


Abstract
We examined the effects of several agents, including dietary flavonoids, on CYP1A1 expression utilizing a recently developed high-throughput screening system for assessing human cytochrome P450 (CYP) induction. HepG2 cells, stably integrated with regulatory regions of human CYP1A1, were treated with resveratrol, apigenin, curcumin, kaempferol, green tea extract (GTE), (−)-epigallocatechin gallate (EGCG), quercetin, and naringenin. Of these flavonoids, resveratrol produced the greatest increase in CYP1A1-mediated luciferase activity (10-fold), whereas GTE, apigenin, curcumin, and kaempferol produced 2- to 3-fold increases in activity. Compared with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), omeprazole, or benzanthracene, where increases in luciferase activity ranged from 12- to 35-fold, these flavonoids exhibited weak agonist activity. The remaining compounds, EGCG, quercetin, and naringenin, produced negligible effects. Cotreatment of cells with TCDD and GTE, naringenin, and apigenin resulted in 58, 77, and 74% reductions, respectively, in TCDD-mediated CYP1A1 induction, indicating that these flavonoids exhibit potential antagonist activity toward the aryl hydrocarbon (Ah) receptor. Furthermore, results also suggest that GTE and apigenin possess Ah receptor antagonist and weak agonist activities. Thus, we have shown that a 96-well plate assay allowing high-throughput screening for P450 induction in less than 24 h was efficient in determining the effects of flavonoids on human CYP1A expression. Signal-to-noise ratios were low, and well-to-well and replicate variability was below 10%, allowing induction to be easily detected in this system. These features illustrate the reliability and feasibility of this high-volume screening system for identifying CYP inducers. Furthermore, results produced with the stable cell line were corroborated in HepG2 cells and primary cultures of human hepatocytes, suggesting that stably integrated cell lines harboring enhancer elements of P450 genes may be highly conducive to high-throughput screening.
The ability of new molecular entities to induce cytochrome P450s (CYP1) and other related drug-metabolizing enzymes can be assessed in a variety of ways to predict drug-drug interactions (Rodrigues, 1997, 1998)2. One method for assessing enzyme induction involves the use of animal models. Animals can be treated with candidate drugs, and hepatic drug-metabolizing enzyme activities and concentrations can be measured. Unfortunately, in many cases information obtained from laboratory animals cannot be readily extrapolated to humans because there are evident species differences in the induction of several CYP genes (Barwick et al., 1996; Shih et al., 1999). Thus, alternative procedures are being explored for directly monitoring induction of human drug-metabolizing enzymes.
Primary cultures of human hepatocytes can be used to predict enzyme induction (Maurel, 1996; Li et al., 1999; LeCluyse et al., 2000). However, there are limitations associated with this system. Results obtained from human hepatocyte cultures frequently show marked sample-to-sample variability in response to P450 enzyme inducers, thereby making it important to screen samples from several individuals (LeCluyse et al., 2000). Other disadvantages of using hepatocytes include cost and the need for fresh human livers, which are available sporadically. Furthermore, the time required between the acquisition of hepatocytes and the generation of results can vary from 1 to 2 weeks, depending on the experiments used to monitor enzyme induction and the xenobiotics being tested (Runge et al., 2000). Finally, in some cases minimal differences are observed between treated and untreated cells due to loss of cellular integrity or polymorphisms. These minimal alterations in enzyme levels make interpretation of results difficult. Therefore, a system that more readily predicts induction of P450 enzymes in a timely and less costly manner is needed. Such a system has recently been developed in our laboratory and was used to examine the interactions between the CYP1A1 inducer 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and dietary flavonoids.
Flavonoids are naturally occurring, polyphenolic compounds that are widely distributed in fruits, vegetables, whole grains, and beverages such as red wine and tea (Shih et al., 2000). Moreover, extracts of many flavonoids are now available in health food stores. Epidemiological studies and in vivo animal studies have suggested that flavonoids inhibit carcinogen-induced tumors in a variety of organs (Huang et al., 1988; Stoner and Mukhtar, 1995; Weisburger, 1999). The protective effects of flavonoids have been attributed to a wide variety of mechanisms, including prevention of xenobiotic-mediated induction of enzymes that activate or detoxify carcinogens (Canivenc-Lavier et al., 1996; Mukhtar and Ahmad, 1999). Flavonoids may alter the expression of the CYP1A gene products by interacting with the Ah receptor (AhR) pathway. Ligands (e.g., TCDD) bind the AhR, resulting in its translocation to the nucleus. The AhR then forms a heterodimer complex with the AhR nuclear translocator protein (ARNT), which functions as a transcriptional activator by binding to consensus sequences called dioxin response elements present in the 5′-flanking region of numerous genes, including CYP1A1 (Denison and Whitlock, 1995). Interaction of a flavonoid with AhR could prevent expression of CYP1A1 and ultimately decrease the metabolic activation of some carcinogens (Turesky et al., 1991).
