


Abstract
Flutamide (2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide), a nonsteroidal antiandrogen, is used in the treatment of prostate cancer but is occasionally associated with hepatic dysfunction. In the present study, the metabolism of flutamide including the formation of the possible reactive toxic metabolites was investigated using human liver microsomes and 10 isoforms of recombinant human cytochrome P450 (P450). 2-Hydroxyflutamide (OH-flutamide) and 4-nitro-3-(trifluoromethyl)phenylamine (FLU-1) were the main products of flutamide metabolism in human liver microsomes. The formation of OH-flutamide was markedly inhibited by ellipticine, an inhibitor of CYP1A1/1A2, and was mainly catalyzed by the recombinant CYP1A2. FLU-1 was also produced from OH-flutamide, but its metabolic rate was much less than that from flutamide. An inhibitor of carboxylesterase, bis-(p-nitrophenyl)phosphoric acid, completely inhibited the formation of FLU-1 from flutamide in human liver microsomes. A new metabolite, N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine (FLU-1-N-OH), was detected as a product of the reaction of FLU-1 with human liver microsomes and identified by comparison with the synthetic standard. The formation of FLU-1-N-OH was markedly inhibited by the addition of miconazole, an inhibitor of CYP3A4, and was mediated by recombinant CYP3A4. Furthermore, FLU-1-N-OH was detected mostly as the conjugates (glucuronide/sulfate) in the urine of prostate cancer patients collected for 3 h after treatment with flutamide. The formation of FLU-1-N-OH, however, did not differ between patients with and without abnormalities of hepatic functions among a total of 29 patients. The lack of an apparent association of the urinary excretion of FLU-1-N-OH and hepatic disorder may suggest the involvement of an additional unknown factor in the mechanisms of flutamide hepatotoxicity.
Flutamide (2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide), a nonsteroidal antiandrogenic drug, is widely used for the treatment of prostate cancer. The combination of flutamide with luteinizing hormone-releasing hormone agonist or orchidectomy showed significant prolongation of survival of prostate cancer patients (Prostate Cancer Trialists' Collaborative Group, 2000; Schmitt et al., 2001). Flutamide, however, occasionally causes a temporary slight increase in transaminase markers, and a certain population of prostate cancer patients suffers from severe hepatic dysfunction (Gomez et al., 1992; Matsuzaki and Nishikawa, 2003).
After oral administration, flutamide is rapidly absorbed and extensively metabolized in the liver (Schulz et al., 1988) and is excreted in the urine predominantly as metabolites (Brogden and Chrisp, 1991). We previously extensively investigated the metabolism of flutamide and proposed possible metabolic pathways of flutamide, as shown in Fig. 1 (Asakawa and Yamashita, 1995). A major metabolite in plasma is 2-hydroxyflutamide (OH-flutamide), whose formation from flutamide is catalyzed by CYP1A2 (Shet et al., 1997; Watanabe et al., 2001). In addition, another metabolite, 4-nitro-3-(trifluoromethyl)phenylamine (FLU-1), was detected as a major metabolite in plasma (Schulz et al., 1988), whereas the main metabolite in urine is 2-amino-5-nitro-4-(trifluoromethyl)phenol (FLU-3), which accounts for 50 to 90% of urinary excretion (Asakawa and Yamashita, 1995). It is suspected that FLU-3 is formed from FLU-1, although it has not been proven yet. Therefore, a considerable amount of FLU-1 is expected to be generated in the process of flutamide metabolism.

Metabolic pathways of flutamide (Asakawa et al., 1995, permitted from Antibiotics and Chemotherapy).
Flutamide-induced hepatic dysfunction is considered to be a metabolic idiosyncrasy (Zimmerman, 1999) that is associated with chemically reactive electrophilic metabolites formed by human CYP3A or CYP1A subfamilies (Berson et al., 1993; Fau et al., 1994). Although the covalent binding of these metabolites to microsomal proteins has been demonstrated (Berson et al., 1993), the reactive metabolite responsible for liver injury has yet to be identified.
N-Oxidative metabolites of arylamines and heterocyclic amines are reported to cause hepatic damage or mutagenesis (Kato and Yamazoe, 1994). Although no report is available on the formation of N-hydroxy derivatives of flutamide and its metabolites, we presumed that N-oxidative metabolites might be involved in the flutamide hepatotoxicity. Four possible N-oxidative derivatives of flutamide and its metabolites [OH-flutamide, FLU-1, and N-[4-nitro-3-(trifluoromethyl)phenyl]-acetamide (FLU-2)] were chemically synthesized, and their cytotoxicities were examined in vitro. Only N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine (FLU-1-N-OH) exhibited strong growth inhibition of primary cultures of rat hepatocytes (D. Nagai, R. Goda, E. Ichimura, A. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript in preparation). In the present study, the metabolism of flutamide leading to the formation of the N-oxidative metabolites was studied using human liver microsomes and recombinant human P450 to determine the enzymes involved. Furthermore, we investigated urinary metabolic profiles of prostate cancer patients, including N-oxidative metabolites. As a result, we made the novel finding of an N-oxidative metabolite of FLU-1 in human liver microsomes and also demonstrated its presence in the urine of prostate cancer patients.
