


Abstract
The effects of hepatic uptake and efflux transporters on erythromycin (ERY) disposition and metabolism were examined by comparing results from rat hepatic microsomes, freshly isolated hepatocytes, and in vivo studies. Uptake studies carried out in freshly isolated rat hepatocytes showed that ERY and its metabolite (N-demethyl-ERY) are substrates of Oatp1a4 and Oatp1b2. Whereas rifampin and GG918 [GF120918: N-{4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)-ethyl]-phenyl}-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamine] exerted minimal effects on metabolism in microsomes, rifampin (2.5 μM) and GG918 (0.5 μM) significantly decreased and increased ERY metabolism in hepatocytes, respectively. Concentration-time course studies further demonstrated that, compared with the intracellular N-demethyl-ERY control area under the curve (AUC) (0.795 ± 0.057 μM · min), a decreased AUC (0.513 ± 0.028 μM · min, p < 0.005) was observed when ERY was coincubated with rifampin, and an increased AUC (2.14 ± 0.21 μM · min, p < 0.05) was found when GG918 was present. The results of the i.v. bolus studies showed that, compared with the ERY clearance of the controls (47.2 ± 12.5 ml/min/kg for the rifampin group and 42.1 ± 5.7 for the GG918 group), a decreased blood clearance, 29.8 ± 6.1 ml/min/kg (p < 0.05) and 21.7 ± 9.0 ml/min/kg (p < 0.01), was observed when rifampin or GG918, respectively, was coadministered. When either inhibitor was codosed with ERY, volume of distribution at steady state was unchanged, but t1/2 and mean residence time significantly increased compared with the controls. Hepatic uptake and efflux transporters modulate intracellular concentrations of ERY, thereby affecting metabolism. The interplay of transporters and enzymes must be considered in evaluating potential drug-drug interactions.
Liver is a major site of metabolism of many xenobiotics and endogenous molecules. The process of metabolism starts with the uptake of the molecules via passive diffusion and/or active transport. Hepatic uptake transporters are localized on the sinusoidal membrane of the hepatocytes and are members of the solute carrier (SLC) family (Meier et al., 1997). The uptake of xenobiotics is predominantly mediated by the isoforms of organic anion-transporting polypeptides (OATP in human/Oatp in rats), organic anion transporter 2 (OAT2/Oat2), and organic cation transporter 1 (OCT1/oct1) (Meier et al., 1997; van Montfoort et al., 2003; Hagenbuch and Meier, 2004). Once a substrate enters the cell, it is subject to metabolism by phase I and/or phase II enzymes. These enzymes are responsible for the conversion of nonpolar lipophilic compounds to their more polar hydrophilic metabolites before excretion. Phase I enzymes, such as cytochromes P450 (P450s), are involved in oxidation and reduction reactions, whereas phase II enzymes such as UDP-glucuronosyltransferase and glutathione S-transferase are involved in bimolecular conjugation reactions. The substrate and its metabolites can either exit back to the blood via diffusion and/or basolateral efflux transporters or exit to the bile through diffusion and/or canalicular efflux transporters such as P-glycoprotein (P-gp) and MRP2/Mrp2 (Ambudkar et al., 1999; Konig et al., 1999). The canalicular efflux transporters are members of the ATP-binding cassette superfamily; essentially, they mediate the extrusion of substrates that are neutral, or positively or negatively charged from inside the cell to the bile (Ambudkar et al., 1999). Uptake and/or efflux transporters may facilitate or limit the hepatic metabolism of substrate compounds.
Altered pharmacokinetics due to hepatic enzyme-based drug-drug interactions have been extensively described in the literature; however, less is known about interactions associated with hepatic transporters. Recently, several significant drug-drug interactions, such as cyclosporin A with bosentan (Treiber et al., 2004), cyclosporin A with cerivastatin (Shitara et al., 2003, 2004a), and gemfibrozil with cerivastatin (Shitara et al., 2004b), have been attributed in part to inhibition of hepatic uptake transporters. Sun et al. (2004) reported that uremic toxins accumulated in the circulation of late stage renal failure patients also inhibit hepatic uptake transporters, leading to an increase in systemic exposure of several drugs. Recently, we have shown that the interplay of membrane transporters and metabolizing enzymes is important in drug disposition and metabolism (Benet et al., 2003, 2004). In the intestine, CYP3A and P-gp function synergistically to limit substrates entering the blood from the lumen (Cummins et al., 2002, 2003, 2004).
Previously, we have shown that hepatic transporters play important roles in drug disposition and metabolism in freshly isolated rat hepatocytes and in the isolated perfused rat liver system, where the integrity of hepatic transporters is intact compared with rat liver microsomes, where the transporters are absent (Lam and Benet, 2004; Lau et al., 2004, 2006a; Sun et al., 2004). In addition, rifampin (an inhibitor of Oatps and Oat) significantly decreased and GG918 (a P-gp inhibitor) significantly increased the metabolism of digoxin in hepatocytes, while exhibiting little effect on metabolism in microsomes (Lam and Benet, 2004). Based on these previous results, we further hypothesize that metabolic drug-drug interactions at the hepatic uptake or efflux transporter level, independent of enzymatic activity, alter metabolism and pharmacokinetics (PK) of substrate compounds. In this study, our hypothesis was tested in vitro and, for the first time, in vivo in rats.
The model compound chosen to examine the hypothesis was erythromycin (ERY), an antibiotic effective against infections caused by both Gram-positive and Gram-negative bacteria. In humans, ERY is partially metabolized by CYP3A4 to its major metabolite, N-demethyl-ERY, but ERY is excreted primarily unchanged in the bile. Eighty percent of ERY is bound to plasma protein, and the total clearance of ERY in humans is 9.1 ml/min/kg (Chambers, 2001). It is well demonstrated that ERY and N-demethyl-ERY are excellent substrates of P-gp (Schuetz et al., 1998; Sun et al., 2004). Recently, ERY has been shown to be a good substrate of Oatp1a4, and the estimated saturable process of the total uptake in rat hepatocytes is approximately 56% (Sun et al., 2004) at concentrations much less than Km (72 μM). In the present study, the relative contributions of hepatic uptake and efflux transporters to the metabolism and the pharmacokinetics of ERY were evaluated. We examined trends observed from in vitro studies with that from in vivo studies in rats.
Materials and Methods
ERY, dirithromycin, rifampin, digoxin, [SO3H)27]-cholecystokinin amide fragment 26-33 (CCK8), and quinidine, as well as HPLC-grade dimethyl sulfoxide, tert-butyl-methyl-ether (MTBE), and acetonitrile (ACN) were purchased from Sigma-Aldrich (St. Louis, MO). Erythrocin lactobionate-I.V. (Abbott laboratories, North Chicago, IL) and rifampin (Bedford Laboratories, Bedford, OH) for i.v. infusion were purchased from the University of California, San Francisco (UCSF) pharmacy for research use only. N-demethyl-ERY standard was purchased from U.S. Pharmacopoeia (Rockville, MD). GG918 (GF120918) was graciously supplied by GlaxoSmithKline (Research Triangle Park, NC). Pooled male Wistar rat liver microsomes were acquired from Xenotech (Lenexa, KS). Male Wistar rats (200–350 g) from Charles River Laboratories (Wilmington, MA) were housed in the UCSF animal care facility with a 12-h light/dark cycle and allowed free access to water and food. Approval of the described studies reported here was obtained from the Committee on Animal Research, UCSF.
Microsome Incubations. The incubation conditions were as described previously (Salphati and Benet, 1999). In brief, each reaction mixture contained 0.5 mg/ml microsomes, 1 mM NADPH, various concentrations of inhibitors, 2 μM ERY, and phosphate buffer. The total DMSO concentration used to solubilize ERY and all inhibitors was less than 1% for all in vitro studies. The total reaction volume was 250 μl. The reaction period was 5 min at 37°C. For each sample, the reaction was stopped via protein precipitation by addition of an equal volume of ACN containing the internal standard (IS), dirithromycin (1 μg/ml). The supernatants were stored at –80°C for LC-MS analysis.
Hepatocyte Isolation. Anesthesia was induced by intraperitoneal injection with a 1 ml/kg dose of ketamine/xylazine (80 mg/ml:12 mg/ml) before surgery. The portal vein was cannulated with an i.v. catheter (catalog number 2007-04; Becton Dickinson, Sandy, UT) and perfused with oxygenated liver perfusion buffer (Invitrogen, Carlsbad, CA) for 5 min at 30 ml/min, followed by perfusion with an oxygenated hepatocyte washing buffer (Invitrogen) modified with 2 mM l-glutamine, 10 mM HEPES, and 1.2 U/ml collagenase (Sigma-Aldrich) for 5 min at 20 ml/min. The digested liver was excised and broken down. Hepatocytes were then washed twice with an ice-cold hepatocyte wash buffer containing 2 mM l-glutamine and 10 mM HEPES and were centrifuged at 50g for 2 to 3 min. Cell viability was determined using the trypan blue exclusion method. Cells with viability of greater than 90% were used for further studies.