The present investigation focuses on the effects of a variety of inducers of human CYP1A gene expression in an effort to develop a system better suited to screening for human P450 induction than current procedures. A high-throughput screening system to monitor CYP1A1 induction via the AhR/ARNT pathway was developed and used to determine whether known CYP1A1 inducers and/or flavonoids mediate induction of this P450. The advantages of this system are that results can be obtained within 6 to 18 h of cell treatment and many agents can be examined in a single assay. In addition, HTS, when expanded to include analyses of the induction of other CYP gene promoters, can be used to predict not only interactions involving xenobiotics and naturally occurring compounds, but also drug-drug interactions.
Materials and Methods
Cell Cultures and Treatment.
The 101L cell line, derived from the human hepatoma cell line HepG2, was stably transfected with the human CYP1A1 promoter and 5′-flanking sequences linked to the luciferase reporter gene (Postlind et al., 1993). Briefly, the 101L cell line was established by stably transfecting a plasmid, containing the human CYP1A1 promoter (−3275 to +89) linked to the firefly luciferase reporter gene, into the human hepatoma cell line HepG2. The CYP1A1 promoter region contains three dioxin response elements, and the cell line was estimated to contain two copies of the integrated plasmid.
The 101L cell line was grown as monolayers in media composed of Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA), 50 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM nonessential amino acids (Invitrogen), 0.4 mg/ml G418 (Invitrogen), 10% fetal bovine serum (FBS, Hyclone Laboratories, Logan, UT) and maintained in an atmosphere of 5% CO2 and 95% air at 37°C. Cells were initially seeded in flasks containing medium without G418. After an overnight incubation, the cultures were changed into medium containing G418 for antibiotic selection. After 3 to 5 days, cells were removed from the flasks by trypsinization and replated on either 24-well plates, at a density of 3.5 × 105 cells/well, or 96-well plates, at a density of 7.5 x104 cells/well, in DMEM containing 0.1% FBS and lacking G418 or the indicator (phenol red). The next day, media containing 0.1% FBS and G418 was added to the cultures. After 24 h, cells containing stably integrated reporter constructs were treated with 0.1% DMSO (control), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, Chemsyn, Lenexa, KY), 3-methylcholanthrene (3-MC, Sigma, St. Louis, MO), benzanthracene (Sigma), omeprazole (Astra-Zeneca, Södertälje, Sweden), rifampicin (Sigma), quercetin (Sigma), green tea extract (GTE, Sigma), resveratrol (Sigma), apiginen (Sigma), curcumin (Sigma), kaempferol (Sigma), (−)-epigallocatechin gallate (EGCG, Sigma), or naringenin (Sigma) in fresh media containing 0.1% FBS and G418 without indicator. For the antagonist experiments, cells were cotreated with a flavonoid and 2 nM TCDD. All inducers were dissolved in DMSO, and this solvent was added to control cells at 0.1%. The cells were treated with various doses and at various times (6–18 h). After treatment, the media containing the compound were removed by aspiration and replaced with 100 μl of DMEM per well for direct analysis of luciferase activity. Experiments were performed on 101L cells from frozen stocks of the initial derivation, and the passage number was limited to 30. The later passages exhibited responses similar to those of the earliest passage.
Luciferase Assays.
Luciferase assays were performed as specified by the manufacturer (LucLite system, Packard Instrument Co., Meriden, CT). Activity was determined using a Packard LumiCount luminometer (Packard Instrument Co.), and results were expressed as relative light units or -fold increase above control (DMSO-treated cells).
HepG2 Cultures and Treatment.
HepG2 cells were obtained from American Type Culture Collection (Manassas, VA). Cells were grown in DMEM supplemented with 10% fetal bovine serum. Twenty-four hours after cells were plated and grown to confluency, they were treated with a bioflavonoid, TCDD or β-naphthoflavone (Sigma). All inducers were dissolved in DMSO, and this solvent was added to control cells at 0.1%.
Primary Cultures of Human Hepatocyte and Treatment.
