


Abstract
Flutamide, a widely used nonsteroidal antiandrogen drug for the treatment of prostate cancer, has been associated with rare incidences of hepatotoxicity in patients. It is believed that bioactivation of flutamide and subsequent covalent binding to cellular proteins is responsible for its toxicity. A novel N-S glutathione adduct has been identified in a previous bioactivation study of flutamide (Kang et al., 2007). Due to the extensive first pass metabolism, flutamide metabolites such as 2-hydroxyflutamide and 4-nitro-3-(trifluoromethyl)phenylamine (Flu-1) have achieved plasma concentrations higher than the parent in prostate cancer patients. In vitro studies in human liver microsomes were conducted to probe the cytochrome P450 (P450)-mediated bioactivation of flutamide metabolites and identify the possible reactive species using reduced glutathione (GSH) as a trapping agent. Several GSH adducts (G1, Flu-1-G1, Flu-1-G2, Flu-6-Gs) derived from the metabolites of flutamide were identified and characterized. A comprehensive bioactivation mechanism was proposed to account for the formation of the observed GSH adducts. Of interest were the formation of a reactive intermediate by the desaturation of the isopropyl group of M5 and the unusual bioactivation of Flu-1. Studies using recombinant P450s suggested that the major P450 isozymes involved in the bioactivation of flutamide and its metabolites were CYP1A2, CYP3A4, and CYP2C19. These findings suggested that, in addition to the direct bioactivation of flutamide, the metabolites of flutamide could also be bioactivated and contribute to flutamide-induced hepatotoxicity.
2-Methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide (flutamide), a widely prescribed nonsteroidal antiandrogen drug, has been shown to increase survival time of prostate cancer patients in combination therapy with luteinizing hormone-releasing agonists or orchiectomy (Brogden and Clissold, 1989; McLeod, 1993; Schmitt et al., 2001). However, a number of hepatotoxicity cases were reported to be associated with the clinical use of this drug, such as temporary increases in transaminase markers and rare incidences of severe liver dysfunction (Gomez et al., 1992; Wysowski and Fourcroy, 1996; Cetin et al., 1999; Nakagawa et al., 1999; Thole et al., 2004; Osculati and Castiglioni, 2006). Several cases of blood eosinophilia have been observed in patients treated with flutamide (Hart and Stricker, 1989), which indicates an immune-mediated mechanism in some patients.
The mechanism of flutamide-induced hepatic dysfunction is currently unknown; however, bioactivation of flutamide has been considered to be the cause of flutamide-induced hepatotoxicity. When metabolically activated by P450s (CYP3A and CYP1A), flutamide has been shown to be covalently bound to microsomal and hepatocyte proteins (Berson et al., 1993; Fau et al., 1994). Ichimura and coworkers have also illustrated the necessity of enhanced flutamide metabolism for development of severe hepatotoxicity (Ichimura et al., 1999). More recently, Matsuzaki and coworkers have demonstrated flutamide-induced toxicity in CYP1A2 knockout SV129 mice administered with 400 mg/kg dose after the mice were fed with an amino acid-deficient diet for 2 weeks, which reduced the glutathione (GSH) content to 27% of the initial content (Matsuzaki et al., 2006). In vitro conjugation with GSH is widely used in the characterization of reactive metabolites in probing the mechanism of bioactivation (Samuel et al., 2003). Recently, a GSH conjugate of hydroxylated flutamide has been detected in human liver microsomal and hepatocyte incubations (Soglia et al., 2006; Kostrubsky et al., 2007). Similarly, Tevell and coworkers (Tevell et al., 2006) have detected a mercapturic acid conjugate of hydroxylated flutamide in the urine of prostate cancer patients. In a previous study of the bioactivation of flutamide in human liver microsomes, a novel N-S glutathionyl adduct was detected and fully characterized by MS and NMR, which is formed by the direct bioactivation of the parent drug (Kang et al., 2007).
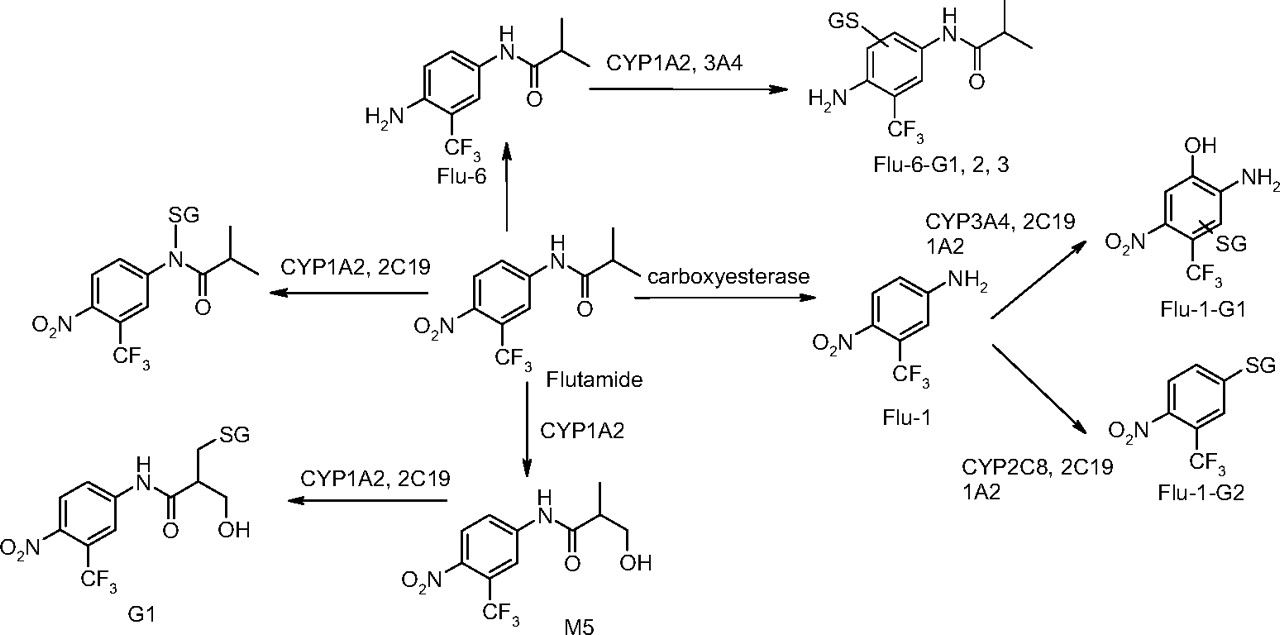
Due to extensive metabolism of flutamide in the liver, some flutamide metabolites such as 2-hydroxyflutamide and Flu-1 achieved even higher plasma concentrations than the parent in prostate cancer patients. Steady-state plasma concentrations of 635 and 210 ng/ml have been determined for 2-hydroxyflutamide and Flu-1, respectively, 3 h postdose in patients with prostate cancer, whereas that of flutamide was only 23.6 ng/ml (Aizawa et al., 2003). Thus, it is highly probable that metabolites of flutamide can also be bioactivated and contribute to flutamide-induced hepatotoxicity. Major metabolites of flutamide identified in vitro or in vivo so far are shown in Scheme 1 (Goda et al., 2006; Tevell et al., 2006). In addition to 2-hydroxyflutamide, other oxidative metabolites are monohydroxylated flutamide M5, dihydroxylated flutamide M7, and trihydroxylated flutamide M8. Recently, a new N-oxidized metabolite of flutamide, N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine (Flu-1-N-OH), possibly derived from Flu-1, has been identified in the urine of prostate cancer patients and in human liver microsomal incubations (Aizawa et al., 2003; Goda et al., 2006). Additionally, a nitro-reduced metabolite, 2-methyl-N-[4-amino-3-(trifluoromethyl)phenyl]-propanamide (Flu-6), was found in human serum (Katchen and Buxbaum 1975; Takashima et al., 2003). Although studies with flutamide in liver microsomes failed to generate this metabolite, other reductive pathways exist in vivo to produce Flu-6. An acetylated metabolite of Flu-1, N-[4-nitro-3-(trifluoromethyl)phenyl]-acetamide (Flu-2), was also reported in the plasma samples of men who were treated with flutamide (Katchen and Buxbaum, 1975).
The present study describes the bioactivation of flutamide metabolites (2-hydroxyflutamide, M5, Flu-1, Flu-2, and Flu-6) in human liver microsomal incubations supplemented with GSH and NADPH. We herein report the detection and characterization of several GSH adducts (G1, Flu-1-G1, Flu-1-G2, Flu-6-G1) derived from the metabolites of flutamide. A comprehensive bioactivation mechanism has been proposed to account for the formation of the observed GSH adducts.
Materials and Methods
Materials. Flutamide, Flu-1, Flu-2, and glutathione (reduced form, GSH) were purchased from Sigma-Aldrich (St. Louis, MO). Human liver microsomes were prepared from human livers (BD Gentest, Woburn, MA) using standard protocols and were characterized using P450-specific marker substrate activities. Aliquots from the individual preparations from 56 individual human livers were pooled on the basis of equivalent protein concentrations to yield a representative microsomal pool with a protein concentration of 20.4 mg/ml. Recombinant P450 isozymes CYP1A2, CYP3A4, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 Supersomes were obtained from BD Gentest. All other commercially available reagents and solvents were of either analytical or HPLC grade.