Hepatocyte Incubations. Hepatocyte incubations were carried out immediately after cell isolation. Cells were resuspended and diluted to 2 × 106 per ml in Krebs-Henseleit buffer (pH 7.4) containing 0.21 g/l sodium bicarbonate and supplemented with 1% BSA and 10 mM glucose. Cell suspensions for all hepatocyte incubations were prewarmed for 5 min in a 37°C shaking water bath before initiation of incubations.
For the uptake studies, ERY (2 μM) or N-demethyl-ERY (2 μM) with and without 50 μM rifampin, 50 μM digoxin, 30 μM CCK8, 50 μM quinidine, or 0.5 μM GG918 was added to the cell suspensions that were shaking in a 37°C water bath. At 2 min, the reactions were terminated by transferring 106 hepatocytes into a centrifuge tube containing 700 μl of a mixture of silicone oil and mineral oil (Shitara et al., 2003) and centrifuged at 13,000g for 10 s. After removing the buffer layer and the oil layer, each cell pellet was resuspended in 100 μl of water and sonicated for 15 min to ensure a thorough cell lysis. This was followed by adding 200 μl of ACN containing IS and spinning at 13,000g for 15 min to precipitate protein. The supernatant was then transferred into a HPLC vial (Hewlett Packard, Palo Alto, CA) for LC-MS analysis.
For evaluating transporter inhibition on metabolism, ERY (2 μM) and various concentrations of rifampin or GG918 were coincubated for 5 min in a 37°C shaking water bath. The reaction was stopped by transferring 1 ml of cells to a fresh glass tube containing MTBE and IS followed by vortexing. All samples were spun down at 2000g for 10 min. After quick-freezing the aqueous layer in a methanol/dry ice bath, the organic layer was poured into a new tube and evaporated under nitrogen gas. Each sample was reconstituted with 300 μl of ACN/water (50:50, v/v) for LC-MS analysis.
For the time course studies, 15 ml of hepatocytes (2 × 106/ml) were warmed in a 50 ml flask shaking in a 37°C water bath for 5 min. Each study was initiated by concomitantly adding ERY (2 μM) with DMSO (control), 2.5 μM rifampin, or 0.5 μM GG918. At 5, 10, 15, 20, 30, and 45 min, 0.5 ml of cells were transferred into a centrifuge tube containing 700 μl of a mixture of silicone oil and mineral oil and centrifuged at 13,000g for 10 s to stop the reaction. Ten seconds later, 1 ml of cells was transferred into a glass tube containing MTBE and IS followed by vortexing to stop the reaction. Sample preparation for the intracellular measurement of ERY metabolism was the same as for the uptake studies. Sample preparation for the measurement of ERY metabolism was the same as that for the inhibition studies.
Surgery and Pharmacokinetic Study in Vivo in Rats. To determine the influence of rifampin and GG918 on ERY metabolism, 24 rats were divided equally into four groups. Since rifampin (25 mg/ml) was dissolved in saline and GG918 (0.25 mg/ml) was dissolved in 10% DMSO, 3% BSA in saline, two sets of vehicle controls were needed for each treatment group. Male Wistar rats (300–350 g) were put under anesthesia induced by intraperitoneal injection with a 1 ml/kg dose of ketamine/xylazine (80 mg/ml:12 mg/ml; Sigma, St. Louis, MO) before surgery and several times during the 4-h study to ensure complete anesthetization. The femoral vein and the femoral artery were cannulated using a 10-cm PE-10 tube (i.d. 0.28 mm, o.d. 0.61 mm; BD Intramedic, Sparks, MD) and a 10-cm SP-35 tube (i.d. 0.5 mm, o.d. 0.9 mm; Natume Co., Tokyo, Japan), respectively. The lines were washed immediately with saline containing 10 units/ml heparin to prevent clotting. The bile duct was cannulated with a 15-cm PE-10 tube, with average bile flow rate of 0.4 ml per 15 min. ERY (10 mg/kg) with vehicle control, or with either rifampin (2.5 mg/kg) or GG918 (0.25 mg/kg), was coadministered through the femoral vein. Blood (150 μl) samples were collected at 3, 5, 10, 15, 30, 60, 90, 120, 180, and 240 min via the femoral artery and stored in K2EDTA-pretreated Microtainer tubes (Becton-Dickinson, Franklin Lakes, NJ). Results from preliminary studies showed that extrapolated areas under the curve (AUCs) were less than 20% of the total AUC over 4 h; thus, in vivo studies were stopped at 240 min. Bile samples were collected at 15-min intervals until 120 min and then, every 30 min. Urine was also collected for the duration of the PK study, and the bladder was emptied at the end of the study. Rat livers were excised and weighed at 240 min. To each 100 μl of blood, bile, urine, and liver homogenate sample, 200 μl of ACN containing IS were added, and samples were centrifuged for 15 min at 13,000g. The supernatants were transferred into HPLC vials and assayed using LC-MS.
Measurement of ERY,N-demethyl-ERY, Rifampin, and GG918. All samples were analyzed with a LC/LC-MS system coupled with a mass selective detector system (Hewlett Packard), which consists of 1100 HPLC components HPLC I and HPLC II as described previously (Christians et al., 2000). The two HPLC systems were connected by a 7240 Rheodyne six-port switching valve mounted on a step motor (Rheodyne, Cotati, CA). The LC/LC-MS system was controlled and the data were integrated by ChemStation software version A.06.01 (Hewlett Packard). Samples (100 μl) were first injected into a 10 × 5 mm Hypersil ODS-1, 5-μm extraction column (Thermo Hypersil-Keystone, Shelton, CT) at a flow rate of 2 ml/min followed by washing with 2 mM ammonium acetate. After 1 min, the switching valve was activated, and the analytes were eluted with ACN in the back-flush mode from the extraction column into a 4.6 mm × 150 mm C18 XTerra MS 5-μm analytical column (Waters, Milford, MA). For the detection of ERY, N-demethyl-ERY, and IS, the analytes were separated isocratically with 60% 2 mM ammonium acetate (pH 9) and 40% ACN at a flow rate of 1.5 ml/min and at 50°C. For the detection of rifampin and GG918, all column and HPLC setups were identical to that in the ERY method; however, the extraction column was washed with water containing 0.1% formic acid, and the inhibitors were eluted isocratically with 70% water containing 0.1% formic acid and 30% ACN.
Data Analysis. The kinetic parameters for the uptake of N-demethyl-ERY were estimated using fitting of the following equation: v = (Vmax × S)/(Km + S) + Pdiff × S, where v is the rate of uptake (pmol/min/106 cells), S is the substrate concentration (μM), Km is the Michaelis-Menten constant (μM), and Pdiff is the nonsaturable diffusion constant (μl/min/106 cells). For the pharmacokinetic studies, terminal half-life (t1/2) was determined from the log-linear regression of the last three concentration measurements. AUC, calculated by the trapezoidal rule, was extrapolated to infinite time by the additions of Cblood, last · t1/2/0.693. Clearance (CL) was calculated by dose/AUC0-inf. Biliary and renal clearances, CLbiliary and CLrenal, respectively, were determined by dividing the amount of ERY excreted into the bile and urine over 240 min by the AUC over that time interval. Renal clearance of the metabolite (CLrenal, met) was calculated using the 240-min blood and urine measurements of the N-demethyl-ERY. CLother was calculated as CL – CLbiliary – CLrenal for ERY measurements. Volume of distribution at steady state (Vss) and mean residence time (MRT) were calculated using moments (Benet and Galeazzi, 1979). Student's t test was used to analyze differences between two groups such as in the in vivo studies. Analysis of variance was used to analyze differences among more than two groups, such as in the in vitro studies. The significance between two means in these groups was evaluated using Bonferroni's multiple comparison test. The p value for statistical significance was set at <0.05.
Results
Uptake of ERY and Its Major MetaboliteN-demethyl-ERY by Freshly Isolated Rat Hepatocytes. The estimated saturable portion of the ERY uptake in the freshly isolated rat hepatocytes has been described previously by Sun et al. (2004) to be 55%. In this study, we examined the uptake kinetics for N-demethyl-ERY. Various concentrations of N-demethyl-ERY were incubated with hepatocytes for 2 min. Both saturable and nonsaturable components were observed (data not shown). The fitted kinetic parameters were 25.0 ± 1.0 pmol/min/106 cells for Vmax, 31.7 ± 4.6 μM for Km, and 0.696 ± 0.014 μl/min/106 cells for Pdiff. The estimated transporter-mediated uptake for N-demethyl-ERY (Vmax/Km) is 53% of the total uptake at the relevant in vitro and in vivo concentrations.

