albani et al., 1993; Sketris et al., 1996; Gorski et al., 1998). An illustration of this effect is the interaction between oral clarithromycin and midazolam administered orally and i.v. (Gorski et al., 1998). Clarithromycin increased the oral and i.v. AUCs of midazolam by 6- and 3-fold, respectively. Numerous additional examples of drug interactions between clarithromycin and other CYP3A substrates can also be found in the literature. In contrast, fluoxetine is a selective serotonin reuptake inhibitor for which few clinical interactions have been reported with CYP3A substrates. However, a recent report has revealed that fluoxetine prolongs alprazolam half-life from 17 to 20 h and reduces alprazolam clearance by 20 to 30% (Greenblatt et al., 1992).
In an attempt to predict their potency as inhibitors of CYP3A in vivo, the equilibrium inhibition constants (Ki) for competitive inhibition of CYP3A have been determined to be about 60, 10, and 50 μM for diltiazem, clarithromycin, and fluoxetine, respectively, using human liver microsomes (Pichard et al., 1990;Jurima-Romet et al., 1994; Ring et al., 1995). However, the steady-state plasma concentrations of diltiazem, clarithromycin, and fluoxetine are approximately 0.3, 0.9, and 1.0 μM, respectively (Ring et al., 1995; Azie et al., 1998; Gorski et al., 1998). In view of the low plasma concentrations of these compounds relative toKi, standard competitive models fail to predict that diltiazem, clarithromycin, or fluoxetine will inhibit metabolism of CYP3A substrates in vivo (Rodrigues and Wong, 1997). Nonetheless, diltiazem and clarithromycin have been reported to increase the AUCs of coadministered CYP3A substrates by severalfold and fluoxetine by 1.3-fold. Efforts to explain the in vivo inhibition for these compounds using competitive models are further confounded when it is considered that only approximately 20, 28, and 10% of diltiazem, clarithromycin, and fluoxetine, respectively, can be found unbound in plasma (Bloedow et al., 1982; Davey, 1991; Ring et al., 1995). The disparity between the prediction of competitive inhibition models and the observations in the above-cited clinical studies strongly suggests that some type of CYP3A inhibition is ongoing rather than simple, reversible inhibition.
Based on the above-cited clinical studies, concentrations of 0.1 μM are within the range of clinically expected unbound plasma concentrations for fluoxetine, clarithromycin, and the primary metabolite of diltiazem. Our model suggests that these compounds at unbound plasma concentrations of 0.1 μM will increase the AUC of a coadministered CYP3A substrate by 1.4-, 2.6-, or 4.7-fold, respectively. These predicted results are in good qualitative agreement with reported clinical data, especially when one considers that simple competitive models completely fail to predict any interaction.
An important assumption in our predictive approach is that only hepatic first-pass metabolism is affected by an inactivator. However, for substrates of CYP3A4, it is clear that significant first-pass metabolism may occur in the epithelium of the small intestine. Consequently, the ratio of oral area under the curves (AUC) will be determined by both hepatic intrinsic clearance and intestinal wall availability, in the absence (FG) or presence (FG′) of inactivator (eq. 13).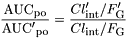 Equation 13Oral administration of clarithromycin for a week resulted in an AUC ratio for i.v. midazolam of 0.33, but the ratio was 0.16 for oral midazolam (Gorski et al., 1998). This greater extent of interaction for oral administration is consistent with the intestinal availability of approximately 0.4 for midazolam and a complete inactivation of intestinal CYP3A by clarithromycin (Gorski et al., 1998). Such a complete inactivation of intestinal CYP3A is consistent with our estimates of kinact andKI (Table 1) and the relatively high concentrations of clarithromycin expected in the intestinal lumen. In general, the impact of inactivation of intestinal wall CYP3A will depend on the values of the inactivation parameters, the residence time of the inhibitor at the site of CYP3A expression, and the relative contribution of intestinal and hepatic metabolism to the first-pass elimination of a given substrate. At present, the poor definition of these properties for common substrates and inhibitors of CYP3A precludes a clear understanding of the contribution that inactivation at the intestinal wall makes to drug-drug interactions.
Equation 13Oral administration of clarithromycin for a week resulted in an AUC ratio for i.v. midazolam of 0.33, but the ratio was 0.16 for oral midazolam (Gorski et al., 1998). This greater extent of interaction for oral administration is consistent with the intestinal availability of approximately 0.4 for midazolam and a complete inactivation of intestinal CYP3A by clarithromycin (Gorski et al., 1998). Such a complete inactivation of intestinal CYP3A is consistent with our estimates of kinact andKI (Table 1) and the relatively high concentrations of clarithromycin expected in the intestinal lumen. In general, the impact of inactivation of intestinal wall CYP3A will depend on the values of the inactivation parameters, the residence time of the inhibitor at the site of CYP3A expression, and the relative contribution of intestinal and hepatic metabolism to the first-pass elimination of a given substrate. At present, the poor definition of these properties for common substrates and inhibitors of CYP3A precludes a clear understanding of the contribution that inactivation at the intestinal wall makes to drug-drug interactions.