Synthesis of 2-Methyl-N-(4-amino-3-trifluoromethyl-phenyl)-propanamide. Flutamide (0.505 g, 1.83 mmol) was dissolved in 5 ml of methanol. The reaction flask was placed in an oil bath at 55°C, and a mixture of sodium sulfide nonahydrate (1.15 g, 4.79 mmol) and sodium bicarbonate (0.355 g, 4.23 mmol) in 5 ml of water was added over 5 min. The solution began to turn from yellow to orange upon addition of the reducing agent. After addition of approximately 1 ml of the aqueous solution, precipitate began to form. Addition of 15 ml of methanol dissolved most of the precipitate, and the reaction mixture was heated in an oil bath at 60°C for 1 h. The reaction appeared to be 50% complete (an aliquot was diluted with methanol and was examined by LC/MS). More of the mixture of sodium sulfide nonahydrate (1.15 g, 4.79 mmol) and sodium bicarbonate (0.355 g, 4.23 mmol) in 5 ml of water was added over 3 min. After 20 min, water (10 ml) was added and the methanol was removed in vacuo. The remaining aqueous layer was extracted with dichloromethane (DCM) twice. The organic layer was dried over sodium sulfate and concentrated in vacuo producing 419.6 mg of a yellow solid. Purification was accomplished by radial chromatography (2-mm plate, DCM to 90 DCM to 10 ethyl acetate, sample loaded in DCM). The second UV active band produced 90.5 mg of the target compound (20% yield) as a very light yellow solid. LC/MS >95% (254 nm); 1H-NMR (400 MHz, MeOD) δ 7.63 (1 H, d, J = 2.5 Hz), 7.40 (1 H, dd, J = 8.8, 2.5 Hz), 6.79 (1 H, d, J = 8.8 Hz), 2.57 (1 H, dt, J = 13.6, 6.8 Hz), 1.17 (6 H, d, J = 6.8 Hz). 1H NMR (400 MHz, CDCl3) δ 7.54 (1 H, d, J = 2.3 Hz), 7.46 (1 H, dd, J = 8.6, 2.3 Hz), 7.14 (1 H, s), 6.69 (1 H, d, J = 8.6 Hz), 4.02–4.13 (2 H, m), 2.46 (1 H, dt, J = 13.7, 6.9 Hz), 1.23 (6 H, d, J = 7.1 Hz). 13C-NMR (100 MHz, CDCl3) δ 175.4, 142.2, 128.6, 125.9, 124.5 (q, J = 273 Hz), 119.0, 117.6, 113.8 (q, J = 30 Hz), 36.3, 19.5. HRMS - C11H13N2OF3 Theoretical (M+H): 247.10527; Found: 247.10545. Elemental Analysis Theory C, 53.66%; H, 5.32%; F, 23.15%; N, 11.38%; Found C, 53.59%; H, 5.32%; F, 23.02%; N, 11.36%.
Microsomal Metabolism. Flutamide, Flu-1, or Flu-2 (5–100 μM) was incubated for 1 h at 37°C in an incubation system consisting of 100 mM potassium phosphate buffer (pH 7.4), 2 mg of human liver microsomes, 5 mM GSH, and 1 mM NADPH in a final volume of 1 ml. Reactions were terminated by the addition of 6 ml of acetonitrile. Samples were mixed on a vortex mixer and centrifuged for 5 min. The supernatants were transferred into conical glass tubes for evaporation to dryness under N2 at 30°C. The residues were reconstituted in 200 μl of 30:70 (v/v) methanol/20 mM ammonium acetate (pH 4), and aliquots (40 μl) were injected into an HPLC-MS system. Metabolite profiling was performed on an Agilent 1100 HPLC system (Agilent Technologies, Palo Alto, CA) coupled with a Finnigan LCQ-Deca ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Separation was achieved using a Kromasil C4 100A column (3.5 μm, 150 × 2.0 mm; Phenomenex, Torrance, CA) at a flow rate of 0.2 ml/min. A gradient of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid was as follows: initiated with 1% B for 5 min and then increased in a linear manner to 30% at 20 min and to 50% at 25 min, held at 50% until 28 min, changed linearly to 90% at 40 min, maintained at 90% for up to 43 min, and then decreased to 1% at 45 min. The column was allowed to equilibrate at 1% solvent B for 5 min prior to the next injection. The HPLC effluent going to the mass spectrometer was directed to waste through a divert valve for the initial 5 min after sample injection. Major operating parameters for the ion-trap ESI-MS method were as follows: capillary temperature, 270°C; spray voltage, 5.0 kV; capillary voltage, -14 V; sheath gas flow rate 90; and axillary gas flow rate, 30 (arbitrary value). For the detection of Flu-6 GSH adducts the major operating parameters for the ion-trap ESI-MS method were set in the positive mode: capillary temperature, 270°C; spray voltage, 4.5 kV; capillary voltage, 4.5 V; sheath gas flow rate, 90; and axillary gas flow rate, 30. The mass spectrometer was operated with data-dependent scanning. The ions were monitored over a full mass range of m/z 125 to 1000. For a full scan, the automatic gain control was set at 5.0 × 108, maximum ion time was 100 ms, and the number of microscans was set at 3. For MSn scanning, the automatic gain control was set at 1.0 × 108, maximum ion time was 400 ms, and the number of microscans was set at 2. For data-dependent scanning, the default charge state was 1, default isolation width was 3.0, and normalized collision energy was 45.0.
Flutamide metabolites of M5 were isolated from human liver microsomal incubation of flutamide. Further incubation of M5 in human liver microsomes and HPLC-MS analysis were the same as above.
Incubations with cDNA-Expressed Human P450 Enzymes. Flutamide, Flu-1, and Flu-6 (50 μM) were incubated for 1 h at 37°C in an incubation system consisting of 100 mM potassium phosphate buffer (pH 7.4), recombinant P450 CYP1A2, CYP3A4, CYP3A5, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 Supersomes (50 pmol), 5 mM GSH, and 1 mM NADPH in a final volume of 0.5 ml. After 3 min of preincubation, incubations were initiated by the addition of NADPH. Reactions were terminated by the addition of 1 ml of acetonitrile. Nilutamide was added as an internal standard. Formation of the glutathionyl adduct was quantified using a Shimadzu LC-10AD VP with binary pumps (Shimadzu, Columbia, MD) coupled with a Q-Trap 4000 (Applied Biosystems/MSD Sciex, Concord, ON, Canada). The adduct was separated by a Kromasil C4 100A column (3.5 μm, 150 × 2.0 mm) at a flow rate of 0.2 ml/min. A gradient of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid was as follows: initiated with 0% B for 3 min and then increased in a linear manner to 90% at 15 min and then decreased to 0% at 17 min. The column was allowed to equilibrate at 0% solvent B for 3 min prior to the next injection. The HPLC effluent going to the mass spectrometer was directed to waste through a divert valve for the initial 3 min after sample injection. The Q-trap 4000 electrospray-MS was operated in the negative ionization mode by applying to the capillary a voltage (IS) of -4.5 kV. Nitrogen was used as curtain gas (CUR), as well as a nebulizing (GS1) and turbo spray gas (GS2, heated at 450°C), with the optimum values set, respectively, at 36, 50, and 40 (arbitrary values). Collision-activated dissociation was performed at 6 (arbitrary value) with nitrogen as collision gas. Declustering potential was set at -90 V, whereas entrance potential was set at -10 V; collision energy was optimized at -34 eV. The multiple reaction monitoring transitions used were 596→323 for G1, 526→253 for Flu-1-G1, 495→222 for Flu-1-G2, and 316→205 for internal standard nilutamide, respectively. For Flu-6-G1, the mass spectrometer was operated in a positive ion mode with a capillary voltage of 4.5 kV. The source temperature was set at 450°C, declustering potential at 65 V, and the entrance potential at 10 V. The collision-activated dissociation was performed using a collision energy of 40 V. Flu-6-G1 and the internal standard buspirone were monitored in multiple reaction monitoring using the transitions of 552→423 for Flu-6-G1 and 386 →122 for buspirone.
Isolation of GSH Adducts and NMR Characterization. The same human liver microsomal incubation with flutamide was carried out on a 10-ml scale. G1, 2-hydroxyflutamide, M5, and M7 were isolated with the following LC conditions. Separation was achieved using a COSMOSIL 5PYE column (150 × 4.6 mm; Waters, Milford, MA) at a flow rate of 1.0 ml/min with an Agilent 1100 HPLC system (Agilent, Wilmington, DE). A gradient of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid was as follows: initiated with 100% A for 5 min, changed to 80% A from 5 to 10 min, changed to 50% A from 10 to 60 min, changed to 10% A from 60 to 70 min, held at 10% A from 70 to 75 min, changed to 100% A from 75 to 76 min, and held at 100% A from 76 to 80 min for the column to be equilibrated. All NMR spectra were acquired on a Bruker-Biospin AV700 spectrometer running TopSpin 1.3 software and equipped with a Bruker 5-mm TCI z-gradient Cryoprobe (Bruker, Rheinstetten, Germany). 1D 1H spectra were acquired with water suppression using a Watergate W5 pulse sequence with gradients and a double echo. 2D COSY and HSQC spectra were acquired without solvent suppression using gradient pulses for coherence selection. Chemical shifts are reported in ppm relative to tetramethylsilane.
Flu-1-G2 was isolated from the incubation of Flu-1 with rat liver microsomes on a 10-ml scale using the method as described above. 1D and 2D NMR spectra were acquired similarly as above.

UV and extracted ion chromatograms (XIC) of Flu-1-G1, Flu-1-G2, and G1 in human liver microsomal incubations of flutamide. A, UV chromatogram at 306 nm; B, XIC of G1 at m/z 596 ([M-H]-); C, XIC of Flu-1-G2 at m/z 495 ([M-H]-); D, XIC of Flu-1-G1 at m/z 526 ([M-H]-).