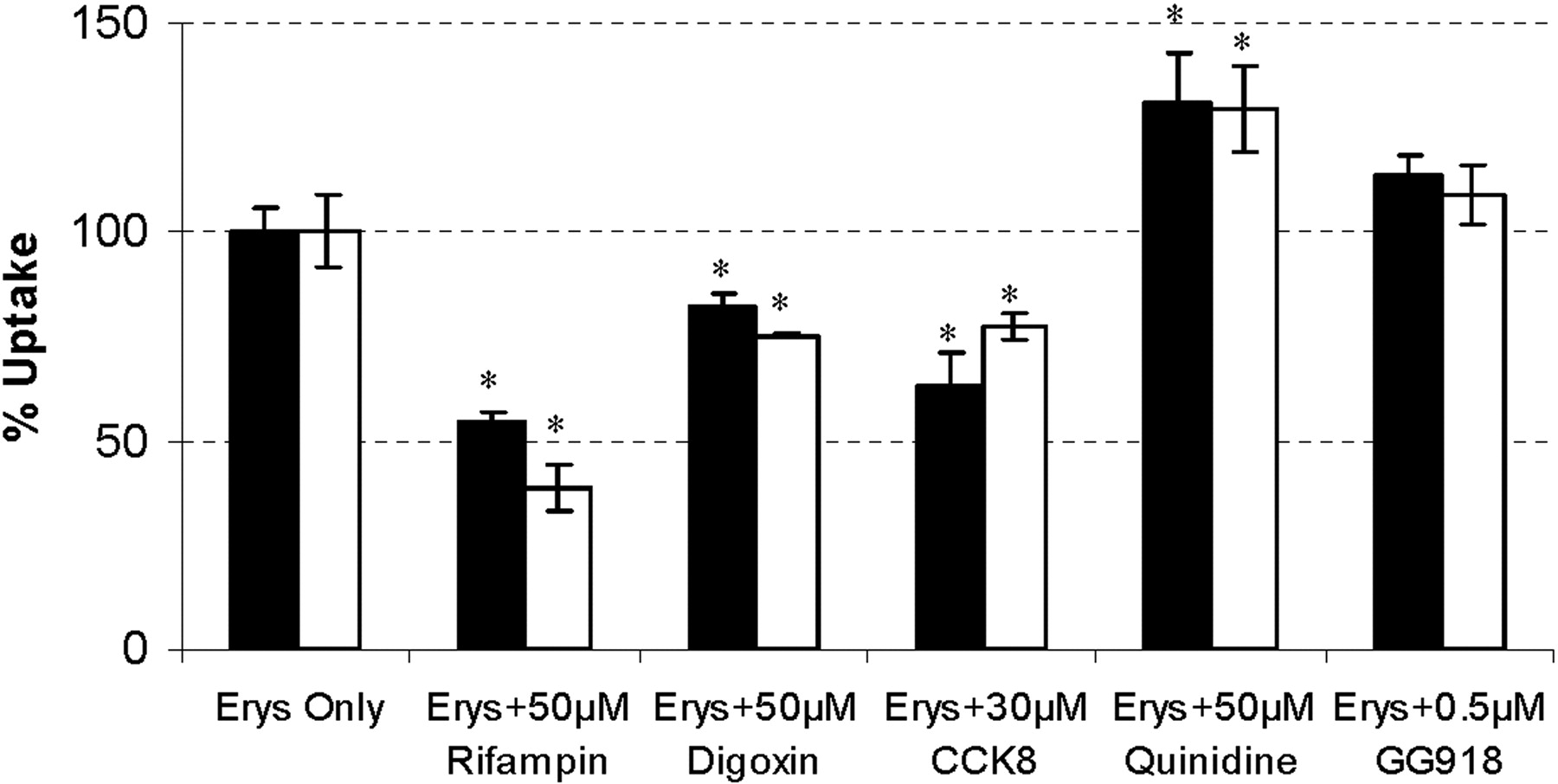
Uptake study of ERY and N-demethyl-ERY in hepatocytes. Concentrations of ERY (2 μM; black bars) and N-demethyl-ERY (2 μM; white bars) were incubated for 2 min with various concentrations of transporter inhibitors. Data are depicted as the mean ± S.D., n = 3. *, p < 0.05, significantly different from control.
To determine which major isoforms of the hepatic uptake transporters are responsible for the uptake of ERY and its metabolite, 2 μM ERY and N-demethyl-ERY were coincubated with various concentrations of transporter inhibitors for 2 min. The substrate concentration (2 μM) was in the linear range of uptake without saturating the uptake transporters (data not shown). Rifampin is a general inhibitor of Oatps and Oats, whereas digoxin and CCK8 differentially inhibit Oatp1a4 and Oatp1b2, respectively (Ismair et al., 2001; Shitara et al., 2002). Quinidine inhibits both Oatp1 and P-gp (Shitara et al., 2002). We have previously shown that GG918 is also an inhibitor of Oatp1a4, but this could only be clearly seen at concentrations much higher than 0.5 μM (Lam and Benet, 2004). The results are depicted in Fig. 1. At a concentration of 50 μM, rifampin reduced the intracellular concentration of ERY and N-demethyl-ERY, respectively, to 54.4 ± 2.7% and 38.8 ± 5.6% of the controls. Digoxin (50 μM) and CCK8 (30 μM) significantly decreased the uptake of ERY and N-demethyl-ERY to 84.7 ± 9.5% and 75.4 ± 0.6%, and 63.5 ± 7.3% and 77.3 ± 3.2%, respectively. Sun et al. (2004) also showed significant uptake inhibition at 100 μM digoxin. Quinidine (50 μM) did not reduce the uptake of ERY and its metabolite but increased their intracellular concentrations by 130 ± 12% and 130 ± 10%, as might be expected if cellular efflux was inhibited. GG918 (0.5 μM) had no significant effects on the uptake of ERY and N-demethyl-ERY, 113 ± 5% and 108 ± 7%, respectively, which indicates a possible increase due to efflux inhibition countered by possible uptake inhibition. This inhibition profile suggests that ERY and N-demethyl-ERY are substrates of Oatp1a4 (Oatp2) and Oatp1b2 (Oatp4) but not Oatp1a1 (Oatp1) in rats.