Six-well plates containing human hepatocytes were obtained from the Liver Tissue Procurement and Distribution System (University of Minnesota, Minneapolis, MN). Upon arrival, media were replaced with that containing Human Hepatocyte Maintenance Media (Cambrex Life Sciences, North Brunswick, NJ) (Runge et al., 2000) and maintained in an atmosphere of 95% air/5% CO2at 37°C. The following day, cells were treated for 24 h with 0.1% DMSO (control), 50 μM benzanthracene, 2 nM TCDD, 20 μM kaempferol, 20 μM resveratrol; or 20 μM naringenin, 10 μM apigenin, 0.1 mg/ml GTE; or cotreated with TCDD and a flavonoid. All inducers were dissolved in DMSO and added to media at a 0.1% final concentration of solvent. After treatment, medium was removed, and cells harvested for RNA isolation.
RNA Isolation and Northern Blot Analysis.
Total RNA from hepatocytes or HepG2 cells was isolated using Trizol reagent (Invitrogen) and quantified by measuring absorbance at 260 nm; purity was assessed by determining the 260:280-nm ratio. Northern blot analysis was performed by electrophoresis of total RNA (10 μg) through a 1% agarose/2.2 M formaldehyde gel, followed by blotting onto a nylon membrane (Molecular Simulations, Inc., Westboro, MA) (Shih et al., 1999). RNA was cross-linked to the membranes using a UV Crosslinker (Stratagene, La Jolla, CA) and the membranes hybridized to random-primed cDNA probes encoding human CYP1A1. The cDNA probe for human CYP1A1 has previously been described (Shih et al., 1999). A cDNA probe for human 18S RNA probe (Ambion, Austin, TX) was used to normalize the amount of RNA loaded in each lane. Hybridization of blots was performed as previously described (Quattrochi et al., 1985). Autoradiographs of Northern blots were quantified by densitometry using a Model GS-670 Imaging Densitometer equipped with Molecular Analyst (Mac version 1.1.1. Image Analysis software) (Bio-Rad, Hercules, CA) or by scanning autoradiograms with a ScanMaker II (Microtek) and digitizing with Un-Scan-It software (Silk Scientific, Orem, Utah). Exposure times used were in the linear range for the film, Kodak XAR-5 (Eastman Kodak Co., Rochester, NY).
Data Analysis.
Student's t test was used for the statistical analysis of data. Statistical significance was defined at a level ofp < 0.05. Data are expressed as the mean ± S.D.
Results