Results
To assess the bioactivation of flutamide metabolites, the major flutamide metabolites identified in vivo and in vitro were obtained and incubated with human liver microsomes supplemented with NADPH and GSH.
Incubation of 2-Hydroxyflutamide with Human Liver Microsomes. When the major circulating metabolite 2-hydroxyflutamide was incubated in human liver microsomes supplemented with NADPH and GSH, no GSH adducts could be detected.
Formation of GSH Adducts from Flu-1. In the presence of GSH, two GSH adducts were detected in the NADPH-supplemented human liver microsomal incubation of Flu-1 or flutamide (Fig. 1). The first GSH adduct (Flu-1-G1) showed a molecular ion at m/z 526 ([M-H]-) in the negative ion mode, 305 Da greater than that of M4, a hydroxylated metabolite of Flu-1 (Scheme 1). Subsequent MS/MS of Flu-1-G1 produced two major fragment ions at m/z of 253 and 272 (Fig. 2). The first ion at m/z of 253 was derived from cleavage of sulfur-carbon bond of the glutathionyl moiety and appeared to retain the sulfur atom. The second ion at m/z of 272 was part of glutathionyl moiety and was produced from the same sulfur-carbon bond cleavage. The neutral loss of 129, corresponding to elimination of the pyroglutamate, was also observed in the MS/MS spectrum of Flu-1-G1. The fragmentation pathway suggested that GSH was added to the aromatic ring of M4 (Fig. 2).
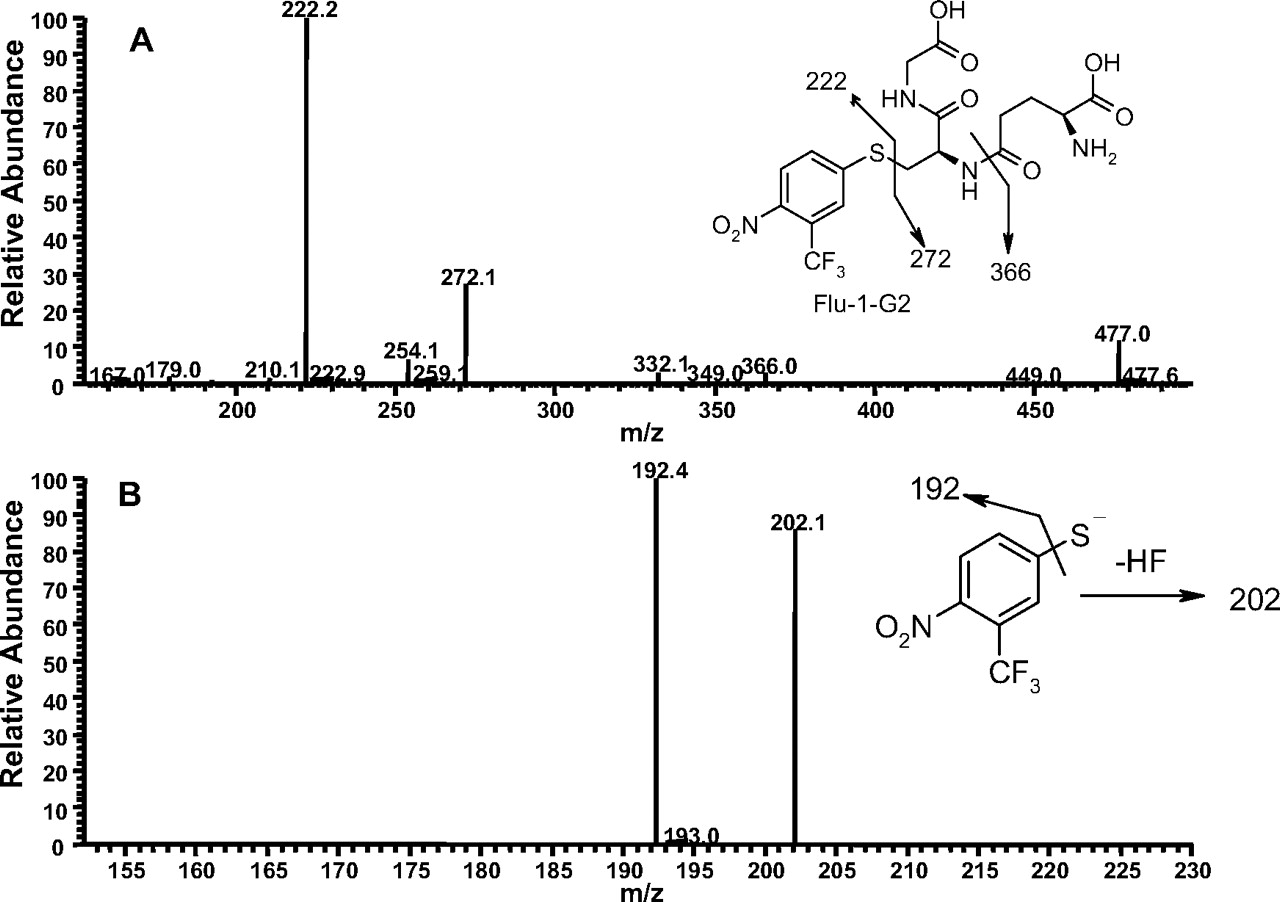
The second GSH adduct (Flu-1-G2; Fig. 1D) from Flu-1 showed a molecular ion at m/z 495 ([M-H]-), an addition of 290 amu to Flu-1 (m/z 205, [M-H]-), which corresponded to the addition of a glutathionyl moiety (307 amu) and loss of the amino group (-17 amu) of Flu-1. MS/MS (Fig. 3A) of Flu-1-G2 generated fragment ions at m/z 222 and 272, the latter ion being part of the glutathionyl moiety resulted from sulfur-carbon bond cleavage. Further fragmentation of the ion at m/z 222 resulted in the loss of hydrogen fluoride and sulfur atom (Fig. 3B). The ion at m/z 222 was consistent with substitution of the amino group of Flu-1 by a sulfur atom. Furthermore, a change from odd ion of Flu-1 at m/z 205 to an even ion at m/z 222 implicated loss of the amino group. The loss of pyroglutamate (-129 amu) was also observed in MS/MS of Flu-1-G2.


MS/MS spectrum of Flu-1-G1 at m/z 526 ([M-H]-).
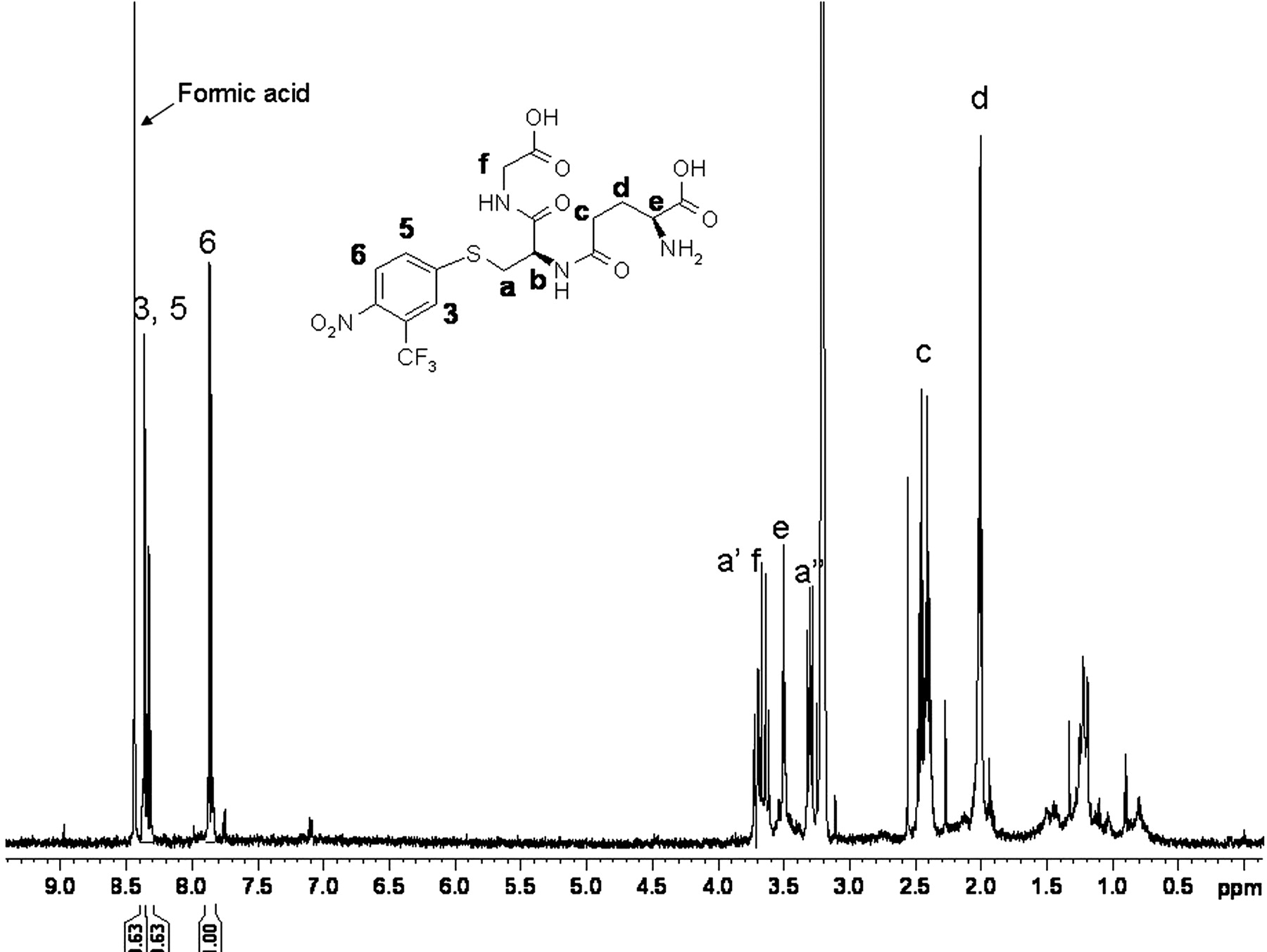
For a more definitive characterization of the structure of Flu-1-G2, the adduct was isolated from the incubation of Flu-1 with human liver microsomes. 1H NMR spectrum of Flu-1-G2 showed (Fig. 4; Table 1) three aromatic proton signals (3, 5, and 6). The presence of a larger J coupling of 9.30 Hz between 5 and 6, a smaller J coupling of 2.36 Hz between 3 and 5, and an undetectable coupling between 3 and 6 is consistent with the aromatic substitution pattern of the proposed structure (Fig. 4). This aromatic substitution pattern of Flu-1-G2 was identical to that of flutamide, which strongly suggested replacement of the amino group by a glutathionyl moiety. The proton signals of glutathionyl group appeared in the region from 1.8 ppm to 4.5 ppm in the 1H-NMR spectrum. The assignments of all the glutathionyl protons were achieved by a 1H-1H COSY experiment (data not shown).