Materials and Methods
Chemicals and Reagents. Flutamide and its metabolites OH-flutamide, FLU-1, FLU-2, FLU-3, FLU-4, FLU-5, and FLU-6 were supplied by Schering-Plough (Bloomfield, NJ). Four possible N-oxidized metabolites, FLU-N-OH, OH-FLU-N-OH, FLU-1-N-OH, and FLU-2-N-OH, as well as bicalutamide [(2RS)-4′-cyano-3-(4-fluorophenylsulphonyl)-2-hydroxy-2-methyl-3′-(trifluoromethyl)propionanilide], were synthesized in our laboratory, and their structures were confirmed by their 1H NMR spectra, 13C NMR spectra, and electrospray ionization (ESI) mass spectra. NADPH was purchased from Oriental Yeast Co. (Tokyo, Japan). β-Glucuronidase (from Helix pomatia, type H-2, containing sulfatase activity), ellipticine, miconazole, and bis-(p-nitrophenyl)phosphoric acid (BNPP) were obtained from Sigma Chemical Co. (St. Louis, MO). All other chemicals were of the highest commercial grade.
Microsomal preparations of 10 recombinant human P450 isoforms (i.e., CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4) expressed in a human B lymphoblastoid cell line, AHH-1, and a microsomal preparation of insect cells infected with baculovirus containing CYP3A4 were purchased from Gentest Corp. (Woburn, MA).
Human Liver Microsomes. The preparation of microsomes from human liver samples from 10 Japanese subjects was performed as described previously (Yamazoe et al., 1988). Microsomal suspensions were stored at –80°C until use. Experiments with human samples were approved by the Tohoku University Ethical Committee.
Metabolic Experiment with Human Liver Microsomes. The reaction mixture consisted of 0.5 mg of microsomal protein, 100 μM substrate (flutamide, OH-flutamide, FLU-1, or FLU-2), and 0.1 M potassium-phosphate buffer (pH 7.4) in a final volume of 1 ml was incubated at 37°C. Although clinical plasma concentration ranges of flutamide and its metabolites are 0.05 ∼ 5 μM (Nakagawa et al., 1999; Aizawa et al., 2003), we used 100 μM substrates to detect their metabolic capacities. The reaction was started by the addition of NADPH (1.6 mM, final concentration) and terminated by the addition of 4 ml of ice-cold 99% ethyl acetate containing 0.01% ascorbic acid after incubation at 37°C for 30 min, and 1 μg of bicalutamide (flutamide analog) was added to the mixture as an internal standard. The mixture was centrifuged at 1800g for 5 min, and the resultant organic layer was evaporated. The residue was dissolved in ammonium-acetate buffer (25 mM, pH 5)/methanol (50:50, v/v) and filtered through an Ultrafree-MG (0.2 μm; Millipore, Bedford, MA) filter. Metabolites were quantified by liquid chromatography/mass spectrometry (LC/MS) as described below.
The effects of NADPH and inhibitors of CYP1A1/1A2, CYP3A4, or carboxylesterase on the metabolism of flutamide and FLU-1 were also studied. The inhibitors used were BNPP for carboxylesterase (Slatter et al., 1997), ellipticine for CYP1A1/1A2 (Tassaneeyakul et al., 1993), and miconazole for CYP3A4 (Sakaeda et al., 2005). Flutamide and FLU-1 (100 μM) were incubated as described above, except for omitting NADPH or adding 500 μM BNPP, 1 μM ellipticine, or 10 μM miconazole.
Metabolic Experiment with Purified Recombinant Human P450s. Incubation conditions for the recombinant human P450 isoforms were the same as used for human microsomes as described above, except for the microsomal protein content and the incubation period (i.e., 1 mg and 120 min for human P450 enzymes from B lymphoblastoid cells, and 100 pM and 30 min for human CYP3A4 from insect cells, in accord with the supplier's instructions). Two concentrations of substrates (flutamide or FLU-1) at 1 and 100 μM were used to compare various P450s for their metabolism. In the experiments regarding FLU-1 metabolism, we used only human CYP3A4 obtained from insect cells because of the low metabolic activity for FLU-1 of human CYP3A4 derived from B lymphoblastoid cells (data not shown).
Correction by Relative Content of Human P450s. Each metabolic rate determined using recombinant human P450 isoforms was corrected by the composition ratio of each P450 in human liver reported previously (Shimada et al., 1994) using the following equation: 
Correlation Study. The rate of formation of FLU-1-N-OH was compared with the rate of testosterone 6β-hydroxylation (Sanwald et al., 1995), which is an index for CYP3A4 activity, using microsomes obtained from 10 human livers.
Patient Urine. Urine samples were obtained from 29 prostate cancer patients prescribed flutamide and luteinizing hormone-releasing hormone agonist combination therapy for at least 12 weeks at Jikei University Hospital and Fuji City General Hospital. Four patients showed signs of hepatic dysfunction during the flutamide treatment (Aizawa et al., 2003). Tablets containing 125 mg of flutamide were administered orally three times daily, and urine was collected for 3 h after the oral intake of flutamide in the morning of day 28. The urine volume was recorded and a part of the urine was stored at –80°C until it was analyzed.