101L cells were plated at a density of 3.5 × 105 or 7.5 × 104cells/well in 24- or 96-well plates, respectively. Following exposure to the Ah receptor ligand, benzanthracene, luciferase activity was determined. When results obtained from 96-well plate assays were compared with those from 24-well plates, negligible differences in luciferase activity were detected (Fig.1). These findings alleviated the concern that too few cells per well would produce an inadequate signal. There was also concern that variability between replicate wells would be high. However, the 96-well format exhibited a maximum of 10% well-to-well variability with minimum background noise (Fig. 1) and therefore was used to perform the following experiments.

Effects of plating stable cell lines in 24- and 96-well plates.
101L cells were plated in 24- or 96-well plates and exposed to various doses of the Ah receptor ligand benzanthracene. After 18-h exposures, luciferase activity was assessed. Results are expressed as the mean of three different experiments ± S.D.
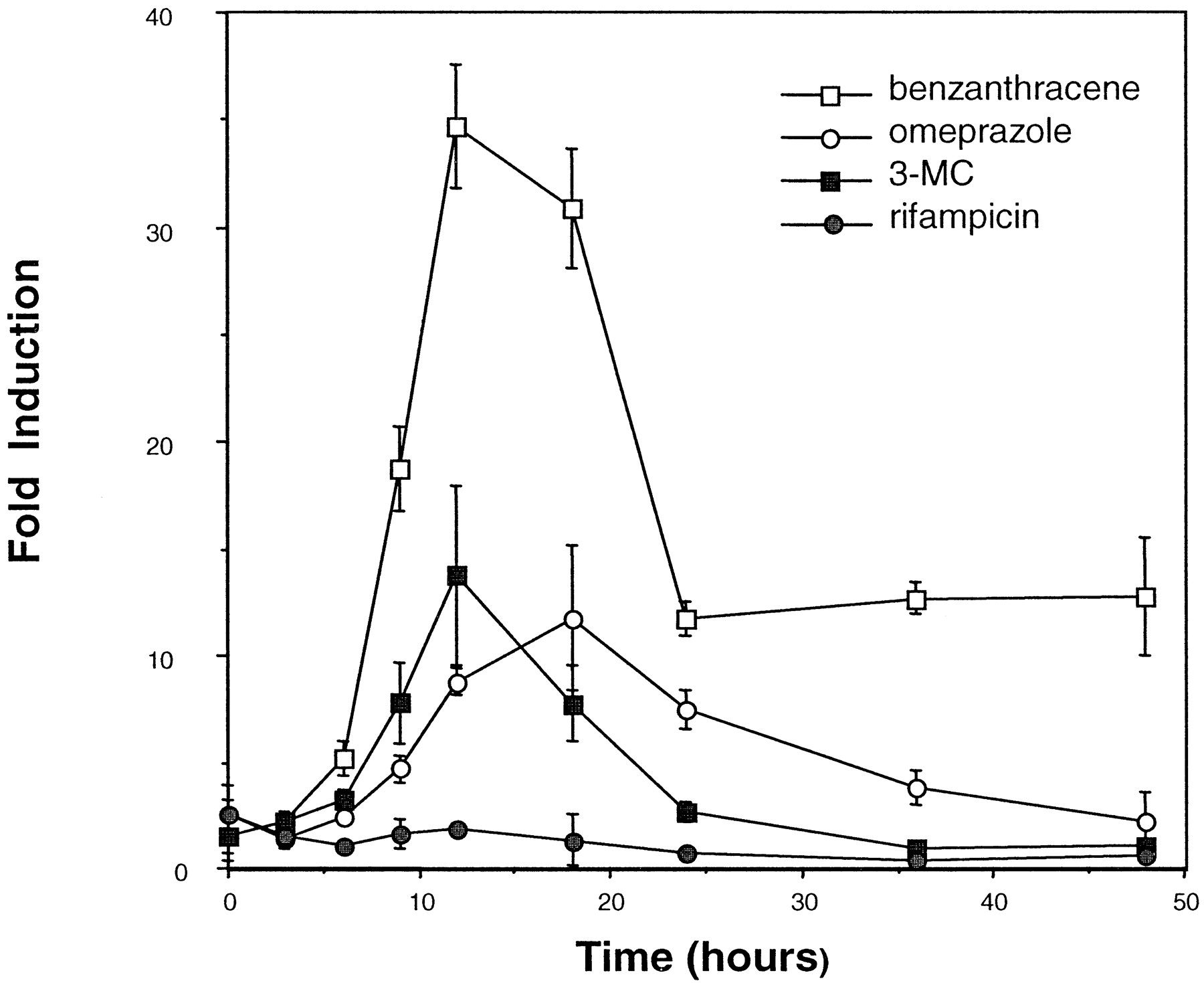
The maximal time period for inducer exposure was determined by establishing a time course of inducer-mediated luciferase activity. Enhanced activity was observed within 6 h of dosing with benzanthracene (100 μM), omeprazole (100 μM), or 3-MC (10 μM) (Fig. 2). Maximum induction by benzanthracene (35-fold) and 3-MC (14-fold) occurred at 12 h, whereas omeprazole-mediated induction was maximal at 18 h (12-fold), after which luciferase activity declined. The decline in inductive response is probably due to the metabolism of the inducer by HepG2 CYP1A1. As expected, induction by rifampicin (100 μM) was negligible, since this antibiotic is not known to be a CYP1A inducer (Kostrubsky et al., 1999). These results suggest that this high-volume screening procedure is effective at monitoring easily detectable induction within a relatively short time period (i.e., less than 24 h). In addition, the concentration-dependent effects of various known CYP1A1 inducers were determined in this system. Dose-response curves ranging from 0.5 to 2.5 nM were generated for TCDD (Fig.3A) and 1 to 200 μM for benzanthracene and omeprazole (Fig. 3B). For benzanthracene and omeprazole, maximum induction (35- and 12-fold, respectively) occurred at 100 μM. The -fold induction by TCDD had not peaked at a dose of 2 nM, and this dose produced a 22-fold increase in luciferase activity.