1H NMR data for flutamide, G1, and Flu-1-G2 in methanol-d4
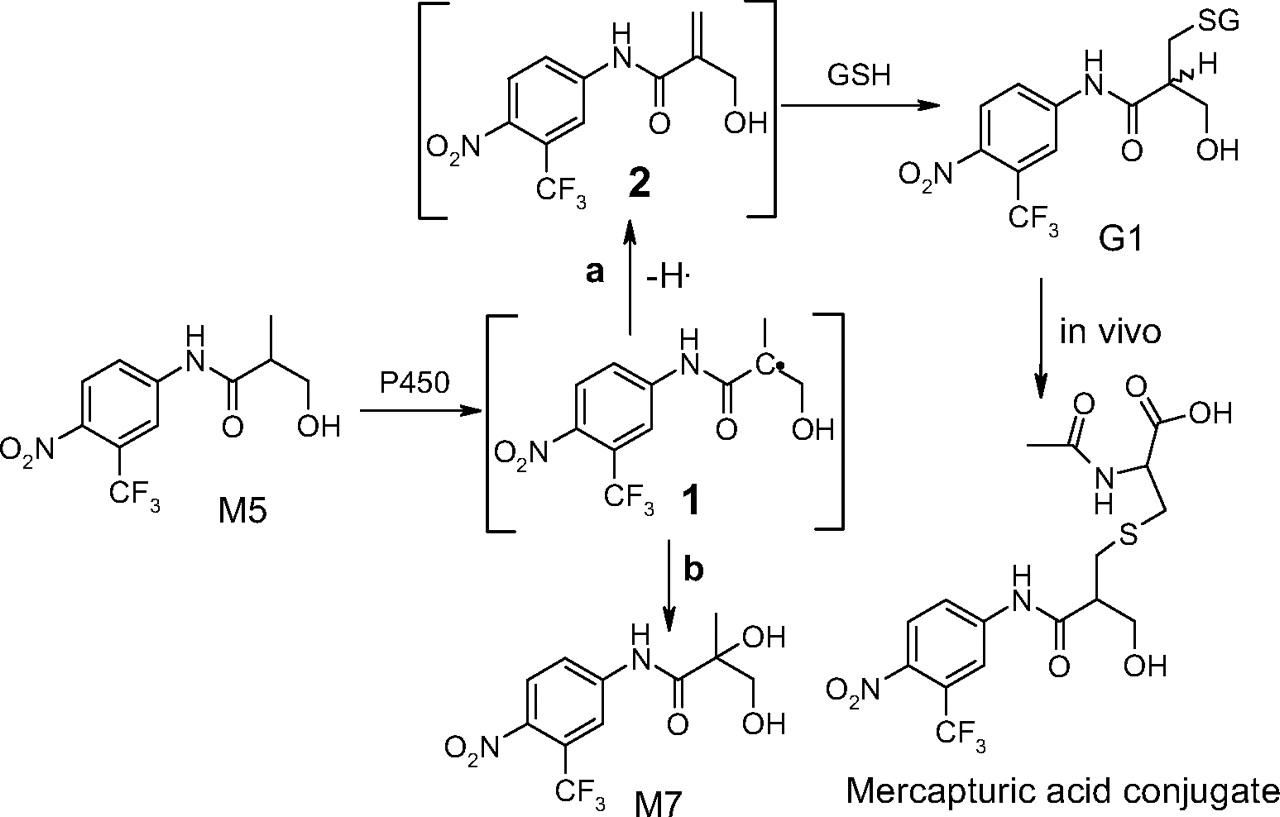
Formation of GSH Adduct G1 from M5. M5 is a minor metabolite and only observed in human liver microsomal incubations. However a closer examination of the NADPH-supplemented human liver microsomal incubations of flutamide with GSH revealed that, in addition to the N-glutathionyl flutamide adduct identified in the previous report (Kang et al., 2007), another major GSH adduct (G1) was also formed, and it was later shown to be derived from M5.
G1 displayed a molecular ion of m/z 596 ([M-H]-) in the MS spectrum (Fig. 1, A and B), 305 Da greater than that of a monooxygenated metabolite of flutamide. Further fragmentation of G1 molecular ions resulted in a neutral loss of 129, corresponding to elimination of the pyroglutamate of GSH. MS/MS spectrum of G1 showed two intense fragment ions at m/z of 323 and 306, respectively (Fig. 5A). The ion at m/z 306 corresponded to the molecular ion of GSH ([M-H]-) as fragmentation of this ion produced diagnostic fragment ions of GSH. The ion at m/z of 323 was formed via cleavage of sulfur-carbon bond of the glutathionyl moiety and still retained the sulfur atom. Further fragmentation of the ion at m/z 323 afforded two ions at m/z of 259 and 205, respectively (Fig. 5B), which suggested attachment of the glutathionyl moiety to the isobutyramide instead of the aromatic group.
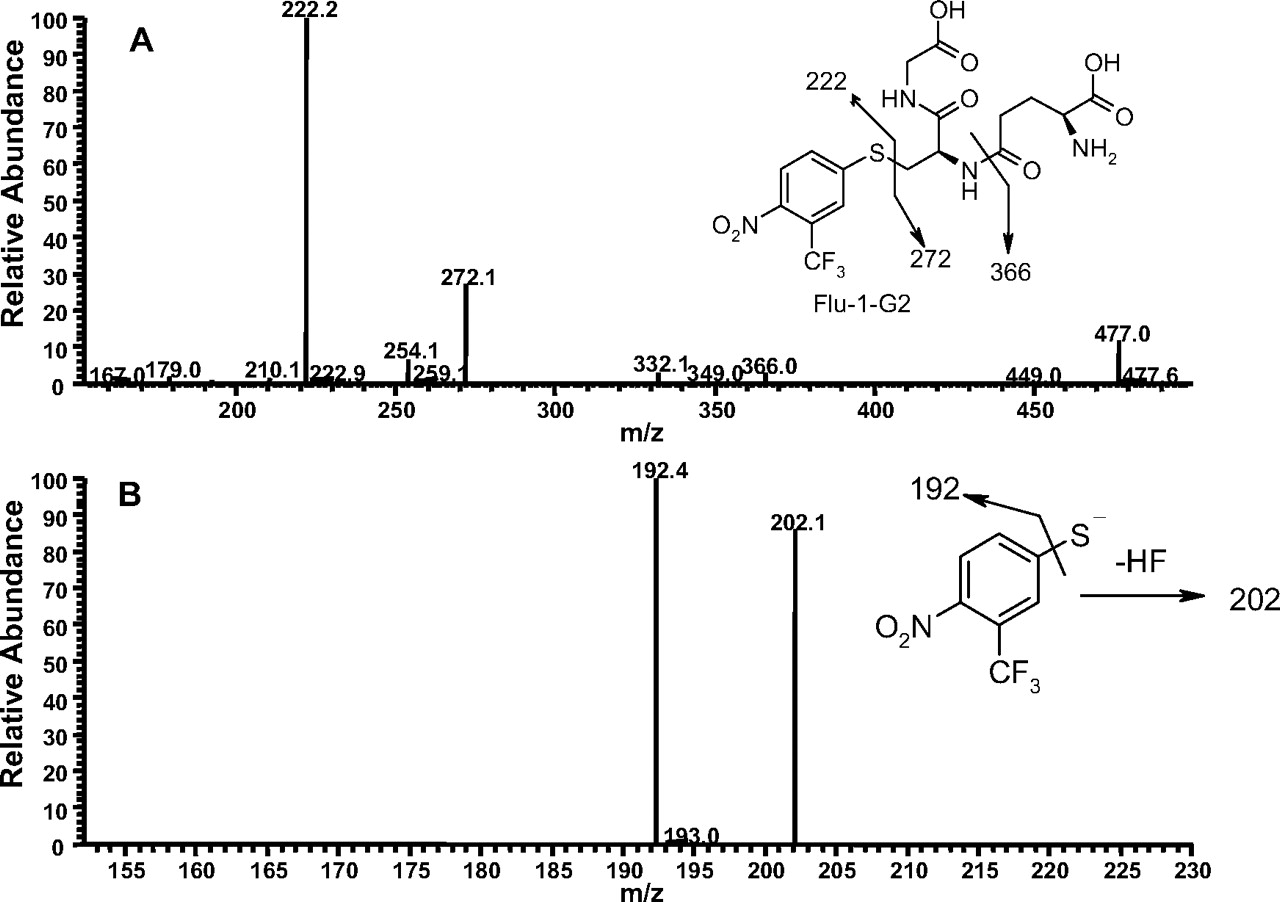
A, MS/MS spectrum of Flu-1-G2 at m/z 495 ([M-H]-). B, MS3 mass spectrum of Flu-1-G2 (the fragment ion at m/z 222) in data-dependent scanning mode on an ion trap mass spectrometer.