The study was approved by the ethics committee of each hospital. Informed consent was obtained from all patients.
Urinary Excretion Analysis. Urine samples (0.5 ml) were mixed with 1 μg of bicalutamide (an internal standard) and 1 ml of 0.2 M acetic acid and then loaded onto a SEP-PAK C18 cartridge (Waters Associates, Milfield, MA), which was preconditioned with methanol and water. Then, the loaded cartridge was washed with 10 ml of water and 0.3 ml of methanol and eluted twice with 10 ml of methanol. The eluent was evaporated to dryness under reduced pressure. The residue was redissolved in a mixture of methanol and ammonium-acetate buffer (25 mM, pH 5) (1:1, v/v), and analyzed by LC/MS as described below.
For the quantification of biologically conjugated forms of flutamide and its metabolites, 0.5 ml of urine sample was mixed with 0.05 ml of 0.2 M acetic acid to adjust the sample to pH 5. After the addition of 1.5 ml of ammonium-acetate buffer (0.2 M, pH 5) containing 0.1 M ascorbic acid, the sample was treated with 11,035 units of β-glucuronidase and 419.4 units of sulfatase at 37°C for 20 h. We confirmed the stability of metabolites under this hydrolysis condition (data not shown). The resultant mixture was then extracted as described above.
After urine concentration quantified by LC/MS was converted to the concentration equivalent to flutamide, urinary excretion ratio of each metabolite was calculated using the following equation: 
Analysis of Metabolites by LC/MS. LC/ESI-MS was used for analyses of metabolites. The LC/MS system consisted of a GULLIVER SERIES PU-980 (Jasco International, Tokyo, Japan), PLATFORM (Micromass, Manchester, UK) and CAPCELL PAK C18 SG-120 column (4.6 × 150 mm, 5 μm; Shiseido, Tokyo, Japan) with a gradient mobile phase of methanol and ammonium-acetate buffer (25 mM, pH 5). The MS conditions were as follows: negative ion mode; gas, nitrogen; capillary, 3.5 kV; cone, 50 V; source heater, 180°C. Detection was performed by selected ion monitoring. The [M-H]– ions, m/z 275 for flutamide, m/z 291 for OH-flutamide, m/z 205 for FLU-1, m/z 247 for FLU-2, m/z 221 for FLU-3, m/z 217 for FLU-4, m/z 261 for FLU-5, m/z 245 for FLU-6, m/z 291 for FLU-N-OH, m/z 307 for OH-FLU-N-OH, m/z 221 for FLU-1-N-OH, and m/z 263 for FLU-2-N-OH, were monitored for quantification of each metabolite.
Statistical Analysis. Data are presented as means ± S.D. Statistical comparisons between two groups were made using Student's t test. A value of p < 0.05 was considered to be statistically significant.
Results
Metabolism in Human Liver Microsomes. In the course of flutamide study to elucidate the mechanism of its hepatotoxicity, we assumed that toxic metabolite might be produced and cause hepatic cell damage. For example, chemically synthesized FLU-1-N-OH exhibited growth inhibition of primary cultures of rat hepatocytes (D. Nagai, R. Goda, E. Ichimura, A. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript in preparation). To assess whether the postulated toxic compound was produced biologically, we incubated FLU-1 with human liver microsomes. When the incubated mixture was extracted in the presence of an antioxidant, a new metabolite was detected by LC/ESI-MS in addition to FLU-3. A representative LC/MS chromatogram is shown in Fig. 2 with that of chemically synthesized authentic samples. The chromatogram of the metabolite showed good agreement with that of a chemically synthesized authentic sample of FLU-1-N-OH. Furthermore, the metabolite showed an LC/ESI mass spectrum characterized by a predominant deprotonated molecular ion at m/z 221 and two less intense ions at m/z 204 and 281. The spectrum was identical to that of authentic FLU-1-N-OH standard (data not shown).
The production of FLU-1-N-OH was examined with 10 different microsomal preparations. As shown in Fig. 3C, FLU-1-N-OH was detected in the reaction products of seven of 10 human liver microsomes by LC/MS. The metabolic rate for FLU-1-N-OH from FLU-1 was less than 0.1 nmol/mg/min, which was much slower than that for FLU-3 production.

LC/ESI-MS chromatograms of FLU-1 metabolites formed by human liver microsomes. Representative chromatographic profiles of authentic samples of FLU-1-N-OH (A), FLU-3 (B), and reaction products formed by incubation of FLU-1 with human liver microsomes at 37°C for 30 min (C).