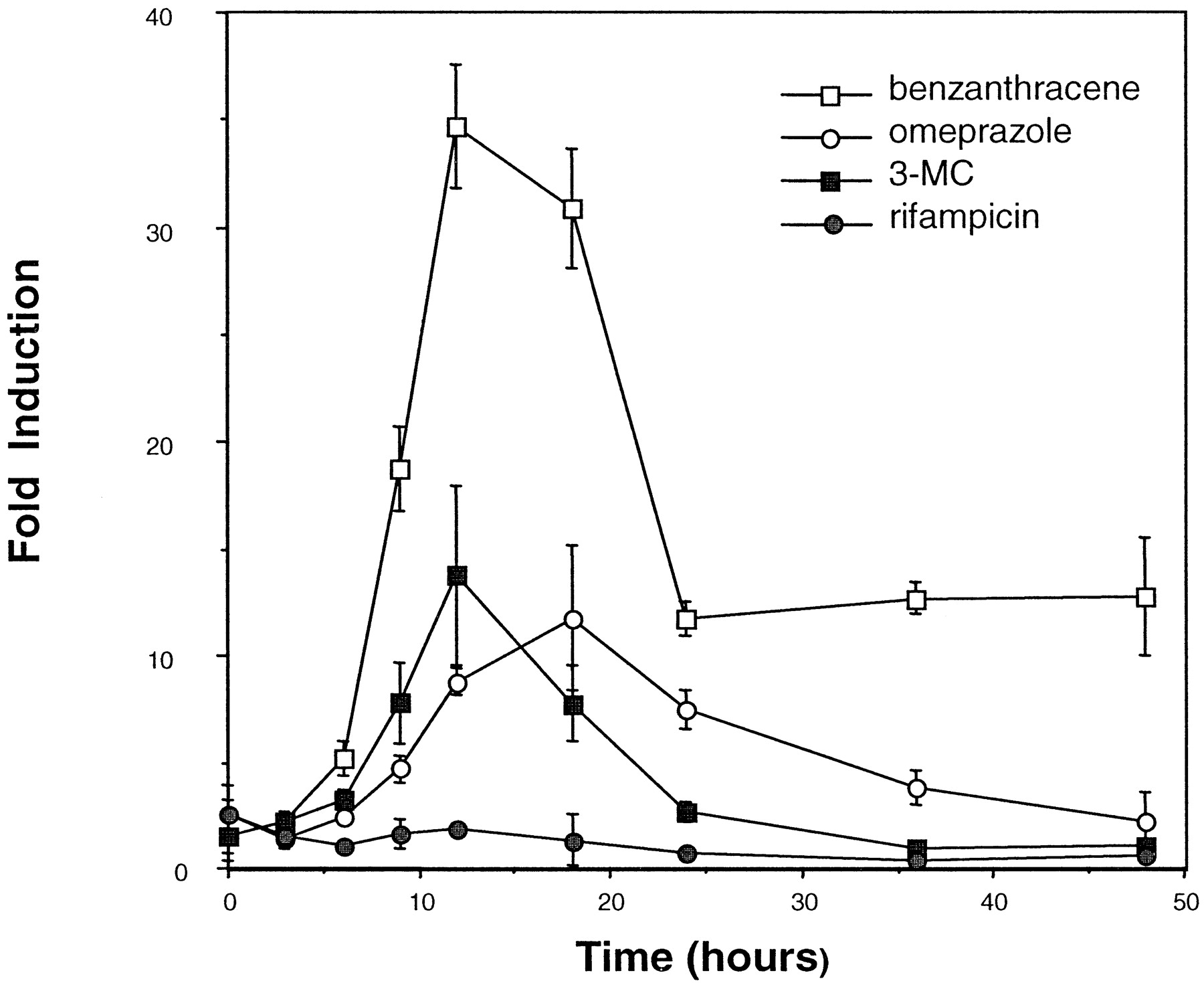
Time-response curve of various CYP1A1 inducers.
The maximal time period for inducer exposure was determined by establishing a time course of inducer-mediated luciferase activity in 101L cells and with the 96-well plates. Enhanced activity was observed within 6 h of dosing with benzanthracene (100 μM), omeprazole (100 μM), and 3-methylcholanthrene (10 μM). Cells were also treated with rifampicin (100 μM) as a negative control. Each point represents the mean of results from three experiments ± S.D.

Dose-response curve of various known CYP1A1 inducers.
The effects of various CYP1A1 inducers were determined using the 96-well plate format and the 101L cells. Dose-response curves were generated for TCDD (0.5–2.5 nM) (A), benzanthracene, and omeprazole (1–200 μM) (B). Each point represents the mean of results from three experiments ± S.D.
The feasibility of using HTS for mechanistic studies was also explored. To determine a mechanism that may be involved in flavonoid prevention of chemical carcinogenesis, we examined the effects of several dietary flavonoids on CYP1A1 induction. Results could indicate whether the flavonoid exhibited Ah receptor agonist and/or antagonist activities. Initial studies examined the ability of various naturally occurring flavonoids to induce CYP1A1 promoter-mediated reporter gene activity in the 101L cell line. Dose-response curves for GTE, EGCG, quercetin, curcumin, kaempferol, naringenin, apigenin, and resveratrol were determined. Of these flavonoids, resveratrol (20 μM), produced the greatest induction of CYP1A1 (10-fold). The second most effective flavonoid inducers were apigenin, quercetin, and curcumin (3-fold). A 3-fold elevation in luciferase activity was observed with 5 μM apigenin treatment, whereas higher doses of quercetin and curcumin (20 μM) were required for similar levels of induction. Doses higher than 5 μM apigenin produced a decline in CYP1A1 induction, which would most likely indicate cytotoxicity. GTE (0.1 mg/ml) (Fig.4, inset) and kaempferol also produced a slight induction (2–2.5-fold). The remaining flavonoids exhibited negligible effects (<2-fold) on CYP1A1 promoter-mediated induction of luciferase activity at concentrations ranging from 1 to 20 μM (Fig.4).