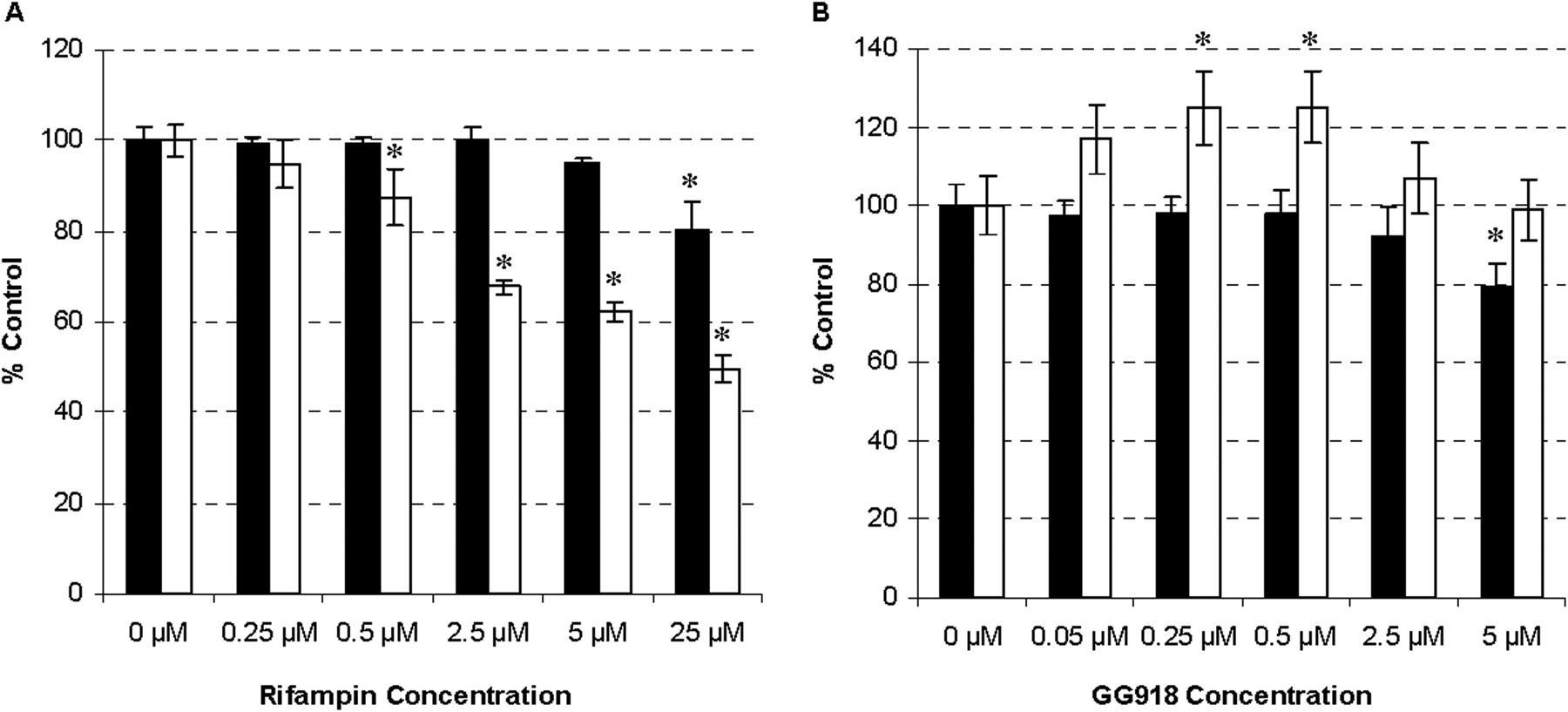
Effects of Rifampin and GG918 on ERY Metabolism in Rat Liver Microsomes and in Freshly Isolated Hepatocytes. Various concentrations of rifampin (Fig. 2A) and GG918 (Fig. 2B) were coincubated with ERY in microsomes and in hepatocytes for 5 min. Concentrations of rifampin less than 25 μM did not show a significant effect on ERY metabolism in microsomes (Fig. 2A, black bars); however, at concentrations of 0.5 μM or higher, rifampin exhibited a significant dose-dependent inhibition of ERY metabolism in the hepatocytes (Fig. 2A, white bars). At concentrations of GG918 less than 5 μM, ERY metabolism was not affected in microsomes (Fig. 2B, black bars). In contrast, a significant increase in ERY metabolism was observed in a dose-dependent manner up to 0.5 μM GG918 in hepatocytes, even though no increase in intracellular concentrations was observed after 2-min incubations at this 0.5 μM GG918 concentration (Fig. 1). However, at GG918 concentrations higher than 0.5 μM, there was a decrease in metabolism in hepatocytes, which suggests inhibition of Oatp1a4 at higher GG918 concentrations, as we have shown previously (Lam and Benet, 2004).

Microsome (black bars) and hepatocyte (white bars) 5-min incubations of ERY with inhibitors. Alteration of N-demethyl-ERY formation by rifampin (A) and GG918 (B). ERY concentration used was 2 μM. Data are depicted as the mean ± S.D., n = 3. *, p < 0.05, for significantly different from control.
Time Course Studies of Effects of Rifampin and GG918 on ERY Metabolism in Hepatocyte Suspension. For the time course studies, 2.5 μM rifampin and 0.5 μM GG918 were used based on the results from Fig. 2, which showed that these two concentrations had no effect on microsome metabolism but a significant effect on hepatocyte metabolism. When 2.5 μM rifampin was coincubated with 2 μM ERY over 45 min, a nonsignificant increase in ERY concentration (cell and media) (Fig. 3A) but a significant decrease in metabolite formation compared with that of ERY-only control (Fig. 3B) were observed. In contrast, when 0.5 μM GG918 was coincubated with ERY, a nonsignificant decrease in ERY concentration (Fig. 3A) but a significant increase in N-demethyl-ERY formation were observed compared with controls (Fig. 3B). AUCs of ERY and N-demethyl-ERY were calculated (Table 1). Mass balance was also determined (Table 1). Compared with the control (100 ± 5%), the mass balance was 101 ± 4% for the rifampin treatment group and 99.3 ± 3.5% for the GG918 treatment group, which indicates that N-demethyl-ERY is the major metabolite formed in the hepatocyte system, and no other metabolites needed to be monitored.