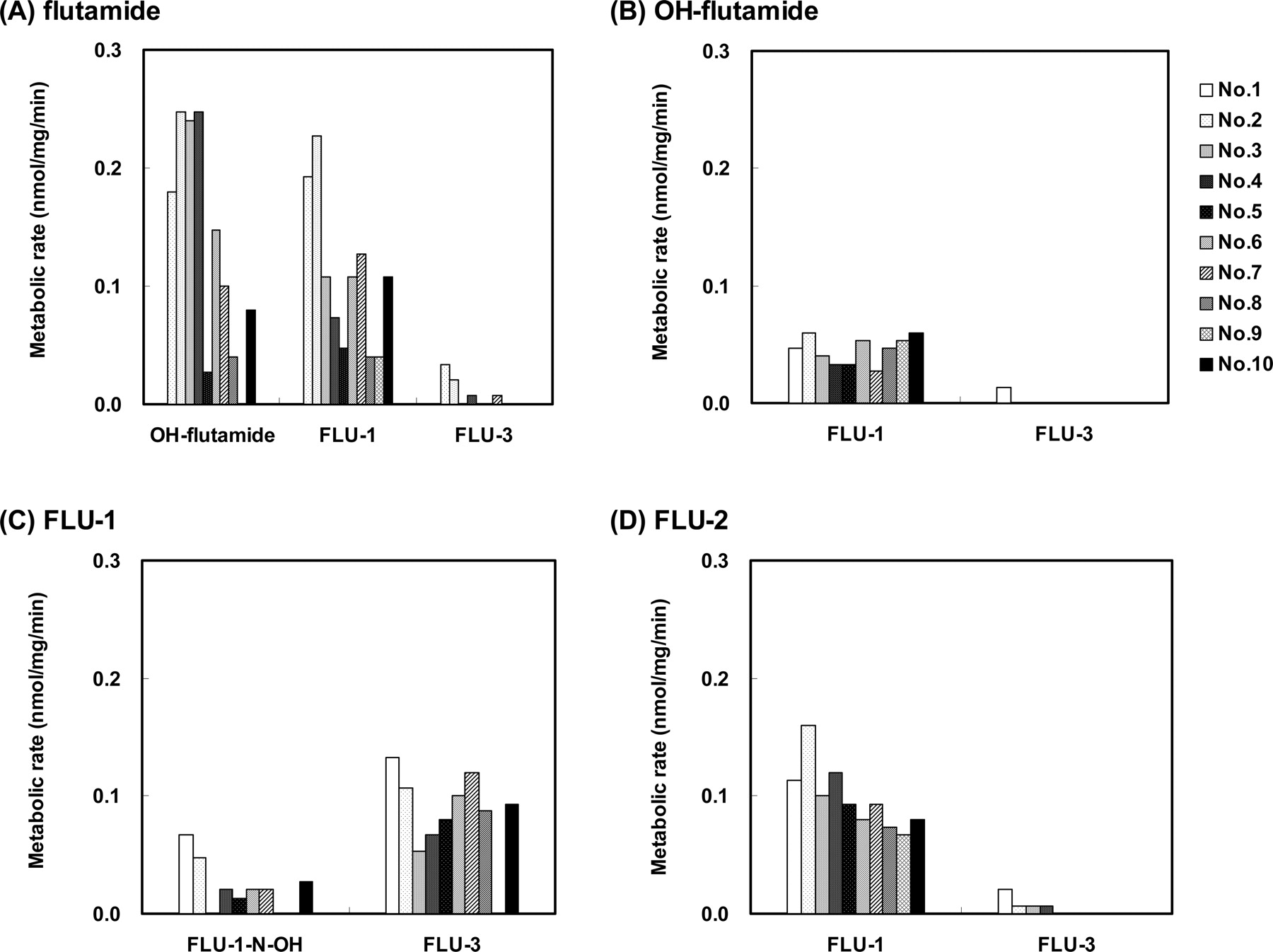
The production of other N-oxidative products from flutamide, OH-flutamide and FLU-2, as well as FLU-1, as a result of incubation with 10 individual human liver microsomes was also examined (Fig. 3, A, B, and D). None of the three N-oxidative metabolites FLU-N-OH, OH-FLU-N-OH, or FLU-2-N-OH were detected in the microsomal reaction mixtures. Two major metabolites, OH-flutamide and FLU-1, and one minor metabolite, FLU-3, were products of flutamide. Only one metabolite, FLU-1, was formed from OH-flutamide except in one microsome preparation, and FLU-1 was also a major metabolite of FLU-2. FLU-1 produced FLU-3 as a major metabolite in addition to FLU-1-N-OH.
Detection of FLU-1-N-OH in Urine of Prostate Cancer Patients. The presence of urinary flutamide and its metabolites was examined in 25 patients with no sign of hepatic dysfunction and in four patients with hepatic dysfunction as a result of flutamide therapy (Fig. 4). Low but significant amounts of FLU-1-N-OH were detected in the urine of prostate cancer patients treated with flutamide. No other N-oxidative metabolites were detected. Most of the FLU-1-N-OH existed in the form of conjugates. FLU-1-N-OH was not detected in the plasma of these patients (less than 1 nM; data not shown). In addition, no significant difference in the excreted amount of FLU-1-N-OH was observed between the patients with and without hepatic disorder (p > 0.05). As for the excreted amount of other metabolites, statistical significances were not also attained. The patterns of excreted metabolites and the ratios of the conjugated forms in the two patient groups were not markedly different from each other. In urine, FLU-3, rather than OH-flutamide, was the main metabolite excreted. OH-flutamide, FLU-1, FLU-4, FLU-5, and FLU-6 were also detected. Flutamide and FLU-2 were not detected in the urine.