Despite the accuracy of our predictions, the model includes a weakness that should be addressed. The kdegrad value on which we have based our calculations corresponds to the rate of degradation for rat CYP3A rather than human CYP3A (Watkins et al., 1986). The choice to use rat data was not made for lack of available human data. For example, Pichard et al. (1992) reported akdegrad of 0.00026 min−1, which corresponds to at1/2 of 44 h, for CYP3A determined using a human hepatocyte model. However, Correia (1991) warns strongly that hepatocyte models are unsuitable for examiningt1/2 values for constitutive CYPs such as CYP3A and explains that investigations into truet1/2 values should always be conducted in intact animals. To our knowledge, no such data is available for human CYP3A. Thus, although the value reported by Watkins et al. (1996) is for rat CYP3A, their value has the advantage that it was determined in an intact animal. If in fact the value reported by Pichard et al. (1992) more closely reflects the true in vivo rates for human CYP degradation, our model predicts that exposure to fluoxetine, clarithromycin, or the primary metabolite of diltiazem would inhibit the CYP3A pathway more severely than has been reported in the literature. This dilemma demonstrates a need for more firmly establishing in vivo rates for human CYP degradation.
One other simplifying assumption employed in our model should be borne in mind. To simplify the equation describing the new steady-state enzyme concentration for the situation depicted in Scheme FS3, we assumed that ksynth andkdegrad remain constant in the presence of the mechanistic inactivator. In fact, the presence of xenobiotics is known to affect these rates (Gillum et al., 1993, 1996; Chien et al., 1997). Therefore, a more flexible description of how dosing with mechanistic inactivator will affect CYP steady-state concentration will require a more complete understanding of other factors that contribute to alterations in CYP turnover.
The irreversible inactivation of CYPs in vitro and in vivo by MIC formation has long been recognized as the mechanism by which some macrolide antibiotics, such as erythromycin and troleandomycin, exert their inhibitory effects. It is also clear that this mechanism of inhibition is not restricted to the macrolides and has been demonstrated to occur for a large number of CYP3A substrates in animal models. The only structural moiety common to these substrates is secondary or tertiary amino group that undergoes CYP3A catalyzedN-dealkylation. These substrates include diltiazem, lidocaine, propoxyphene, tamoxifen, and a number of antidepressants (Franklin, 1977; Bensoussan et al., 1995). The predictive approach we have described is therefore expected to have broad use in predicting drug interactions for many drugs, beyond the three inactivators specifically studied, and is not restricted to inactivation by MIC formation. In addition to providing an explanation for the potent inhibition of CYP3A activity in vivo, irreversible inactivation has clinically important consequences. First, the time-dependent inactivation of CYP3A enzymes is expected to result in nonlinear pharmacokinetics. This is illustrated by the 50 to 100% prolongation of the diltiazem half-life in humans after chronic dosing compared with the single-dose data (Tsao et al., 1990). Second, the extent of a drug interaction will be time-dependent in both onset and offset (Fig. 3). Erythromycin did not significantly inhibit the clearance of alfentanil on the 1st day of coadministration but produced a 25% decrease after 7 days (Bartkowski et al., 1989). In general, the half-life for onset of inactivation is inversely proportional to the efficiency [kinact/(KI +I)] of inactivation (eq. 11; Fig. 3). Thus, the delayed onset of inhibition by erythromycin is a predictable property of a relatively weak inactivator. The delayed offset of CYP3A inhibition is expected to be independent of the inactivating drug and the extent of inhibition (eq. 12; Fig. 3). This time-dependent offset may explain the serious adverse events associated with discontinuation of the irreversible inactivator, mibefradil, and immediate initiation of alternative calcium-channel blocker treatment (Mullins et al., 1998;Prueksaritanont et al., 1999). A mibefradil washout period of 7 to 14 days was subsequently recommended.
In this work, we have demonstrated that an additional pathway of enzyme degradation, namely mechanistic inactivation through MIC formation, can occur at rates that are sufficient to significantly diminish steady-state levels of CYP in vivo. Many drugs are mechanism-based inactivators of CYP, and we therefore speculate that mechanism-based inactivation plays a larger role in drug-drug interactions than has been previously recognized.
Footnotes
-
Send reprint requests to: Stephen D. Hall, Ph.D., Indiana University School of Medicine, Division of Clinical Pharmacology, Wishard Memorial Hospital, OPW 320, 1001 West 10th St., Indianapolis, IN 46202. E-mail: sdhall{at}iupui.edu
-
This work was supported by Public Health Service Grant AG13718.
- Abbreviations used are::
- CYP
- cytochrome P450
- AUC
- area under the plasma concentration-time curve
- b5
- cytochromeb5
- Clint
- intrinsic clearance
- kdegrad
- rate constant for endogenous enzyme degradation
- kinact
- rate constant for mechanistic inactivation
- ksynth
- rate constant for endogenous enzyme synthesis
- Ki
- dissociation constant for reversible inhibition
- KI
- inactivator concentration that supports half the maximal rate of mechanistic inactivation
- λ
- apparent rate constant for enzyme degradation
- MA
- N-desmethyl diltiazem
- MIC
- metabolic intermediate complex
- GC
- gas chromatography
- MS
- mass spectroscopy
- Received December 2, 1999.
- Accepted May 22, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}