AUC of total measurements (intracellular and incubation media) and intracellular measurements of ERY and N-demethyl-ERY
Data are depicted as the mean ± S.D., n = 3. Mass balances were determined. AUC ratios of N-demethyl-ERY to ERY (Met/ERY ratio) were also calculated.
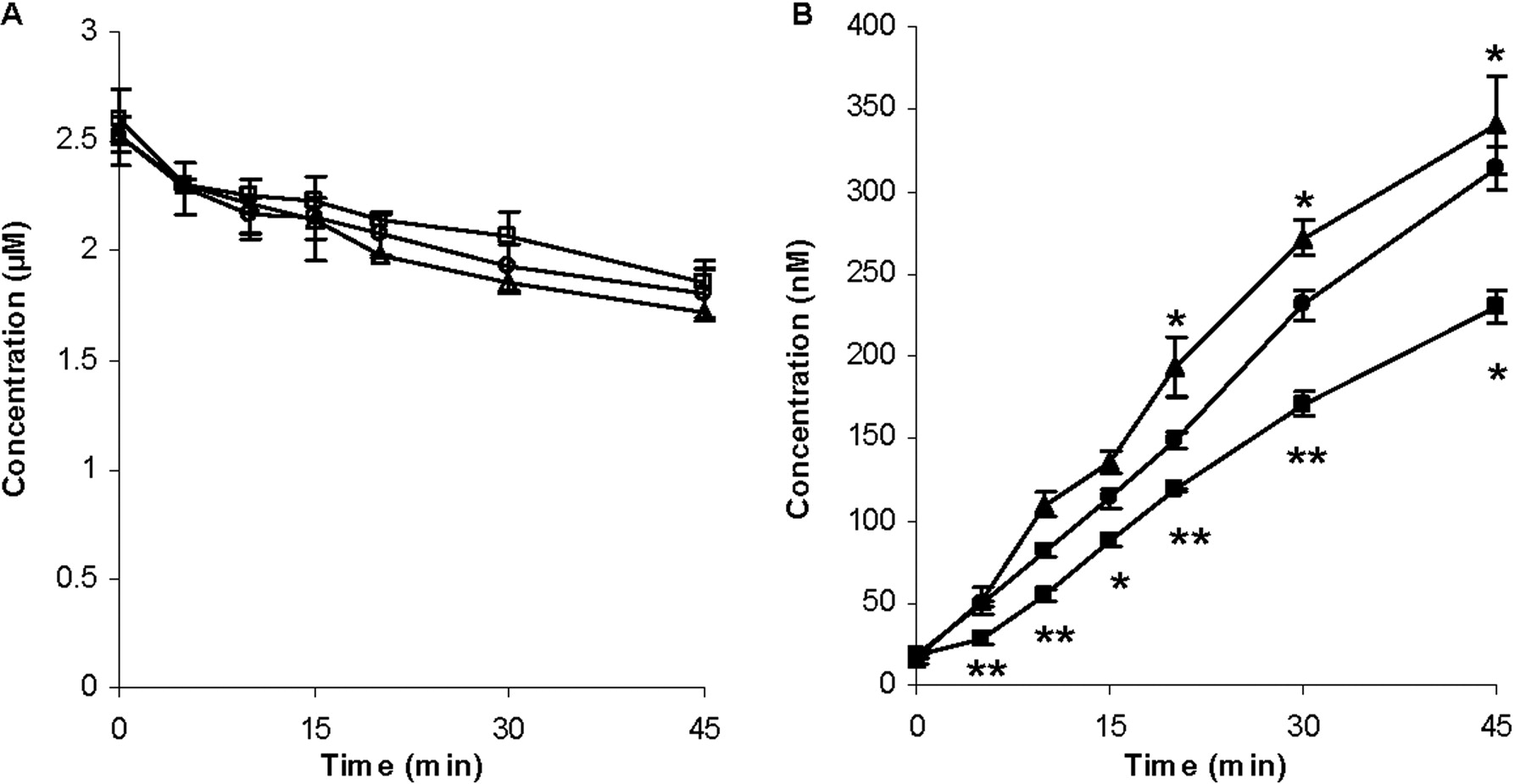
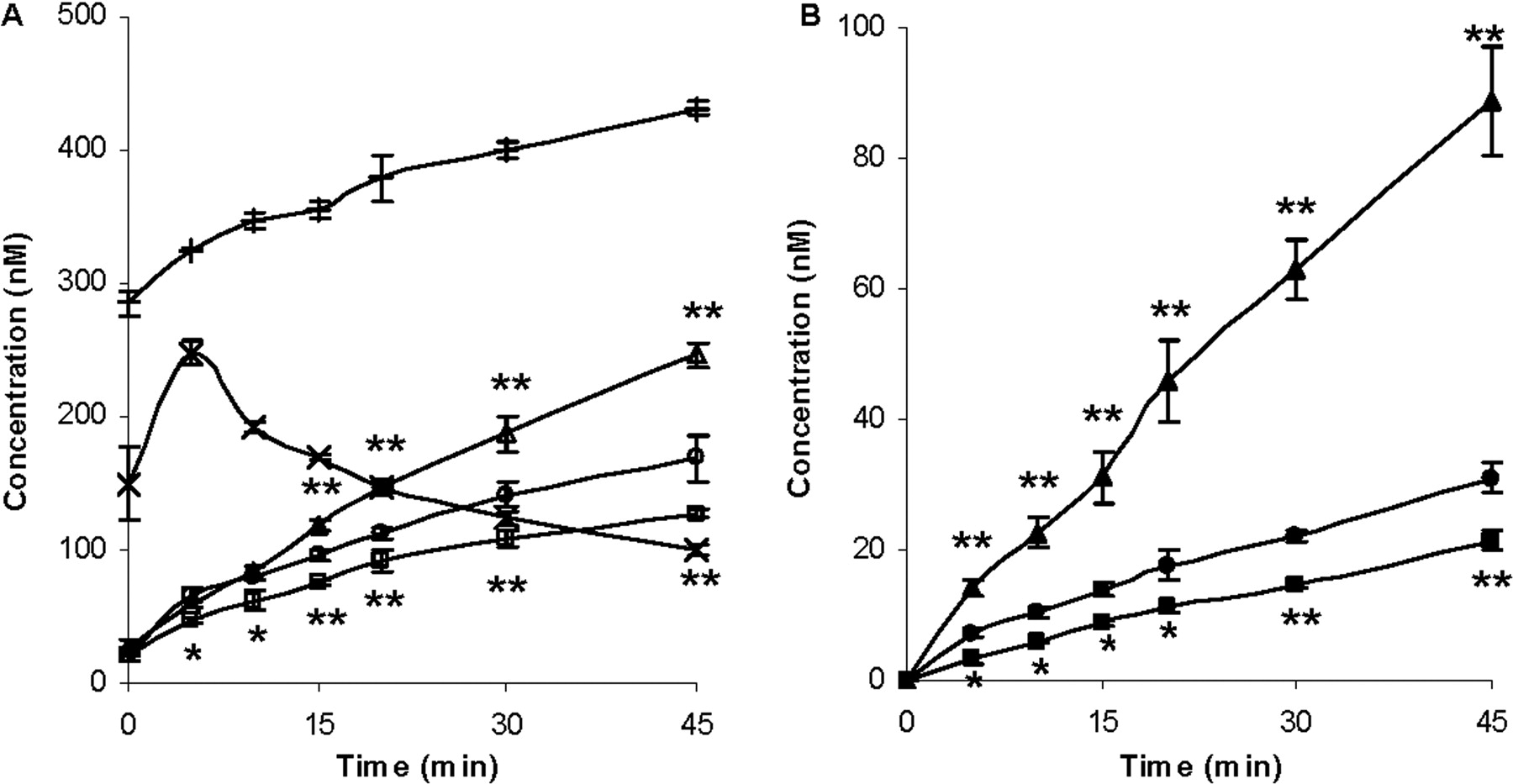
Concentrations of ERY (A) and N-demethyl-ERY (B) in the cells and media over 45 min. Circles represent the ERY control group; squares represent the coadministration of ERY and rifampin group; triangles represent the coadministration of ERY and GG918 group, n = 3. *, p < 0.05; **, p < 0.01, significantly different from control. Lines are drawn point-to-point.
Changes in AUC became significant when the intracellular concentrations of ERY and N-demethyl-ERY were measured (Fig. 4). Inhibition of uptake transporters by rifampin led to a reduction in intracellular concentration of ERY (Fig. 4A) and resulted in a decreased metabolism in comparison with the controls (Fig. 4B). Conversely, when the efflux transporters were inhibited by GG918, an accumulation of ERY resulted, and an increase in metabolism was observed compared with the controls. Intracellular rifampin and GG918 concentrations were also measured. For rifampin, there was a slight increase in concentrations over time, whereas for GG918, a brief increase followed by a steady decrease in concentrations was observed. AUCs and AUC ratios of ERY and N-demethyl-ERY were calculated (Table 1). Coincubation of GG918 with ERY significantly increased the metabolite to parent AUC ratio from 0.155 (control group) to 0.320. In contrast, no significant change in the metabolite to parent ratio was observed when rifampin was coincubated with ERY.
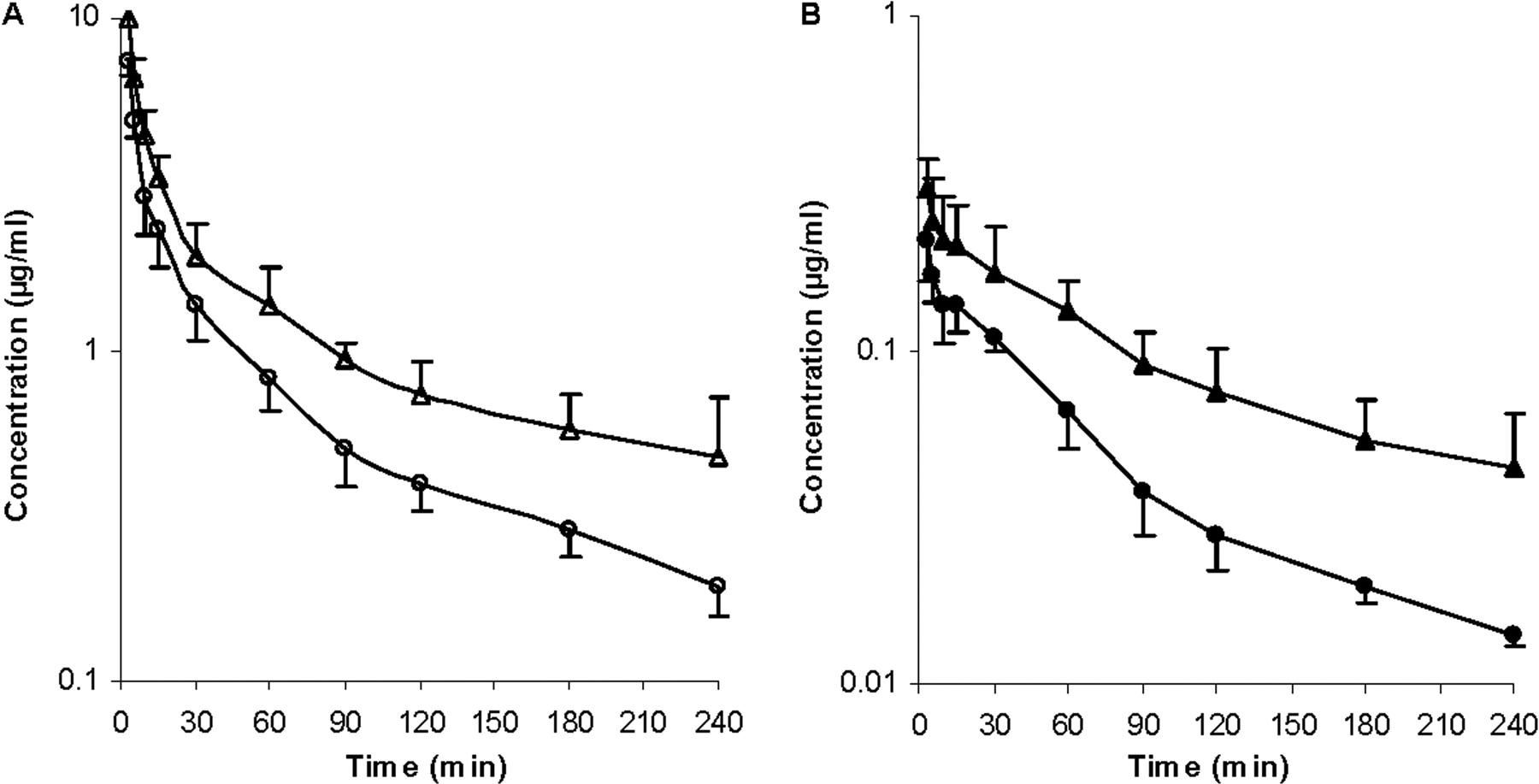
Effects of Rifampin and GG918 on the Pharmacokinetics of ERY in Rats. To determine the pharmacokinetics of ERY and N-demethyl-ERY with and without transporter inhibitors, i.v. bolus studies were carried out in Wistar rats. The blood concentrations of drugs were measured to obviate correction of plasma values, since the blood to plasma concentration ratio is 1.3 for ERY (data not shown). For the rifampin treatment group, the concentration-time profile showed that in the presence of rifampin, there is an increase in AUC of ERY (Fig. 5A) and N-demethyl-ERY (Fig. 5B) compared with that without rifampin. The blood concentrations of rifampin were also measured (Fig. 5A). For the GG918 treatment group, when GG918 was coadministered, there was an increase in AUC of ERY (Fig. 6A) and N-demethyl-ERY (Fig. 6B) compared with that of the controls. Levels of GG918 in the blood were below the detection limit of 250 nM.