The Effects of NADPH and Inhibitors of CYP1A1/1A2, CYP3A4, and Carboxylesterase on Flutamide and FLU-1 Metabolism. FLU-1-N-OH is expected to be formed through the N-oxidation of FLU-1, which is metabolized from flutamide. Therefore, we next investigated the metabolizing enzyme for both flutamide and FLU-1. To verify the involvement of P450 in the metabolism of flutamide and FLU-1, the requirement for NADPH was investigated in each metabolic pathway. As shown in Table 1, the formation of FLU-1 from flutamide was not inhibited at all by the depletion of NADPH, whereas it was completely inhibited by BNPP, an inhibitor of carboxylesterase. Therefore, the pathway from flutamide to FLU-1, in which the amide bond in the side chain is cleaved, might be mediated by carboxylesterase. In contrast, the formation of OH-flutamide from flutamide, and the production of FLU-1-N-OH and FLU-3 from FLU-1 were completely abolished in the absence of NADPH in the reaction mixture. Furthermore, the microsomal oxidation of flutamide to OH-flutamide was partially inhibited by ellipticine, an inhibitor of CYP1A1/1A2, although it was not inhibited by miconazole, an inhibitor of CYP3A4. These data indicate the involvement of CYP1A1/1A2 in this pathway. In contrast, the formation of FLU-1-N-OH and FLU-3 from FLU-1 was only partially inhibited by miconazole and was not affected by the addition of ellipticine.
Effects of NADPH, carboxylesterase inhibitor, and P450 inhibitors on the metabolism of flutamide and FLU-1 in human liver microsomes
Flutamide or FLU-1 (100 μM) was incubated with or without NADPH, or with BNPP, ellipticine, or miconazole at 37°C for 30 min with human microsomes, and the products formed were determined by LC/MS as described under Materials and Methods.

Metabolism of flutamide and three of its metabolites in human liver microsomes. One hundred micromolar flutamide (A), OH-flutamide (B), FLU-1 (C), or FLU-2 (D) was incubated with 10 human liver microsomes preparations at 37°C for 30 min. Metabolites formed were determined by LC/MS as described under Materials and Methods.

Urinary excretion ratio of flutamide and its metabolites against a dose in prostate cancer patients treated with flutamide (125 mg, three times daily). Urine was pooled for 3 h after oral intake of flutamide in the morning of day 28 from 25 patients with normal hepatic function (open columns) and four patients with flutamide-induced hepatic dysfunction (solid columns). LC/MS was used for the determination of each metabolite in nontreated urine (A) and β-glucuronidase-treated urine (B) as described under Materials and Methods. Data represent the mean ± S.D. of each patient group. Data were analyzed statistically using Student's t test with no differences between two patient groups observed.
Metabolism By Recombinant Human P450s. To assess P450 forms responsible for the formation of the three major metabolites, flutamide and FLU-1 were reacted with 10 different recombinant human P450s: CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4. The in vitro metabolism from flutamide to OH-flutamide (Fig. 5), FLU-1 to FLU-1-N-OH (Fig. 6), and FLU-1 to FLU-3 (Fig. 7) was determined at two different concentrations of substrate in each experiment. Each metabolic rate determined in vitro was corrected by the composition ratio of each P450 in human liver reported previously (Shimada et al., 1994) to predict the relative contribution of each P450 moiety in vivo. As shown in Fig. 5, CYP1A2 exhibited the highest catalytic activity for the metabolism to OH-flutamide at both low and high concentrations of flutamide. The activities of CYP1A1, CYP2C9, and CYP2C19 were 1:4, 1:6, and 1:20 of that of CYP1A2, respectively, at 1 μM flutamide, whereas at 100 μM flutamide the activities of CYP1A1, CYP2C9, CYP2C19, and CYP3A4 were 1:10, 1:4, 1:20, and 1:20, respectively, of that of CYP1A2.

Formation of OH-flutamide from flutamide by human P450 isoforms. Flutamide (A, 1 μM; B, 100 μM) was incubated at 37°C for 120 min with microsomes (1 mg/ml) from human B lymphoblastoid cells expressing each of 10 P450 isoforms. The left-hand graphs show net enzymatic reaction velocity without correction. The right-hand graphs show relative velocity corrected by the reported content ratios of P450 isoforms in human liver microsomes (Shimada et al., 1994). The amount of OH-flutamide formed was estimated by LC/MS as described under Materials and Methods. The data are from a representative experiment. N.D., not detected.