1H NMR spectrum of Flu-1-G2. The spectrum was acquired on the isolated Flu-1-G2 dissolved in methanol-d4 with water suppression using a Watergate W5 pulse sequence with gradients and a double echo. The signal of b is not observable due to water suppression.
G1 was isolated from human liver microsomal incubation of flutamide, and NMR analyses allowed the definitive assignment of the structure. The corresponding chemical shifts and coupling constants of G1 are shown in Table 1. The presence of glutathionyl moiety in G1 was confirmed by the proton signals in the region from 1.8 to 4.5 ppm. Three aromatic proton signals appeared from 8.0 to 8.3 ppm. Interestingly, double peaks appeared for aromatic proton 3 and 6 in 1H NMR spectrum of G1 in methanol-d4 (Supplemental data), indicating that G1 was a mixture of diastereomers due to a newly formed chiral center (carbon g in Fig. 5). The proton signals of the two methyl groups of flutamide (d, 6H, 1.22 ppm, J = 4.85 Hz) disappeared, and two new peaks appeared at 3.74 ppm (d, 2H, J = 4.85 Hz) and 2.74 ppm (d, 2H, J = 5.32 Hz), respectively, indicating the modification of both methyl groups in G1. The chemical shifts at 3.74 ppm and 2.74 ppm are consistent with literature chemical shifts of hydroxymethyl and thiol methyl, respectively (Dean, 1987). The methine hydrogen of flutamide (m, 1H, 2.66 ppm, J = 6.85 Hz) shifted downfield to 2.92 ppm in G1 (m, 1H, J = 5.32, 4.85 Hz). Since the three new proton signals of G1 were overlapped with those of glutathionyl moiety, a 1H-1H COSY experiment was conducted to distinguish these mixed signals. As shown in the COSY spectrum of G1 (Fig. 6), the peak at 2.92 ppm was coupled to the two peaks at 3.74 ppm and 2.74 ppm, respectively, whereas the later two peaks were not coupled to each other, thus confirming the assignment of methine hydrogen at 2.92 ppm. The results of NMR experiments have indicated the presence of a hydroxymethyl and a glutathionyl methyl in G1 structure.

A, MS/MS spectrum of flutamide G1, at m/z 596 ([M-H]-). B, MS3 mass spectrum of G1 (the fragment ion at m/z 323) in data-dependent scanning mode on an ion trap mass spectrometer.

Expanded region of the chemical shifts exhibiting the glutathionyl and isopropyl groups in the 1H-1H COSY spectrum of G1 in D2O.
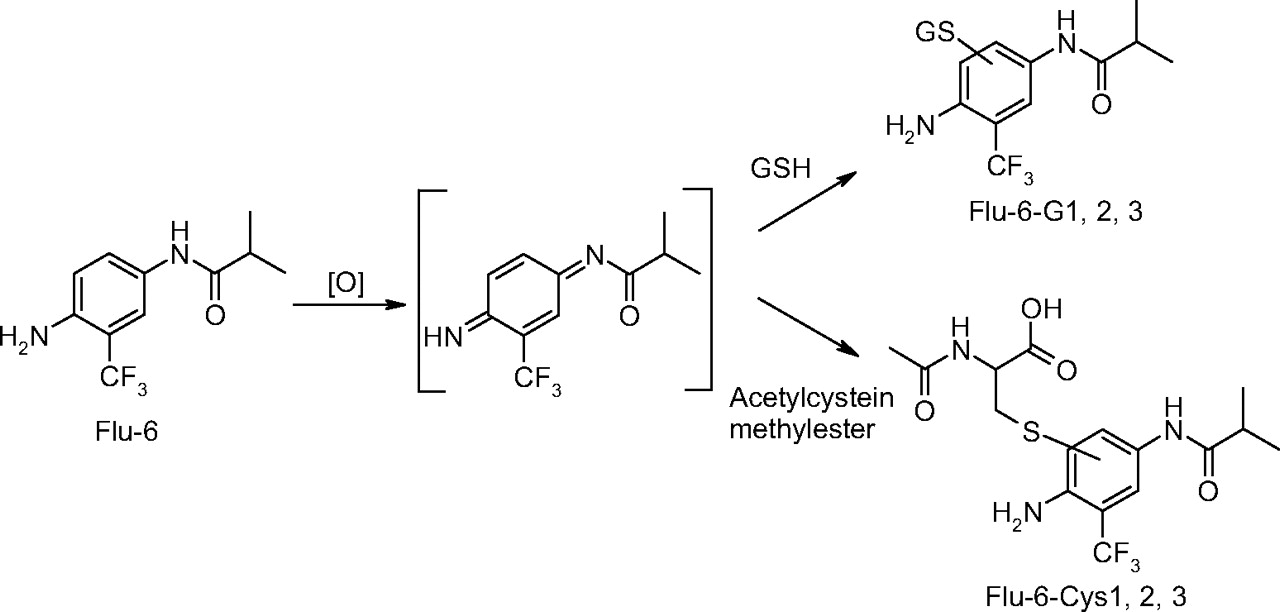
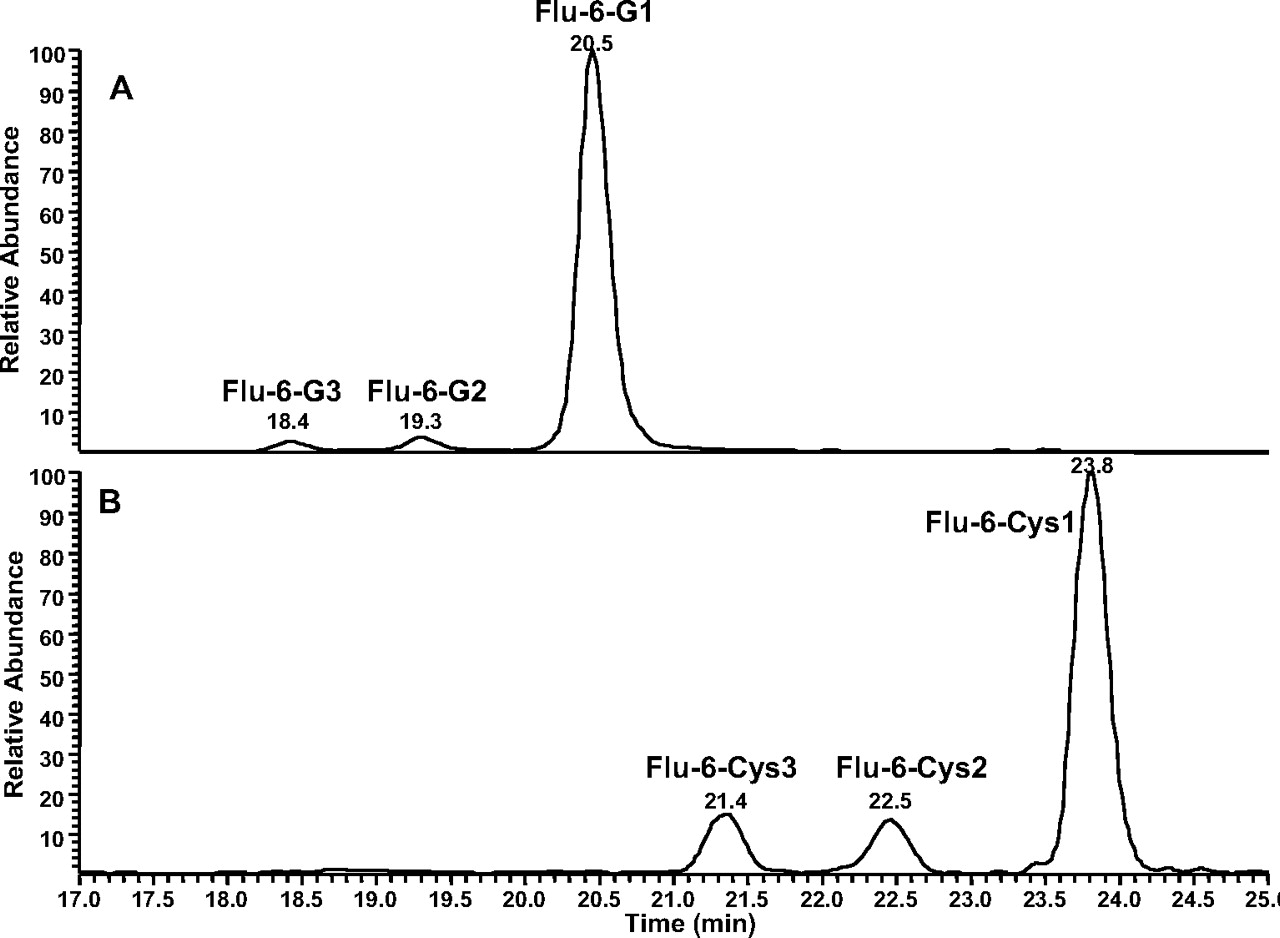
Formation of GSH Adducts from Flu-6. Although Flu-6 was not formed in human liver microsomal incubations of flutamide, it was detected in vivo in human plasma and urine samples (Katchen and Buxbaum 1975; Takashima et al., 2003). This metabolite was synthesized from flutamide by sodium sulfide-mediated reduction of the nitro group. Incubation of Flu-6 in NADPH-supplemented human liver microsomes in the presence of GSH generated three adducts with the same molecular ion at m/z 552 ([M+H]+) in positive ion mode: Flu-6-G1, Flu-6-G2, and Flu-6-G3, corresponding to the attachment of a glutathionyl moiety (305 amu) to FLU-6 (Fig. 7A). The most abundant of the three was Flu-6-G1. MS/MS of the molecular ions at Flu-6-G1 at m/z 552 mainly afforded an ion at m/z 423 from the loss of pyroglutamate (-129) (Fig. 8A). Further fragmentation of this ion yielded fragment ions shown in Fig. 8B. A fragmentation pathway of the ion at m/z 423 is proposed (Fig. 9). The cyclized structure of the ions at m/z 320 and 303 suggested that the glutathionyl could be attached to the aromatic ring and adjacent to either amino or amide nitrogen. The ion at m/z 277 (30 amu higher than FLU-6 molecular ion) should still retain the sulfur atom. Further fragmentation of the ion at m/z gave rise to an ion at m/z 207 from cleavage of the amide bond and retention of the sulfur atom, consistent with attachment of the glutathionyl group to aromatic ring (Fig. 8C). Similar MS spectra were observed for the other two adducts, suggesting the three FLU-6-Gs were regioisomers following attack of GSH on the three unsubstituted positions of the aromatic ring of Flu-6.