Intracellular concentrations of ERY (A, open symbols) and N-demethyl-ERY (B, closed symbols) over 45 min. Circles represent the ERY control group; squares represent the coadministration of ERY and rifampin group; triangles represent the coadministration of ERY and GG918 group; plus signs (+) represent the concentrations of rifampin; crosses (x) represent concentrations of GG918; n = 3. *, p < 0.05; **, p < 0.01, rifampin and GG918 group concentrations significantly different from control. Lines are drawn point-to-point.
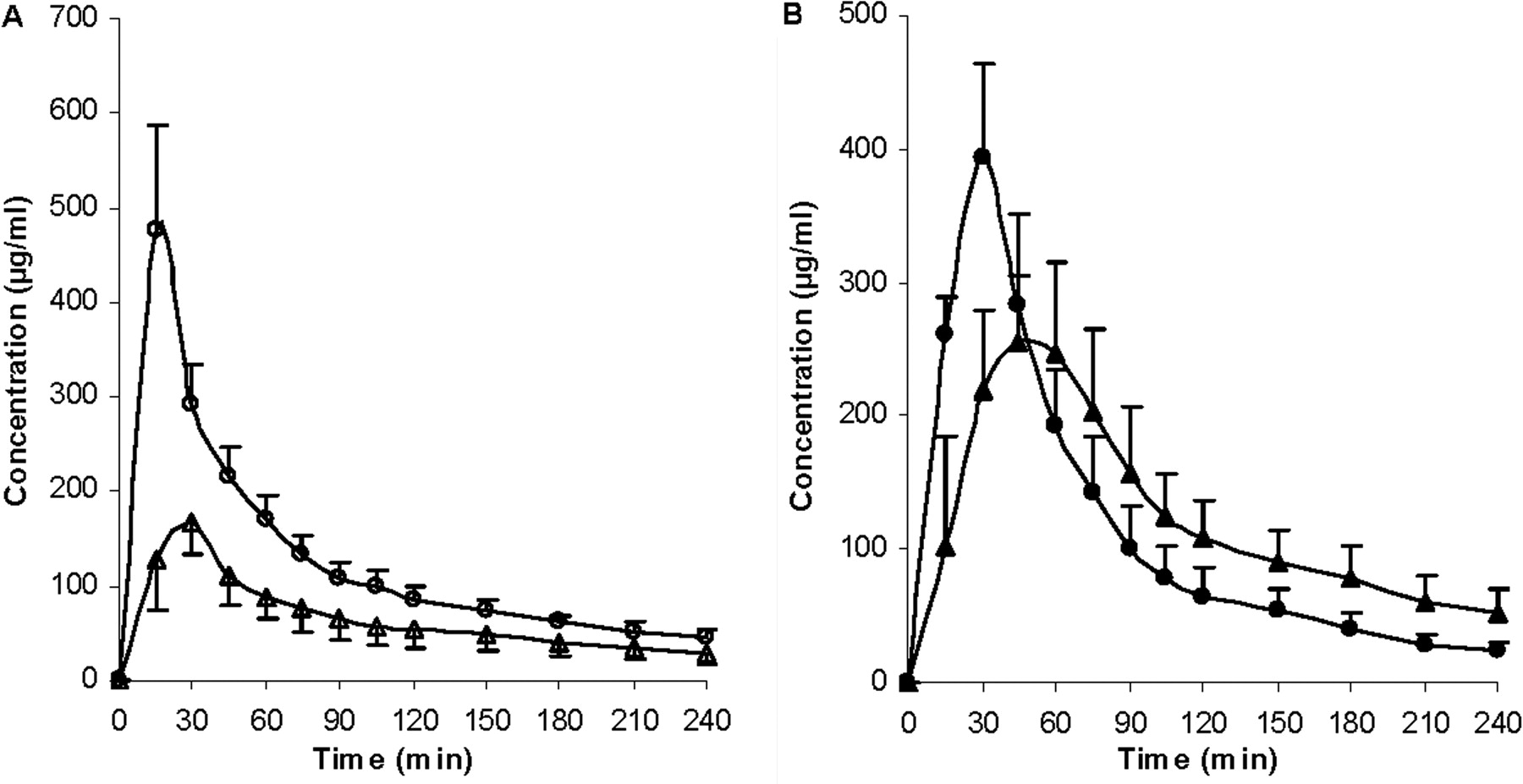
Pharmacokinetic parameters, t1/2, AUC0-inf, CL, MRT, and Vss, were calculated (Table 2). No significant differences were found between the two control groups for t1/2, CLblood and CLother. However, significant differences were observed for Vss, MRT, CLrenal, and CLbiliary, suggesting that the addition of DMSO and BSA to the GG918 dosing solution did not alter overall ERY kinetics but may have affected certain eliminating organs. Compared with their respected controls, t1/2 and MRT were significantly prolonged with either rifampin or GG918 coadministration. The AUC0-inf values of both treatment groups were also increased significantly, which resulted in significant decreases of total clearances relative to the controls (Table 2). Concentrations of ERY and N-demethyl-ERY were measured in the bile (Figs. 7 and 8). Rifampin and GG918 both reduced bile concentrations of ERY and N-demethyl-ERY compared with controls. In the presence of GG918, there was a delay in Cmax of ERY and its metabolite relative to the controls, as would be expected when biliary efflux is inhibited rather than hepatic uptake. Biliary clearances were significantly reduced when rifampin or GG918 were codosed (Table 2). Renal clearances were determined. For the rifampin group, there was no significant change in the renal clearances of ERY or N-demethyl-ERY, whereas a significant decrease in renal clearances was observed for both parent drug and metabolite when GG918 was coadministered with ERY (Table 2). Liver concentrations of ERY, N-demethyl-ERY, rifampin, and GG918 were measured at 240 min (Fig. 9). Parent drug to metabolite concentration ratios were also calculated. For the rifampin group, compared with controls, ERY and N-demethyl-ERY concentrations decreased significantly; however, their ratios were unchanged. For the GG918 group, significant increases of ERY and N-demethyl-ERY concentrations were observed, and their ratios increased in comparison with that of the controls (Fig. 9).
Pharmacokinetic parameters of ERY in rats after i.v. dosing with and without rifampin or GG918 coadministration
Data are depicted as the mean ± S.D., n = 6.
Discussion
We previously reported that hepatic drug metabolism involves the interplay of Oatps, P-gp, and metabolizing enzymes (Lam and Benet, 2004; Lau et al., 2004, 2006a). Here, we compared results from in vitro microsome studies, hepatocyte studies, and rat in vivo pharmacokinetic studies with erythromycin and its primary metabolite to further investigate the significance of transporter-enzyme interplay.

Concentrations of ERY (A, open symbols) and N-demethyl-ERY (B, closed symbols) in the blood over 240 min. Circles represent the ERY control group; squares represent the coadministration of ERY and rifampin group; triangles represent the concentrations of rifampin, n = 6. Lines are drawn point-to-point.

Concentrations of ERY (A, open symbols) and N-demethyl-ERY (B, closed symbols) in the blood over 240 min. Circles represent the ERY control group; triangles represent the coadministration of ERY and GG918 group, n = 6. Lines are drawn point-to-point.
Results from the hepatocyte uptake study here (Fig. 1) and in our previous work (Sun et al., 2004) suggest that ERY and N-demethyl-ERY are probably substrates of Oatp1a4, Oatp1b2, and P-gp. Instead of limiting the uptake of ERYs, quinidine, a good inhibitor of Oatp1a1 and P-gp (Shitara et al., 2002), caused a significant increase in intracellular concentrations of substrates. This is most likely caused by quinidine inhibiting efflux transporters. If that were true, it might be expected that GG918 would also increase intracellular concentrations; however, we suspect that no significant increase was observed since GG918 is also an inhibitor of hepatic uptake (Lam and Benet, 2004). At higher concentrations, GG918 has been shown to inhibit breast cancer resistance protein, but ERY and its metabolite are not substrates for breast cancer resistance protein (data not shown).