As for the metabolism of FLU-1 to FLU-1-N-OH (Fig. 6), CYP3A4 mediated at the highest rate at the lower concentration of FLU-1, whereas CYP1A2 showed a reduced extent of the activity. At the higher concentration of the substrate, this reaction was mediated mainly by CYP2C9 and CYP3A4 at similar rates, whereas CYP1A2 and CYP2A6 showed only marginal activities. In the case of the formation of FLU-3 from FLU-1 (Fig. 7), CYP3A4 showed the highest catalytic activity at the lower concentration of FLU-1. CYP2A6 also had about half the activity of CYP3A4, although the activities of CYP2C9, CYP2D6, and CYP2E1 were very limited. CYP3A4 was also responsible for the metabolism at the high concentration of FLU-1, and CYP2A6 and CYP2C9 showed only slight activities.
Correlation Study. We examined the correlation between the activity of FLU-1-N-OH formation and the metabolism of a specific substrate by CYP3A4 in 10 different human liver microsomes. The formation of FLU-1-N-OH was moderately correlated with the activity of testosterone 6β-hydroxylation (r2 = 0.488) at 100 μM FLU-1.
Discussion
The formation of OH-flutamide and FLU-1, which are generated by oxidation of the isopropyl moiety and cleavage of the amide bond, respectively, are the major routes of the metabolism of flutamide in humans (Schulz et al., 1988). Among these, OH-flutamide was shown to have less cytotoxicity in primary hepatocytes compared with flutamide in our preliminary experiments. Reduction of the arylnitro group is known to yield N-hydroxylamine reactive intermediates. The enterohepatic circulation of amine metabolites, which are produced by enterobacterial reduction of nitro compounds, is proposed to contribute to N-hydroxyl reactive intermediates (Spain, 1995). Most arylnitro compounds are mutagenic for Salmonella typhimurium without the addition of an external metabolic system (e.g., S9). 4-Nitroaniline, however, requires an external metabolic activation system to cause bacterial mutagenicity (Garner and Nutman, 1977). These data suggest to us the possible involvement of N-oxidation in flutamide-induced liver damage. Therefore, we have investigated the metabolism of flutamide metabolites, focusing on the formation of reactive intermediates. Previously reported data showed the metabolic profiles of flutamide in humans and rats (Fig. 1). As a possible mechanism of the hepatotoxicity of flutamide, we hypothesized that reactive toxic metabolites might be produced and cause hepatic cell damage.

Formation of FLU-1-N-OH from FLU-1 by human P450 isoforms. FLU-1 (A, 1 μM; B, 100 μM) was incubated at 37°C for 120 min with microsomes (1 mg/ml) from human B lymphoblastoid cells expressing each of nine P450 isoforms or for 30 min with microsomes (100 pM) from insect cells infected with baculovirus carrying CYP3A4.
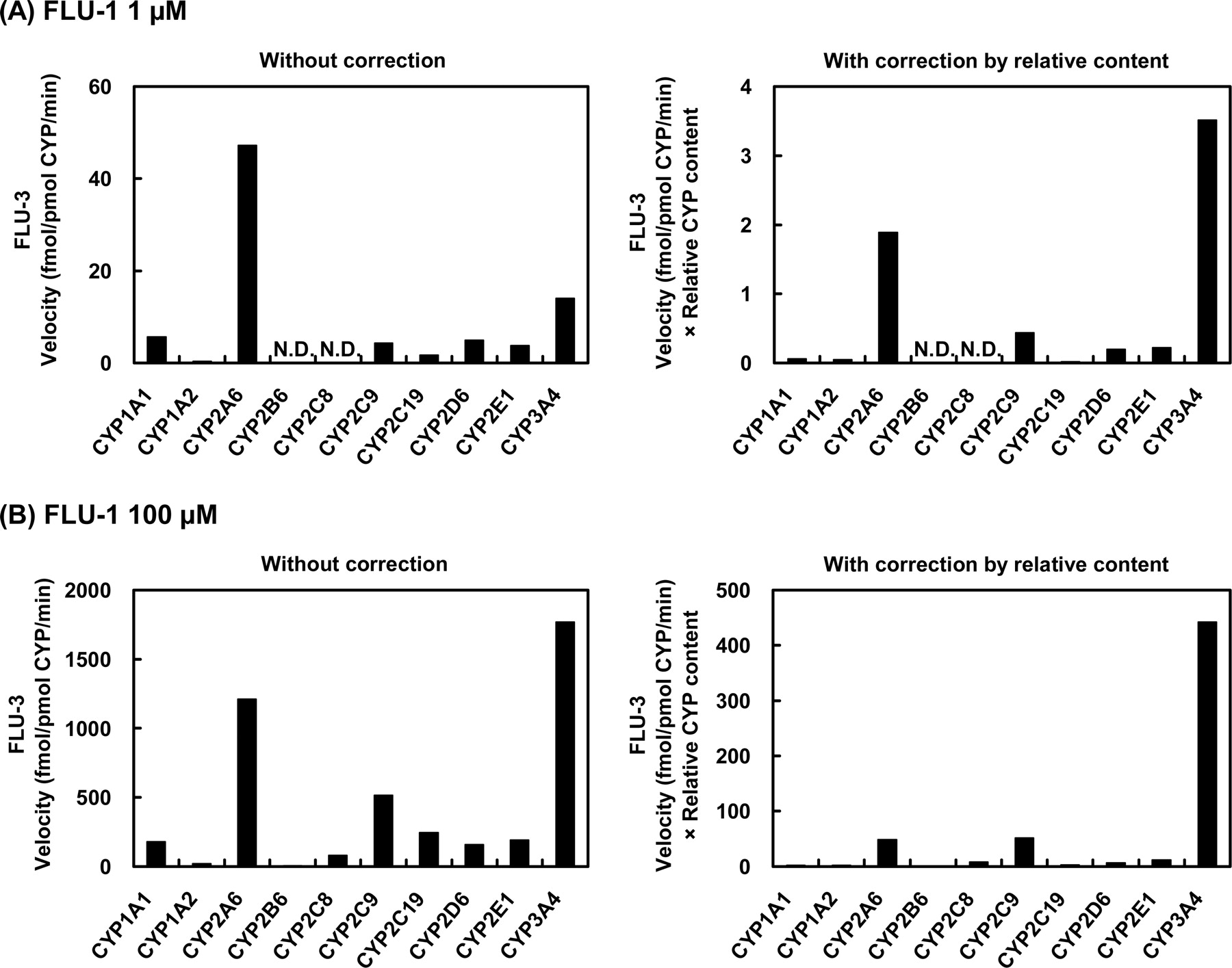
Formation of FLU-3 from FLU-1 by human P450 isoforms. FLU-1 (A, 1 μM; B, 100 μM) was incubated at 37°C for 120 min with microsomes (1 mg/ml) from human B lymphoblastoid cells expressing each of nine P450 isoforms or for 30 min with microsomes (100 pM) from insect cells infected with baculovirus carrying CYP3A4.