Dose-response curves for various flavonoids.
Using the 96-well plate format and the 101L cells, dose-response curves were generated for GTE (inset). Doses ranged from 0.01 to 0.2 mg/ml with 18 h of exposure. Dose-response curves were also determined for EGCG, quercetin, curcumin, kaempferol, naringenin, apigenin, and resveratrol and ranged from 1 to 20 μM. Cells were exposed to each agent for 18 h. Each point represents the mean of results from three experiments ± S.D.
To validate similar inductive responses of the endogenous CYP1A1 gene, HepG2 cells were also treated with the same flavonoids. Enhanced CYP1A1 mRNA expression was observed in cells treated with GTE (10% TCDD-mediated induction), resveratrol (12% TCDD induction), and apigenin (1% TCDD induction) (Table 1). Although increased expression of CYP1A1 mRNA occurred with these flavonoids, the induction was much less than β-naphthoflavone (50% TCDD response). Collectively, GTE, resveratrol, and apigenin appear to be weak agonists for the Ah receptor.
The effect of flavonoids on CYP1A1 mRNA levels in HepG2 cells
The ability of flavonoids to exhibit Ah receptor antagonism activity was also examined using this high-volume screening system. Cotreatment of the 101L cells with TCDD and flavonoids in the 96-well plate assay resulted in decreased TCDD-mediated induction of reporter gene activity by some of the flavonoids, indicating that certain dietary agents exhibited antagonist activity (Fig. 5). When the 101L cells were cotreated with GTE and TCDD, a 58% reduction in luciferase activity was observed compared with cells treated with TCDD alone. Furthermore, the flavonoids naringenin and apigenin produced a 77 and 74% reduction, respectively, in TCDD-mediated induction. The results of these studies demonstrate that these dietary flavonoids are capable of antagonizing TCDD-mediated induction of CYP1A1 promoter activity, with naringenin having the greatest effect (Fig. 5). Based on results where apigenin or GTE alone displayed a 2.5- to 3-fold induction of CYP1A1-mediated reporter gene activity (Fig. 4), these flavonoids appear to exhibit agonist and antagonist activity toward the Ah receptor. The other flavonoids either produced no change in TCDD-mediated induction of luciferase activity or stimulated its effects. Indeed, cotreatment with TCDD and curcumin produced a 1.5-fold stimulation above the effects of TCDD alone, suggesting that mechanisms in addition to those involving the AhR may play a role in the induction of this P450 by curcumin. When HepG2 cells were cotreated with TCDD and individual flavonoids, results similar to those obtained with the reporter gene assay were observed. Resveratrol produced a slight decrease in the TCDD inductive response of CYP1A1 mRNA (14% reduction), whereas apigenin and naringenin produced significant reductions in CYP1A1 mRNA accumulation mediated by TCDD (57–70% decreases) (Table 1). These results corroborate those produced in the 101L cell line and suggest that apigenin and naringenin have AhR antagonist activity.

The effects of cotreatment with TCDD and each flavonoid.
The CYP1A1-containing cell line was treated with 10 μM each flavonoid or 0.1 mg/ml GTE and 2 nM TCDD. Cells were exposed to both agents for 18 h. Results represent the mean of three experiments ± S.D.
To demonstrate whether similar effects would occur in primary cultures of human hepatocytes, Northern analyses were performed on mRNA isolated from these cells treated with TCDD, flavonoids, or a combination of TCDD and individual flavonoids. Results revealed findings similar to those produced by the high-volume screening system. Resveratrol enhanced CYP1A1 mRNA levels to 5 and 12% of TCDD induction in hepatocytes from two individual liver samples (subjects A and C), whereas GTE enhanced CYP1A1 mRNA levels to 34% of TCDD induction in one culture (Table 2, subject A). In comparison, 100 μM benzanthracene produced induction of CYP1A1 mRNA to 50% of that observed with TCDD in all subjects. In hepatocytes from one subject, not only benzanthracene, but also resveratrol, apigenin, and kaempferol produced accumulation of CYP1A1 mRNA (Table 2, subject C). Resveratrol increased expression to 12%, apigenin to 3%, and kaempferol to 10% of that observed with benzanthracene. Hepatocytes from two other subjects (Table 2, subjects B and D) did not display CYP1A1 induction with any of the flavonoids, but did exhibit CYP1A1 mRNA accumulation produced by TCDD and benzanthracene (50% TCDD levels). Human hepatocytes were also cotreated with TCDD and individual flavonoids. Resveratrol produced a 49% reduction in enhanced levels of CYP1A1 mRNA produced by TCDD. Apigenin and naringenin produced 78 and 80% reductions, respectively, in TCDD-mediated increases of CYP1A1 mRNA (Table 2). These results were similar to those obtained from cotreatment of the 101L cell line with TCDD and apigenin or naringenin (Fig. 5).