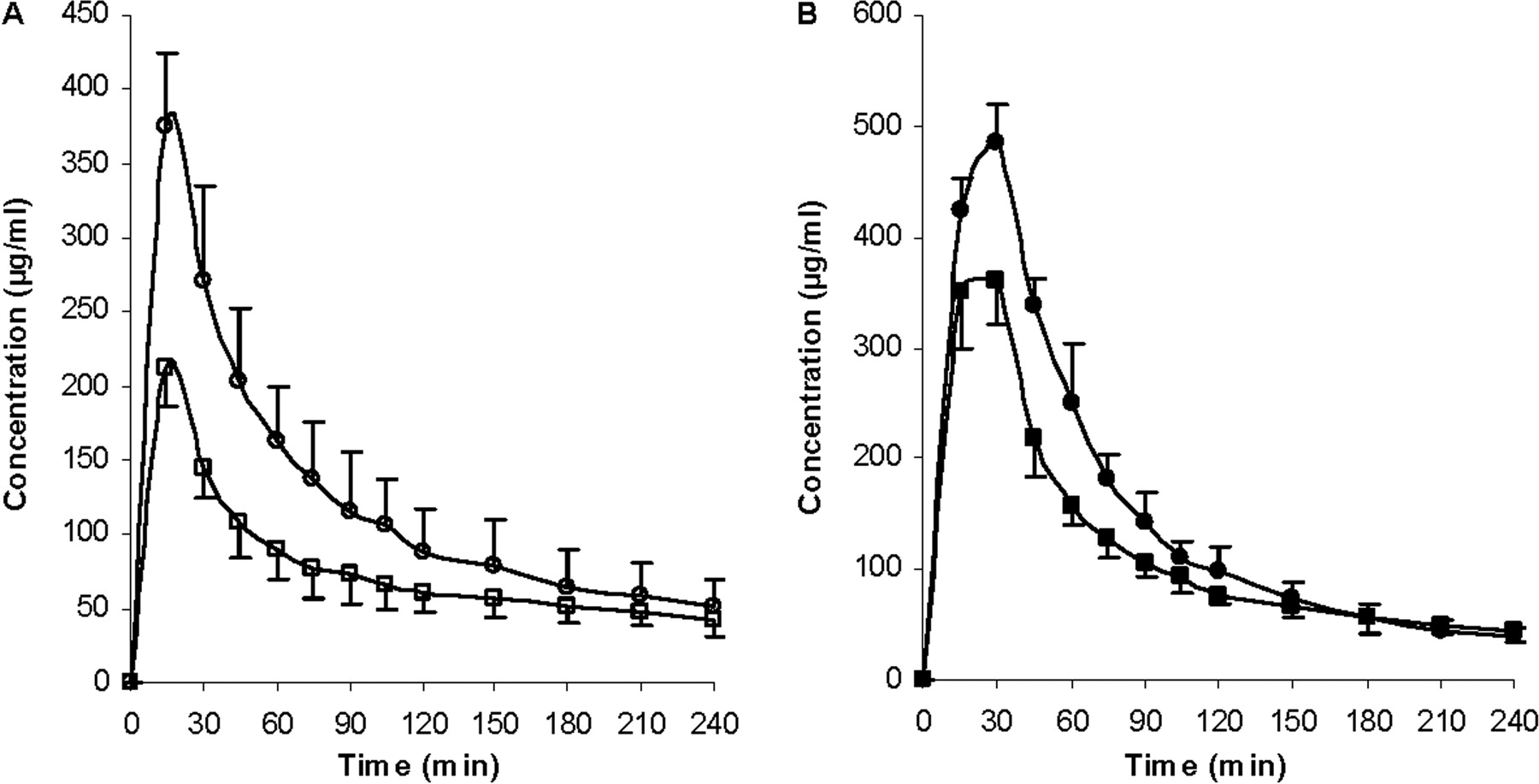
Concentrations of ERY (A, open symbols) and N-demethyl-ERY (B, closed symbols) in the bile over 240 min. Circles represent the ERY control group; squares represent the coadministration of ERY and rifampin group, n = 6. Lines are drawn point-to-point.

Concentrations of ERY (A, open symbols) and N-demethyl-ERY (B, closed symbols) in the bile over 240 min. Circles represent the ERY control group; triangles represent the coadministration of ERY and GG918 group, n = 6. Lines are drawn point-to-point.
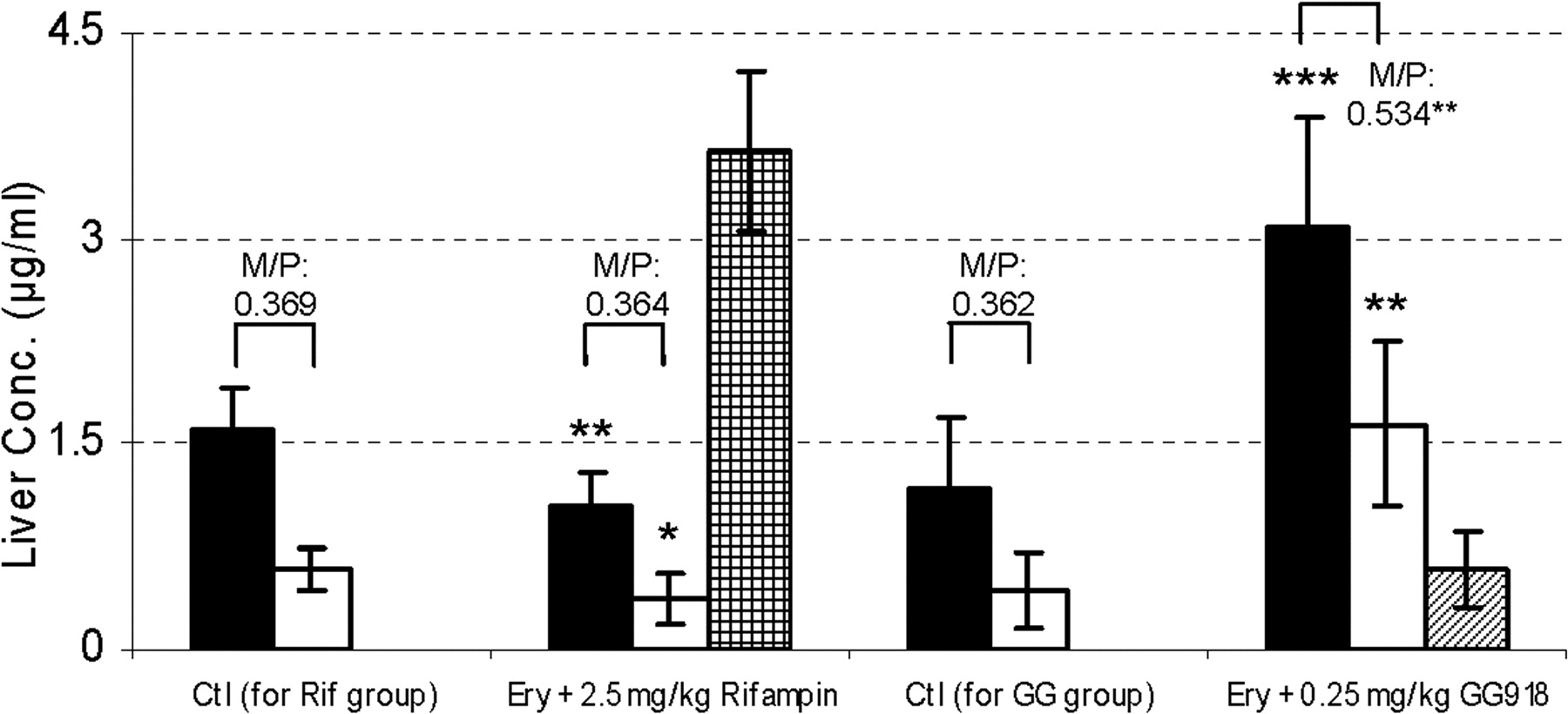
Measurement of drugs in liver. Liver concentrations of ERY (black bars), N-demethyl-ERY (white bars), rifampin (grid bar) and GG918 (striped bar) were measured at 240 min, n = 6. *, p = 0.05; **, p < 0.01; ***, p < 0.001, significantly different from control. M/P represents the concentration ratio of N-demethyl-ERY/ERY.