In the present study, we demonstrated the appearance of a novel N-oxidative flutamide metabolite, FLU-1-N-OH, for the first time both in vitro and in vivo in human specimens. FLU-1-N-OH was recently found by our group to have intense cytotoxicity toward rat hepatocytes among flutamide metabolites, and also to be highly reactive toward glutathione (D. Nagai, R. Goda, E. Ichimura, Y. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript in preparation). Therefore, detection of this metabolite in human liver microsomes is necessary to assess its role in flutamide-induced hepatic dysfunction in prostate cancer patients. Moreover, the present study using human liver microsomes and recombinant human P450 isoforms has shown that FLU-1-N-OH is metabolized from FLU-1 mainly by CYP3A4. This is consistent with previous studies showing that reactive metabolites related to flutamide-induced hepatotoxicity might be products of reactions catalyzed by CYP1A or CYP3A (Berson et al., 1993; Fau et al., 1994). In addition, the covalent binding of reactive metabolites to microsomal proteins was reported to be decreased by an inhibitor of CYP3A and to be increased by an inducer of CYP3A (Berson et al., 1993). The metabolic activation to reactive metabolites through FLU-1 is supported by the clinical finding that the plasma concentration of FLU-1 in patients suffering from hepatic disorders is considerably higher than that in patients with normal liver function (Aizawa et al., 2003).
Flutamide is biotransformed into two main metabolites, OH-flutamide and FLU-1. The formation of OH-flutamide from flutamide in the human liver is predominantly mediated by CYP1A2. This is concordant with earlier studies (Schulz et al., 1988; Shet et al., 1997; Watanabe et al., 2001). In the present study, flutamide was shown to undergo deamidation to FLU-1 by a carboxylesterase. This reaction was completely inhibited by carboxylesterase inhibitor BNPP. Although FLU-1 was also produced from OH-flutamide, its metabolic rate was low in comparison with that from flutamide. This finding suggests that most of the FLU-1 produced in the human liver is formed directly from flutamide and also implies that the formation of FLU-1-N-OH may be increased through enhanced production of FLU-1. Low CYP1A2 activity as measured by the caffeine test is reported to correlate with the onset of flutamide-induced hepatic dysfunction (Ozono et al., 2002). Such patients with low CYP1A2 activity are expected to produce more FLU-1 and may also support consequently the formation of cytotoxic FLU-1-N-OH. Patients who smoke, which induces CYP1A2 activity, were reported to be a low-risk population for developing flutamide hepatotoxicity (Wada et al., 1999). Moreover, it is reported that hepatic injury could be induced in CYP1A2 knockout mice after the oral administration of FLU-1 (Matsuzaki et al., 2006). The activity of CYP1A2 varies greatly depending on diet, personal habits, and chemical exposure and might affect the formation of FLU-1 and indirectly affect the formation of FLU-1-N-OH.
FLU-3 is another product of FLU-1. CYP3A4 is mainly responsible for the formation of FLU-1-N-OH and FLU-3 at lower and higher concentrations of FLU-1, respectively. Although FLU-1-N-OH formation at 100 μM FLU-1 did not show a good correlation with the activity of testosterone 6β-hydroxylation, the data may implicate the contribution of a P450 isoform other than CYP3A4 on the formation of FLU-1-N-OH at higher concentrations of FLU-1. Actually, CYP2C9 exhibited the same catalytic activity of FLU-1-N-OH formation as CYP3A4 at 100 μM FLU-1, although at a low concentration equivalent to clinical level CYP3A4 showed the highest activity. On the other hand, with respect to FLU-3 formation, CYP2A6 revealed itself to be essential to the formation of FLU-3 in addition to CYP3A4 at low concentration.