Incubation of Flu-6 with N-acetyl cysteine methyl ester generated three cysteine conjugates (Flu-6-Cys1, Flu-6-Cys2, and Flu-6-Cys3; Fig. 7B) in a similar fashion, all with a molecular ion at m/z 408 ([M+H]+), suggesting addition of acetyl cysteine to Flu-6 and hydrolysis of the methyl ester. MS/MS of the ion at m/z 408 (Fig. 10A) resulted in the same ion at m/z 320, which, upon further fragmentation, yielded the ion at m/z 303, also present in MS/MS of Flu-6-G1 (Fig. 10B). The fragmentation pathway of the ion at m/z 423 of Flu-6-G1 (Fig. 9) can be applied to account for the formation of fragment ions of the acetyl cysteine adduct of Flu-6.
Formation of GSH Adducts from Flu-2. Incubation of Flu-2 in human liver microsomes mainly generated Flu-1 via hydrolysis of the amide bond, and Flu-G1 and Flu-G2 were the two GSH adducts formed following further bioactivation of Flu-1 (data not shown).
GSH Adduct Formation by cDNA-Expressed Human P450 Enzymes. The formation of G1, Flu-1-G1, Flu-1-G2, and Flu-6-G1 was found to be NADPH-dependent. To examine the possible involvement of P450 isozymes in the formation of these GSH adducts, incubations of flutamide or Flu-1 or Flu-6 in recombinant human P450 isozymes with GSH and NADPH were conducted.
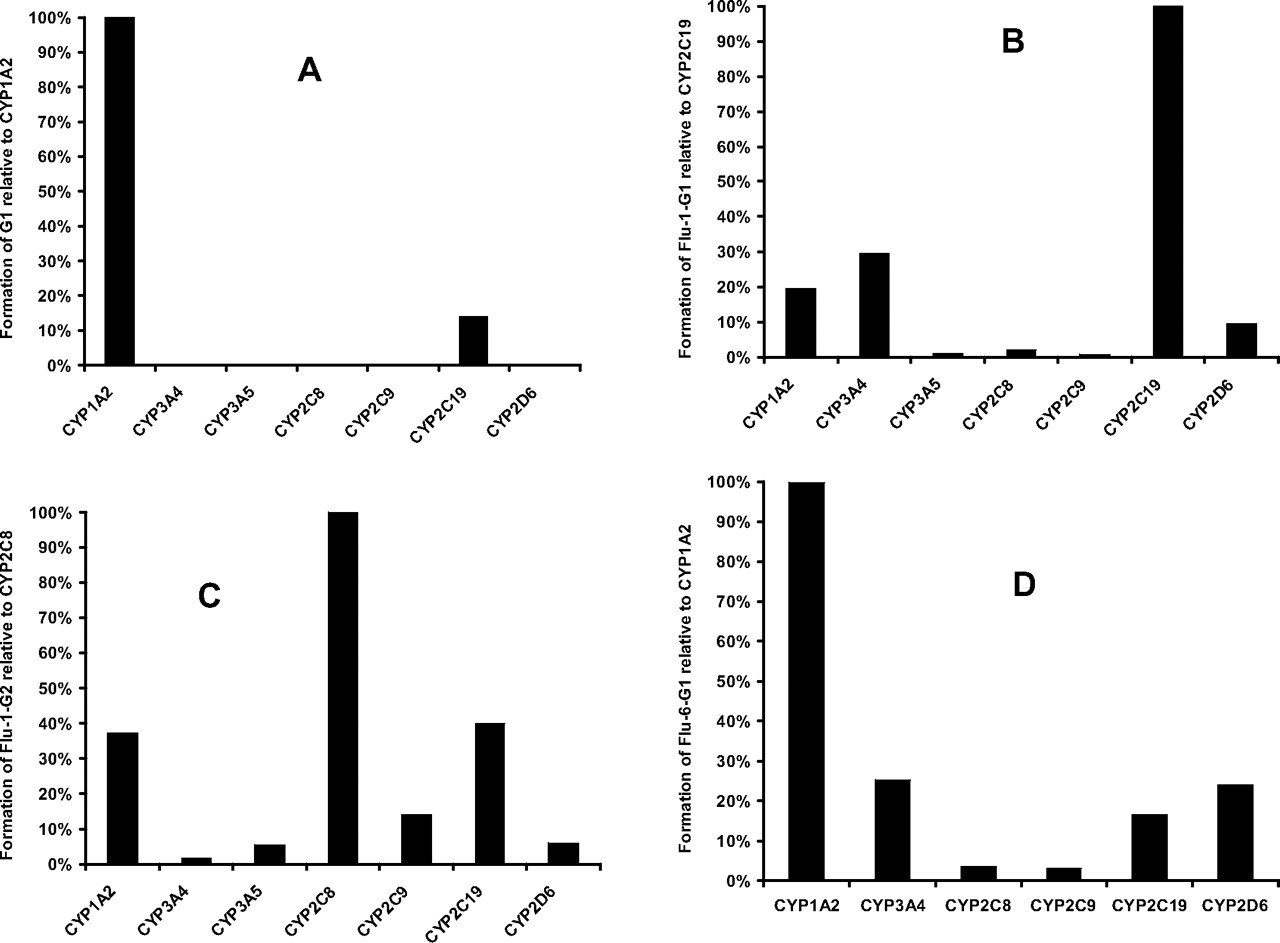
Highest formation of G1 was observed in recombinant CYP1A2 incubation, followed by CYP2C19. G1 was not detectable in CYP3A4, -3A5, -2C8, -2C9, or -2D6 incubations (Fig. 11A). After normalization of relative hepatic abundance of P450 isozymes (Rodrigues, 1999), CYP1A2 turned out to be the major isoform responsible for the formation of G1 (94%). CYP2C19 played a minor role in the bioactivation pathway leading to the observed adduct (6%). An inhibition study in human liver microsomes with a mechanism-based CYP1A2-specific inhibitor, furafylline (25 μM), resulted in a 99% inhibition of G1 formation, confirming the major role played by CYP1A2.
The formation of Flu-1-G1 showed different dependence on P450 isoforms. As shown in Fig. 11B, recombinant CYP2C19 displayed the highest activity. CYP3A4, -1A2, and -2D6 exhibited lower activity toward the formation of Flu-1-G2. After normalization of relative hepatic abundance of P450 isozymes, CYP3A4 accounts for 48% of Flu-1-G2 formation, followed by CYP2C19 (28%) and -1A2 (13%).
Studies of Flu-1-G2 formation by recombinant P450s showed that the highest activity was catalyzed by CYP2C8 (Fig. 11C). Lower activities were found in CYP1A2, -2C9, -2C19, and -2D6. After normalization of relative hepatic abundance of P450 isozymes, CYP2C8 was the major isoform responsible for the formation of Flu-1-G2 (50%). CYP1A2 (13%) and CYP2C9 (11%) played a minor role in the formation of Flu-1-G2.

Extracted ion chromatograms of Flu-6-Gs at m/z 552 ([M+H]+) (A) and Flu-6 acetyl cysteine adducts at m/z 408 ([M+H]+) (B) in human liver microsomal incubations.