The effect of flavonoids on CYP1A1 mRNA levels in primary cultures of human hepatocytes
Discussion
The study described here uses a reporter gene assay and a stable cell line, namely 101L cells (Postlind et al., 1993), to screen potential CYP1A inducers. Stable cell lines harboring P450 enhancers and reporter genes are advantageous for screening applications because the need to continually transfect cells is alleviated, eliminating variability associated with transient transfections. Stably integrated cells also markedly increase sensitivity, allowing induction to be easily assessed. Consistent results are obtained, and the stable cells allow an alternative to other systems that are time-consuming and labor-intensive. Thus, the use of stable cell lines with P450 enhancers can facilitate screening of potential inducers. Indeed, the 101L reporter gene system is an application currently being used in 6-well plate formats by industry to screen environmental samples for the presence of CYP1A1-inducing compounds (Jones et al., 2000).
To develop a high-throughput system with stable cell lines, the previously characterized 101L cells were initially plated in either 24- or 96-well plates and treated with benzanthracene (Fig. 1). Results generated from these experiments indicated that the 96-well plate format was as efficient as the 24-well plates. Furthermore, in the high-throughput (96-well) format, there was minimum background and less than 10% well-to-well variability. In the presence of various CYP1A inducers, maximum induction (12–35-fold) occurred within a 24-h exposure period, similar to that obtained in 6-well plates (Postlind et al., 1993; Quattrochi and Tukey, 1993). To our knowledge, there is only one additional report describing a 96-well plate screening procedure using stable cell lines harboring P450 enhancer elements and reporter genes. The previous investigation (Ziccardi et al., 2000) used a similar format to screen serum samples for Ah receptor ligands. The advantage of the format described here and by Ziccardi et al. (2000) is that several drugs or compounds can be screened on a single plate in a relatively short time period.
To test this high-throughput format further, additional CYP1A inducers were examined. A dose-response curve was established for benzanthracene. Maximum induction in 101L cells in 6-well plates occurred at a dose of 50 μM benzanthracene, as previously reported byJones et al. (2000). The same dose produced maximum CYP1A1-mediated luciferase activity (33-fold) in the study described here with the 96-well plates (Fig. 3B). Other known CYP1A inducers including 3-methylcholanthrene, TCDD, and omeprazole also produced induction of luciferase activity in the 96-well plate format, whereas rifampicin, a CYP3A4 inducer, had no effect (Fig. 1), confirming that this system responds solely to CYP1A inducers. TCDD and/or benzanthracene also induced CYP1A1 mRNA in HepG2 cells (Table 1) and in all four human hepatocyte samples tested. Although not tested here, omeprazole has been shown in previous investigations to induce CYP1As in human hepatocytes (Diaz et al., 1990; Shih et al., 1999). Therefore, when an inducer produces 12-fold or greater increases in luciferase activity in HTS, in all likelihood induction of CYP1A1 by the same agent would occur in human hepatocytes.
To determine whether this high-throughput system could be used to identify novel CYP1A1-inducing agents, we examined the ability of a variety of dietary flavonoids to induce CYP1A1. Of the flavonoids examined, only resveratrol produced a substantial increase (10-fold) in CYP1A1-mediated luciferase activity. However, cells treated with concentrations less than 20 μM resveratrol had negligible effects on luciferase activity, consistent with previous reports that this agent does not induce CYP1A1 mRNA in breast cancer cell lines or HepG2 cells (Ciolino et al., 1998; Casper et al., 1999). When induction observed with the reporter gene assay was compared with CYP1A1 mRNA accumulation in primary hepatocytes and HepG2 cells, resveratrol again produced increases in CYP1A1 mRNA from HepG2 cells and in hepatocytes from two individuals (Tables 1 and 2, subjects A and C). These results suggest that agents producing 10-fold increases in luciferase activity observed in the HTS could also produce CYP1A1 induction in hepatocytes. Those flavonoids producing 2.5-fold induction or greater in the high-throughput system, namely GTE and apigenin, also produced slight increases in the accumulation of CYP1A1 mRNA in HepG2 cells (Table 1). However, they only produced increases in CYP1A1 mRNA in primary hepatocytes isolated from one of three individuals examined here. Similarly, kaempferol, which produced 2-fold increases in luciferase activity, also caused accumulation of CYP1A1 mRNA in hepatocytes from a single individual. In contrast, quercetin and curcumin did not elicit induction of CYP1A1 mRNA in isolated hepatocytes (data not shown), but did produce moderate increases (2.5–3-fold) in luciferase activity. Thus, as described under Results, this disparity between the HTS and human hepatocytes among various agents suggests that when reporter assays exhibit relatively low levels of induction by a particular agent (e.g., 2–3-fold), increases in primary hepatocyte CYP1A1 may or may not occur. Because of the sensitivity associated with reporter gene assays, weak inducers may not exhibit pharmacologically significant elevations of CYP1A1 in vivo. In this situation, final testing in hepatocytes may be needed to determine whether the agent induces CYP1A1 in primary cultures.