Proposed metabolic pathways of flutamide in humans and the responsible enzymes. Single asterisk, catalyzing enzyme with CYP3A4 at higher substrate concentration. Double asterisk, catalyzing enzyme with CYP3A4 at lower substrate concentration.
To examine the relationship between flutamide metabolism and hepatic damage, we analyzed metabolites of flutamide in urine of patients with and without flutamide-induced hepatic dysfunction. Although FLU-1-N-OH was detected as the conjugated form (glucuronide/sulfate) in the urine of patients under flutamide therapy, the amounts of excreted FLU-1-N-OH and the profiles of metabolites in urine were similar between both the patients groups. The present in vivo study was limited, and further studies will be required to assess the association the metabolites with flutamide hepatotoxicity. The following two possibilities, however, are envisaged because FLU-1-N-OH was detected in most of the in vitro and in vivo human samples and conjugation in general works as both detoxification and toxification. First, FLU-1-N-OH might be a proximate metabolite causing hepatic damage. FLU-1-N-OH formed in liver through FLU-1 might be detoxified by glucuronidation and sulfation and excreted gradually into the urine and could thus be detected there. Second, FLU-1-N-OH might not be an ultimate form, despite the direct hepatotoxicity of FLU-1-N-OH in hepatocyte cultures. It is possible that the ultimate metabolite is the N-O-ester of FLU-1-N-OH, because it is known that the metabolic activation pathways of carcinogenic arylamines include N-O-esterification of N-hydroxylamines (Kato and Yamazoe, 1994). Sulfotransferases such as ST1A/C are reported to be responsible for the metabolic activation of N-hydroxylamines. Furthermore, in both cases, glutathione S-transferases may have an important role in detoxifying FLU-1-N-OH or other ultimate reactive metabolites.
According to the present results using human liver microsomes and recombinant human P450 isoforms together with reported information on the metabolism of flutamide, the metabolism of flutamide is summarized in Fig. 8. Flutamide is metabolized to OH-flutamide and FLU-1 by CYP1A2 and carboxylesterase, respectively. FLU-1 is further metabolized to a new toxic metabolite, FLU-1-N-OH, and also to FLU-3, and CYP3A4 is mainly responsible for both reactions. FLU-1-N-OH conjugated in glucuronide and sulfate forms was detected in the urine of prostate cancer patients, although the samples of patients with flutamide-induced hepatic damage were limited, and the relation between the amount of conjugated FLU-1-N-OH and hepatic dysfunction was not clarified. It is possible that alterations of carboxylesterase, CYP1A2, and CYP3A4 activities influence the formation of FLU-1-N-OH. The activities of UDP-glucuronosyltransferases, sulfotransferases, or glutathione S-transferases may also have important roles in flutamide hepatic dysfunction. The higher incidence of hepatic dysfunction in Japanese than in Caucasian patients despite the use of low flutamide doses (Nakagawa et al., 1999; Wada et al., 1999; Yamaguchi et al., 2001) may depend on ethnic differences in the frequency of genetic polymorphism in flutamide-metabolizing enzymes such as carboxylesterase, CYP1A2, CYP3A4, UDP-glucuronosyltransferases, sulfotransferases, and glutathione S-transferases. Further studies in regard to flutamide metabolism and its association to the clinical data will be necessary for elucidating the precise mechanisms of flutamide hepatotoxicity.
Footnotes
-
This work was supported by grants-in-aid from the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Ministry of Health, Labour and Welfare of Japan, and the Japan Health Sciences Foundation.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008623.
-
ABBREVIATIONS: OH-flutamide, 2-hydroxyflutamide; P450, cytochrome P450; FLU-1, 4-nitro-3-(trifluoromethyl)phenylamine; FLU-3, 2-amino-5-nitro-4-(trifluoromethyl)phenol; FLU-2, N-[4-nitro-3-(trifluoromethyl)phenyl]-acetamide; FLU-1-N-OH, N-[4-nitro-3-(trifluoromethyl)phenyl]-hydroxylamine; FLU-4, N-[4-amino-3-(trifluoromethyl)phenyl]-acetamide; FLU-5, 2-hydroxy-2-methyl-N-[4-amino-3-(trifluoromethyl)-phenyl]propanamide; FLU-6, 2-methyl-N-[4-amino-3-(trifluoromethyl)-phenyl]-propanamide; FLU-N-OH, N-hydroxy-N-[4-nitro-3-(trifluoromethyl)-phenyl]isobutyramide; OH-FLU-N-OH, 2,N-dihydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]propionamide; FLU-2-N-OH, N-hydroxy-N-[4-nitro-3-(trifluoromethyl)phenyl]acetamide; ESI, electrospray ionization; BNPP, bis-(p-nitrophenyl)phosphoric acid; LC/MS, liquid chromatography/mass spectrometry.
- Received November 28, 2005.
- Accepted February 15, 2006.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}