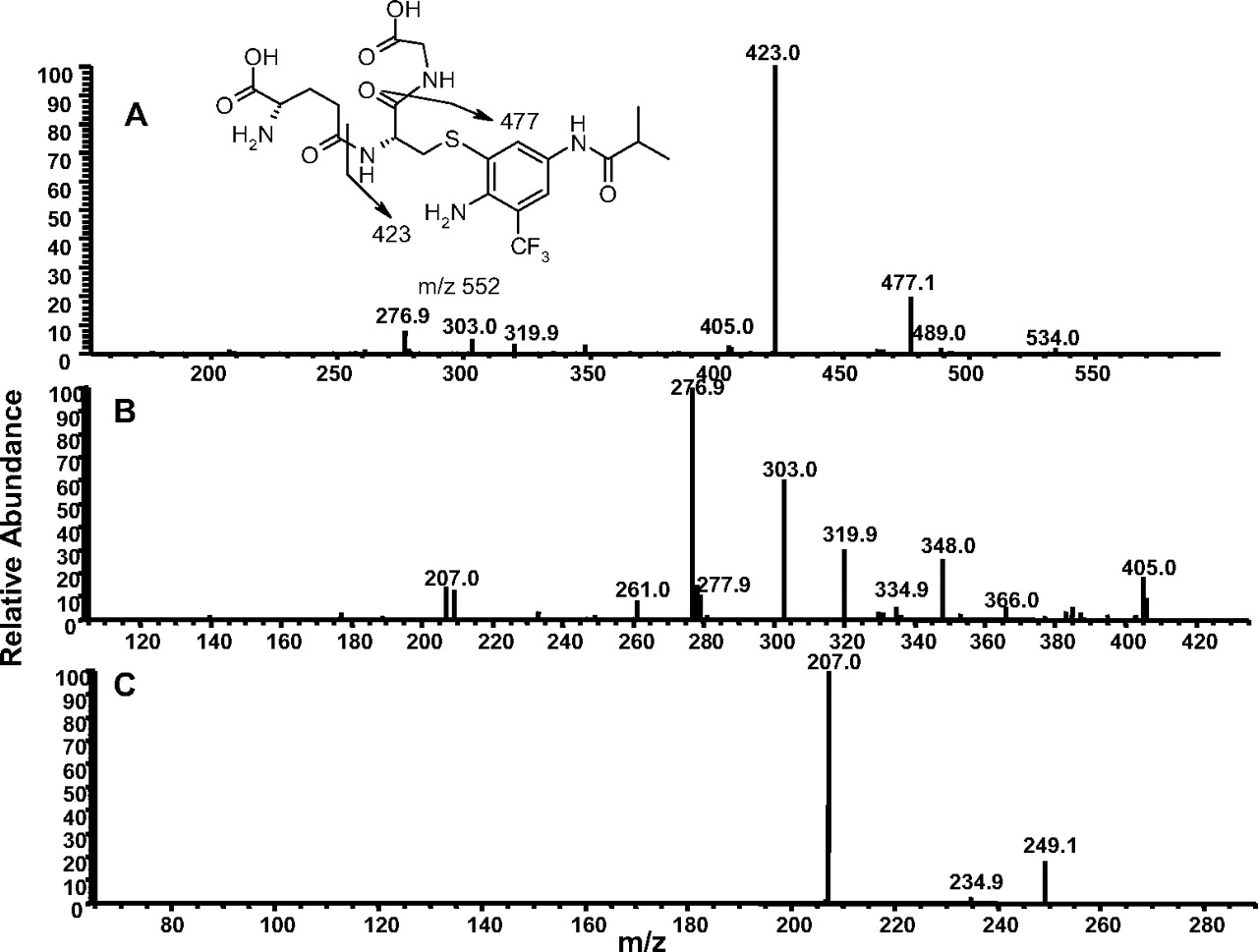
A, MS/MS spectrum of Flu-6-G1 at m/z 552 ([M+H]+). B, MS3 mass spectrum of Flu-6-G1 (the fragment ion at m/z 423). C, MS4 mass spectrum of the fragment ion of Flu-6-G1 at m/z 277.
Among the recombinant P450 isozymes tested, CYP1A2 was also the most active P450 isozyme catalyzing the formation of Flu-6-G1 (55% after normalization of relative hepatic abundance of P450 isozymes), followed by CYP3A4 (33%). CYP2C19 and CYP2D6 were involved to a lesser extent (Fig. 11D).
Discussion
In a previous study of the bioactivation of flutamide in human liver microsomes, a novel N-S glutathionyl adduct was identified. The present study revealed the formation of several GSH adducts (G1, Flu-1-G1, Flu-1-G2, Flu-6-G1) derived from the metabolites of flutamide in human liver microsomal incubations of GSH and NADPH.
G1 was the most abundant GSH adduct observed in human liver microsomal incubation of flutamide (Fig. 1A). Based on its structure, G1 was unlikely to be the result of direct bioactivation of parent flutamide; instead it can be derived from the hydroxylated metabolite/metabolites of flutamide. However, 2-hydroxyflutamide was not the precursor of G1, as incubation of 2-hydroxyflutamide with human liver microsomes failed to show any detectable G1. We explored the origination of G1 from other oxidative metabolites of flutamide. Monohydroxylated metabolite M5 and dihydroxylated flutamide M7 were isolated, and incubation with human liver microsomes was conducted for each isolated metabolite. The incubation of M7 failed to yield any detectable G1. M5 generated significant amounts of G1 in addition to M7 (data not shown). A proposed mechanism for G1 formation from M5 is shown in Scheme 2. Abstraction of the methine hydrogen of M5 by P450 would generate a carbon centered radical 1, oxygen rebound to the radical results in the formation of M7 (pathway b). Alternatively, loss of a hydrogen radical from the methyl group of 1 produces an α, β unsaturated hydroxyflutamide 2 (pathway a), a Michael receptor that can be attacked by GSH to form G1. An interesting feature of 1H NMR spectrum of G1 in methanol-d4 is the appearance of double peaks for protons 3 and 6, indicating two diastereomeric GSH conjugates in G1 due to the newly formed chiral center (carbon g), which would be expected when GSH attacks the proposed intermediate 2 in Scheme 2. P450-mediated desaturation is well documented in the literature (Ortiz de Montellano, 1995). This bioactivation pathway is consistent with the recent finding that a mercapturic acid conjugate of a hydroxylated flutamide was detected in the urine of prostate cancer patients treated with flutamide (Tevell et al., 2006). Although the authors proposed a different structure with a hydroxyl group on the tertiary carbon and a mercapturic acid attached to the primary carbon of the isopropyl group, several common MS fragment ions such as m/z 323, 289, 259, and 205 were present in the spectra of both G1 and the mercapturic acid conjugate, suggesting that the mercapturic acid conjugate was a processed product of G1 in vivo via hydrolysis and acetylation. A deglutamyl derivative of G1, an intermediate to the mercapturic acid conjugate, was also observed in human liver microsomal incubation (data not shown). A GSH conjugate of a hydroxylated flutamide has also been reported in two other studies (Soglia et al., 2006; Kostrubsky et al., 2007), which is most likely G1 based on the fact that only one GSH conjugate of the hydroxylated flutamide is generated in human liver microsomal incubations.

Proposed fragmentation pathway of Flu-6-G1 fragment at m/z 423.
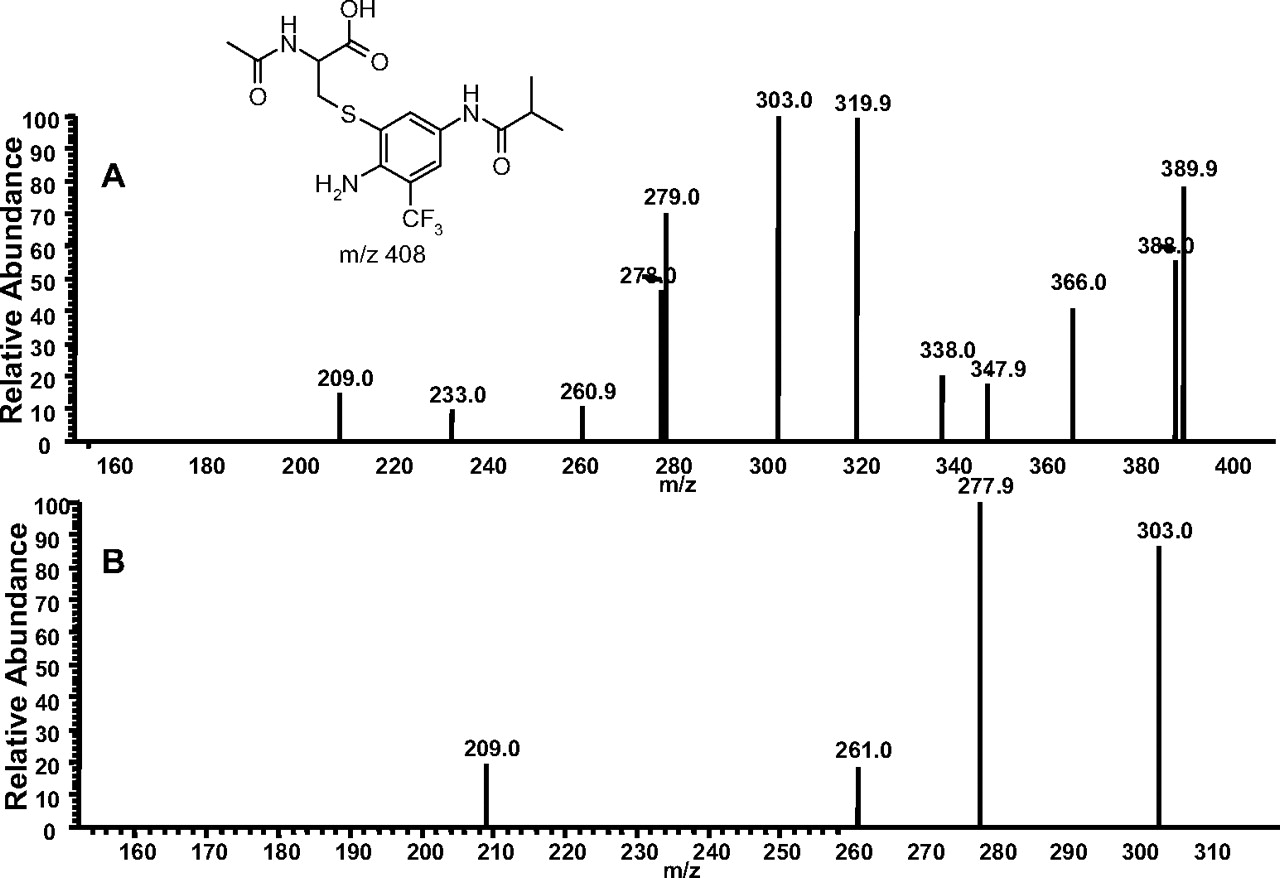
A, MS/MS spectrum of Flu-6-Cys1 at m/z 408 ([M+H]+). B, MS3 mass spectrum of the fragment ion at m/z 320.