Based on our results obtained with the HTS, less than a 2-fold induction of luciferase activity suggests that increased expression of CYP1A1 would be unlikely to occur in primary hepatocytes. The importance of the hepatocyte findings corroborating those of HTS lies in the ability to extrapolate human hepatocyte data to the in vivo situation (Kedderis, 1997; Ito et al., 1998). For example, omeprazole produced induction of CYP1As in both isolated human hepatocytes (Diaz et al., 1990; Shih et al., 1999) and in vivo (Rost et al., 1992). In general, the pharmacokinetics of xenobiotics have been well predicted from studies with isolated hepatocytes (Kedderis, 1997). In our study, we find good agreement between results generated in the stably transfected cells and human liver cells (primary hepatocytes and HepG2 cells), suggesting that cell lines stably transfected with CYP enhancers would be able to predict the in vivo situation.
Consequently, HTS for assessing CYP1A1 induction is useful in identifying agents that might elevate expression of CYP1A1 via the Ah receptor. Furthermore, this system can be used to determine mechanisms involved in CYP induction. We demonstrated that certain flavonoids were identified as exhibiting weak agonist and/or antagonist activity toward the Ah receptor. Regarding the reliability of this high-volume screening system for identifying CYP inducers, signal-to-noise ratios were low, and well-to-well and replicate variability were below 10%, allowing induction to be easily detected in this system. Moreover, results generated with HTS reflected inducer responses obtained in isolated human hepatocytes or HepG2 cells. That extensive variability occurred among hepatocyte samples was also illustrated (Table 2), suggesting the importance of identifying an alternative system for monitoring P450 induction by xenobiotics.
There are limitations that are associated with reporter-based assays, including the induction of endogenous HepG2 CYP1A1 leading to a reduction in inducer-mediated response over time. This was illustrated by the time-response experiments (Fig. 2). Thus, attention must be paid to the length of time that cells are exposed to each inducer. Furthermore, an additional limitation might be the potential for other regulatory sequences within the luciferase construct to modulate or interfere with the induction responses, thereby confounding results. We cannot exclude the possibility that the dietary agents examined here might work through other regulatory sequences or by modulating the induction response via altering other signaling pathways. However, flavonoids tested in this study have been shown by others to function as ligands or to compete with the Ah receptor for TCDD binding (Ciolino et al., 1998; Casper et al., 1999). Overall, stably integrated cell lines harboring enhancer elements of P450 genes appear to be highly conducive to HTS. These cell lines used in a 96-well format can facilitate screening of new chemical entities for induction of human P450 enzymes in a timely and efficient manner.
Footnotes
-
This work was supported by National Institutes of Health Grants GM58287 (S.A.), GM49511 (J.R.), AA08990 (J.R.), and GM54477(L.Q.), by the Liver Transplant, Procurement, and Distribution System (DK62274), and by a grant (99A097) from the American Institute for Cancer Research (to L.Q.). Portions of this work were presented at the International Society for the Study of Xenobiotics (ISSX) meetings in Indianapolis, IN, October, 2000.
-
↵2 U. S. Food and Drug Administration. Guidance for Industry Drug metabolism/drug interaction studies in the drug development process: Studies in vitro. The Drug information Branch, Center for Drug Evaluation and Research. 1998. Ref Type: Unpublished Work.
- Abbreviations used are::
- CYP
- cytochrome P450
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- EGCG
- (−)-epigallocatechin gallate
- GTE
- green tea extract
- HTS
- high-throughput screening
- Ah
- aryl hydrocarbon
- AhR
- Ah receptor
- 3-MC
- 3-methylcholanthrene
- DMSO
- dimethylsulfoxide
- DMEM
- Dulbecco's modified Eagle's medium, FBS, fetal bovine serum
- Received January 26, 2001.
- Accepted April 17, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}