Results from the hepatocyte inhibition studies show that rifampin and GG918, respectively, cause a significant dose-dependent decrease and increase in ERY metabolism (Fig. 2). In contrast, both inhibitors exerted little effect on ERY metabolism in microsomes at the concentrations that caused transporter inhibition. When ERY was incubated with concentrations of GG918 higher than 0.5 μM, a decreased ERY metabolism was noted in hepatocytes, most likely because of inhibition of Oatp1a4 in rats by GG918. This agrees with our previous observation using digoxin as a substrate (Lam and Benet, 2004). In the microsome incubations, at rifampin concentrations of 25 μM and higher and at GG918 concentrations of 5 μM and higher, ERY metabolism decreased significantly. When surveying metabolic drug-drug interactions, hepatocyte incubations should be conducted in addition to microsome incubations. Drug interactions at the transporter level cannot be detected using microsome incubation.
Time course studies demonstrated that over 45 min, ERY metabolism was reduced by rifampin and enhanced by GG918. These results indicate that when uptake transporters were inhibited by 2.5 μM rifampin, less ERY entered the cells (Fig. 4A), fewer metabolites were formed (Figs. 3B and 4B), and more parent drug remained in the cell and in the media (Fig. 3A). In contrast, inhibition of P-gp by 0.5 μM GG918 resulted in an accumulation of ERY intracellularly (Fig. 4A) that led to increased metabolite formation (Figs. 3B and 4B), and less parent drug remained in the cell and in the media (Fig. 3A). AUC ratios of ERY and N-demethyl-ERY were also determined (Table 1). That there was no change in AUC ratio compared with the control when rifampin was present suggests that the metabolic process was not inhibited, only that the drug available to be metabolized was decreased. In contrast, an increase in the AUC ratio relative to control suggests an increase in access of the drug available to be metabolized when GG918 was incubated, inasmuch as both ERY and metabolite intracellular concentrations increased significantly versus control. Note that the increase in intracellular drug and metabolite concentrations in the presence of GG918 at 45 min contrasts with the lack of change noted in the 2-min uptake studies (Fig. 1). Intracellular levels of rifampin and GG918 did not reach concentrations at which cyp3a activities would be inhibited, as seen with microsome studies (Figs. 2 and 4).
For the in vivo studies, the amounts of ERY, rifampin, and GG918 administered were carefully chosen so that levels of N-demethyl-ERY were detectable in the blood and cyp3a activity was not inhibited in any case. ERY serum clearance in rats has been reported to be 73 ml/min/kg using a HPLC method (Duthu, 1985). Since the ERY partition coefficient of blood to plasma is 1.3, our ERY clearance is reasonably consistent with this previous result. For the rifampin coadministered group, the blood concentration-time profile showed that, compared with the control, an increase in AUC was observed leading to a significant (1.5-fold) decrease in total clearance (Fig. 5A). A similar trend in AUC was observed in hepatocytes (Fig. 3A), but it is obvious that in vivo changes in ERY were markedly greater than those seen in hepatocytes over 45 min. As expected, since less compound entered the liver as a result of inhibition of Oatps by rifampin, more ERY remained in the blood. This increase in blood AUC cannot be attributed to inhibition of cyp3a, since the range of rifampin blood concentrations (1.56 μM at 2 min to 0.17 μM at 240 min) should not affect enzymatic activity based on our in vitro inhibition studies. There was an increase in blood concentration of N-demethyl-ERY when rifampin was present (Fig. 5B). We speculate that after the metabolite exits the liver through either passive diffusion or basolateral efflux, it cannot be effectively reabsorbed back into the liver because of the inhibition of the uptake transporters by rifampin, since N-demethyl-ERY is also a substrate for Oatp1a4 and Oatp1b2 (Fig. 1). Ex situ and in vivo increases in metabolites subject to Oatp uptake in the presence of rifampin were also observed by Lau et al. (2006a,b) for atorvastatin.
For the GG918 group, there was an increase in AUC of ERY compared with the control that resulted in nearly a 2-fold decrease in total clearance (Fig. 6). Since ERY and N-demethyl-ERY appear to be substrates for the same transporters, the trend of the systemic metabolite profile follows that of the parent drug. When GG918 was present, the increased blood metabolite concentration was probably a result of increased ERY metabolism. In the liver, ERY and N-demethyl-ERY concentrations increased significantly when P-gp was inhibited by GG918, and the N-demethyl-ERY to ERY ratio increased, indicating that ERY metabolism was increased compared with the control (Fig. 9), although this increase was not reflected in the total clearance or CLother (Table 2). GG918 concentrations in the blood were lower than the detection limit of 250 nM; thus, GG918 is unlikely to inhibit uptake transporters at this low concentration. As a consequence of lowered clearance in both treatment groups and no significant change in volume, the half-lives increased compared with their controls.
Bile concentrations of ERY and N-demethyl-ERY were lower than control for the rifampin group, because of inhibition of uptake transporters (Fig. 7). Cell line studies with MDCK-MDR1 show that rifampin does not inhibit human P-gp (Lau et al., 2006a). Both ERY and metabolite bile concentrations were lower than control for the GG918 group (Fig. 8), we believe as a consequence of inhibiting P-gp biliary elimination, which is consistent with the marked shift of peak time for ERY and N-demethyl-ERY compared with controls. Biliary clearances of both treatment groups significantly decreased versus the controls (Table 2); however, the rifampin effect was caused by inhibition of uptake transporters, whereas the GG918 effect was due to inhibition of efflux transporters.
Renal clearances of ERY and N-demethyl-ERY did not change significantly with rifampin coadministration. However, for the GG918 group, the renal clearances of ERY and its metabolite significantly decreased compared with their respective controls, suggesting inhibition of P-gp in the kidney. This is consistent with previous findings (Hori et al., 1993) demonstrating that known P-gp inhibitors, quinidine and verapamil, decreased the ratio of fraction excreted to filtration fraction of digoxin in a dose-dependent manner in the isolated rat kidney.
Liver concentrations of ERY, N-demethyl-ERY and inhibitors were measured at the end of the PK study (Fig. 9). Compared with the controls, there was a significant decrease and increase, respectively, in liver concentrations of both ERY and N-demethyl-ERY when rifampin or GG918 was coadministered. This correlated well with intracellular measurements of parent drug and metabolite in the hepatocyte studies (Table 1). For the rifampin group, the metabolite to parent drug ratio did not change significantly in the hepatocytes (Table 1) or in the liver (Fig. 9), suggesting that the concentration of rifampin used for the in vitro and in vivo studies did not inhibit cyp3a and only inhibited Oatps. For the GG918 group, as observed for the intracellular hepatocyte measurements (Table 1), the metabolite to ERY ratio increased significantly in the liver, indicating that ERY metabolism increased when P-gp was inhibited. Without affecting enzymatic activities, rifampin and GG918 altered the PK of ERY with similar trends but via very different mechanisms.
In conclusion, hepatic transporters play important roles in the disposition and metabolism of substrate compounds. In this study, we used erythromycin as a model substrate. This class 3 drug (high solubility, low permeability with elimination primarily as unchanged drug in humans), as defined by Wu and Benet (2005), undergoes more extensive metabolism in rats and served as a good substrate for investigating both hepatic uptake and efflux transporter interplay with metabolic enzymes. In addition, since the N-demethyl-ERY metabolite is also a substrate for the same transporters, we could observe in vivo interactions of the metabolite that may not have been obvious from in vitro studies. The studies here should be important primarily for the many class 2 drugs (high permeability, low solubility, extensively metabolized compounds) for which hepatic transporter-enzyme interplay will not be predictable from microsome studies alone.
Acknowledgments
We appreciate the assistance of Alan Wolfe with hepatocyte isolations and hepatocyte incubations.
Footnotes
-
This study was funded in part by National Institutes of Health Grants GM61390 and HD40543, in part through facilities of the UCSF Liver Center (DK 26743), and by an unrestricted grant from Amgen Inc. Dr. Benet serves as a consultant to Amgen. A portion of this work was presented at the Gordon Research Conference on Drug Metabolism, July 10–15, 2005, Plymouth, New Hampshire, and at the November 7–10, 2005 annual meeting of the American Association of Pharmaceutical Scientists, Nashville, Tennessee.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009258.
-
ABBREVIATIONS: OATP/Oatp, organic anion-transporting polypeptide; P-gp, P-glycoprotein; ERY, erythromycin; GG918, GF120918: N-{4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)-ethyl]-phenyl}-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamine; PK, pharmacokinetics; CCK8, [SO3H)27]-cholecystokinin amide fragment 26-33; HPLC, high-performance liquid chromatography; MTBE, tert-butyl-methyl-ether; ACN, acetonitrile; UCSF, University of California, San Francisco; DMSO, dimethyl sulfoxide; IS, internal standard; LC-MS, liquid chromatography-mass spectrometry; BSA, bovine serum albumin; AUC, area under the curve; CL, clearance; Vss, volume of distribution at steady state; MRT, mean residence time.
- Received January 5, 2006.
- Accepted May 5, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}