A, the relative formation of the glutathione conjugate G1 from flutamide mediated by heterologously expressed P450 isoforms. The amounts of G1 produced by P450s were represented by the ratio of peak areas of G1 to that of internal standard, nilutamide. The P450-mediated formation of G1 was normalized to CYP1A2. B, the relative formation of Flu-1-G1 from Flu-1 mediated by heterologously expressed P450 isoforms. The amounts of Flu-1-G1 produced by P450s were represented by the ratio of peak areas of Flu-1-G1 to that of internal standard, nilutamide. The P450-mediated formation of Flu-1-G1 was normalized to CYP2C19. C, the relative formation of Flu-1-G2 from Flu-2 mediated by heterologously expressed P450 isoforms. The amounts of Flu-1-G2 produced by P450s were represented by the ratio of peak areas of Flu-1-G2 to that of internal standard, nilutamide. The P450-mediated formation of Flu-1-G2 was normalized to CYP2C8. D, the relative formation of Flu-6-G1 from Flu-6 mediated by heterologously expressed P450 isoforms. The amounts of Flu-6-G1 produced by P450s were represented by the ratio of peak areas of Flu-6-G1 to that of internal standard, nilutamide. The P450-mediated formation of Flu-6-G1 was normalized to CYP1A2.
Flu-1-G1 is proposed to be derived from Flu-3 as a result of aromatic hydroxylation of Flu-1 followed by oxidation to a quinone imine, which is trapped by GSH (Scheme 3, pathway a). Flu-3 is the major metabolite in the urine of prostate cancer patients treated with flutamide; however, it is mainly in the form of sulfate and glucuronic acid conjugate. Although the current study showed Flu-3 was bioactivated in human liver microsomes, the extensive phase II conjugation pathways could efficiently remove Flu-3 from the body and limit its bioactivation in vivo. On the other hand, the conjugates could potentially transport the reactive metabolite to other organs and cause toxicity locally.
Although the net result of Flu-1-G2 conjugation reaction appeared to be a direct aromatic substitution of the amino group of Flu-1 by GSH, Flu-1-G2 was only formed in NADPH-supplemented liver microsomal incubation. A proposed mechanism (Scheme 3, pathway b) involves initial oxidation of Flu-1 to N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine (Flu-1-N-OH) (Goda et al., 2006), which is further oxidized to a nitroso derivative of Flu-1 (Flu-1-N = O). Due to electron-withdrawing effect of the para nitro group, nucleophilic aromatic substitution of the nitroso group by GSH gives rise to Flu-1-G2. The proposed mechanism is supported by a recent report that chemical reaction of nitrosonitropyrene with GSH yielded a similar adduct via nucleophilic aromatic substitution of the nitroso group (Straube et al., 2005). Nitrosoarenes are reactive intermediates that can react with cellular thiols such as GSH and may play an important role in the biological effects of these compounds. The reaction generally started with formation of a semimercaptal from nucleophilic addition to the nitroso group by GSH. The cleavage of N-O bond generated a cationic sulfenamide intermediate, which can be stabilized by electron-donating substitutes normally found in the nitrosoarenes in these studies (Kazanis and McClelland, 1992; Gallemann and Eyer, 1994; Gallemann et al., 1998a,b). Interestingly, the reaction of the proposed Flu-1-N = O did not undergo nucleophilic addition of GSH to the nitroso group since no such GSH adducts were detected; instead it underwent a nucleophilic aromatic substitution of the nitroso group by GSH. A possible explanation could be the failure to stabilize the cationic sulfenamide intermediate due to electron withdrawing effect of the 4-nitro group of Flu-1. The proposed precursor of Flu-1-N = O, Flu-1-N-OH, was recently observed both in vitro and in vivo in humans and has been found to be cytotoxic toward rat hepatocytes (Goda et al., 2006). This metabolite was also found to be mainly conjugated in vivo.

Proposed mechanism for the formation of G1 from flutamide in human liver microsomal incubations supplemented with NADPH and GSH.

Proposed mechanism for the formation of Flu-1-G1 and Flu-2-G2 from Flu-1 in human liver microsomal incubations supplemented with NADPH and GSH.
Both Flu-1-G1 and Flu-1-G2 were also found in the incubation of parent flutamide with human liver microsomes in the presence of NADPH and GSH. Since flutamide was shown to be hydrolyzed to Flu-1 in liver microsomes, the two GSH adducts were most likely formed from subsequent bioactivation of the hydrolyzed metabolite Flu-1. Flu-1-G1 and Flu-1-G2 were also observed in human liver microsomal incubations of oxygenation metabolites (2-hydroxyflutamide, M5, and M7), where the metabolites were hydrolyzed to yield Flu-1 (data not shown).
The proposed mechanism for the formation of Flu-6-G1, Flu-6-G2, and Flu-6-G3 from Flu-6 involves initial oxidation of Flu-6 to a diimine intermediate (Scheme 4, pathway a) and subsequent nucleophilic attacks by GSH or acetyl cysteine methyl ester (Scheme 4, pathway b) at the three unsubstituted positions of the aromatic group. The mechanism is analogous to that recently reported in the formation of amino acid conjugates of 2,5-[13C]-dimethyl-p-benzoquinonediimine (Eilstein et al., 2006). Flu-6 has been detected in human urine and plasma, albeit at a lower level than those of 2-hydroxyflurtamide and Flu-1.
Proposed mechanism for the formation of Flu-6-G1, 2, 3 and Flu-6-Cys1, 2, 3 from Flu-6 in human liver microsomal incubations supplemented with NADPH and GSH/N-acetyl cysteine methyl ester.

Proposed bioactivation pathways of flutamide and its metabolites.
The formation of the GSH adducts of flutamide metabolites were NADPH-dependent, indicating the involvement of cytochromes P450. CYP1A and CYP3A have been shown to be involved in the covalent binding of flutamide to liver proteins (Berson et al., 1993; Fau et al., 1994). Phenotyping studies indicated that the P450 isozymes involved in the formation G1 and a previously reported N-(glutathio-S-yl) flutamide adduct are mainly CYP1A2 and CYP2C19. The major involvement of CYP1A2 in the formation of G1 could be due to the predominant role of CYP1A2 in catalyzing the formation of G1 precursor M5. A hydroxyflutamide mercapturic acid conjugate was detected in the urine of prostate cancer patients treated with flutamide (Tevell et al., 2006). Based on the similarity of MS data, this mercapturic acid conjugate is most likely derived from G1. It appears that this CYP1A2-mediated bioactivation pathway exists in vivo. However, low CYP1A2 activity has been associated with the onset of flutamide-induced hepatic dysfunction, and induced CYP1A2 activity has been thought to be a risk-lowering factor in smokers for developing flutamide hepatotoxicity (Nakagawa et al., 1999; Ozono et al., 2002). The role of CYP1A2 in flutamide-induced hepatotoxicity remains uncertain. CYP3A is the other major P450 isozyme responsible for the bioactivation of flutamide and subsequent covalent binding to liver proteins (Berson et al., 1993). However, there was only a marginal involvement of CYP3A4 in the formation of G1 and N-(glutathio-S-yl) flutamide adduct (Kang et al., 2007). CYP3A4 must play a major role in the bioactivation of other flutamide metabolites. Indeed, the present study showed that the formation of Flu-1-G1 was mediated by CYP3A4. It has also been reported recently that CYP3A4 is the major P450 isoform catalyzing the formation of Flu-3 and Flu-1-N-OH (Goda et al., 2006), the two proposed precursors of Flu-1-G1 and Flu-1-G2, respectively (Scheme 3), in the liver microsomal incubation of Flu-1. The involvement of CYP2C19 in the bioactivation of flutamide and its metabolite has not been shown in previous studies. Our results suggest further study may be warranted to assess the role of CYP2C19 in catalyzing the bioactivation of flutamide and the associated hepatotoxicity.
A summary of the bioactivation pathways of flutamide and its metabolites and the major P450 isozymes associated with each pathway is shown in Scheme 5. In addition to the one characterized previously for the parent (Kang et al., 2007), we have identified several bioactivation pathways of flutamide metabolites in the present study. These results suggest that, in addition to the direct bioactivation of parent flutamide, flutamide metabolites could contribute to the overall bioactivation of flutamide and potentially cause the flutamide-induced hepatotoxicity.
Footnotes
-
Part of this work was presented at the 8th International ISSX Meeting, Sendai, Japan, October 9–12, 2007.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.020370.
-
ABBREVIATIONS: flutamide, 2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide; P450, cytochrome P450; Flu-1, 4-nitro-3-(trifluoromethyl)phenylamine; Flu-2, N-[4-nitro-3-(trifluoromethyl)phenyl]-acetamide; Flu-1-N-OH, N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine; Flu-6, 2-methyl-N-[4-amino-3-(trifluoromethyl)phenyl]-propanamide; GSH, reduced glutathione; ESI, electrospray ionization; LC/MS, liquid chromatography/mass spectrometry; amu, atomic mass unit; NMR, nuclear magnetic resonance; HPLC, high-performance liquid chromatography; COSY, correlation spectroscopy; DCM, dichloromethane.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. -
↵1 Current affiliation: Genentech, Inc., South San Francisco, CA.
- Received January 7, 2008.
- Accepted April 11, 2008.

- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}