


Abstract
Time-dependent inhibition (TDI) of cytochrome P450 (P450) enzymes caused by new molecular entities (NMEs) is of concern because such compounds can be responsible for clinically relevant drug-drug interactions (DDI). Although the biochemistry underlying mechanism-based inactivation (MBI) of P450 enzymes has been generally understood for several years, significant advances have been made only in the past few years regarding how in vitro time-dependent inhibition data can be used to understand and predict clinical DDI. In this article, a team of scientists from 16 pharmaceutical research organizations that are member companies of the Pharmaceutical Research and Manufacturers of America offer a discussion of the phenomenon of TDI with emphasis on the laboratory methods used in its measurement. Results of an anonymous survey regarding pharmaceutical industry practices and strategies around TDI are reported. Specific topics that still possess a high degree of uncertainty are raised, such as parameter estimates needed to make predictions of DDI magnitude from in vitro inactivation parameters. A description of follow-up mechanistic experiments that can be done to characterize TDI are described. A consensus recommendation regarding common practices to address TDI is included, the salient points of which include the use of a tiered approach wherein abbreviated assays are first used to determine whether NMEs demonstrate TDI or not, followed by more thorough inactivation studies for those that do to define the parameters needed for prediction of DDI.
Pharmacokinetic drug-drug interactions (DDIs) can occur when one drug alters the metabolism of a coadministered drug. The outcome is an increase or decrease in the systemic clearance and/or bioavailability, and a corresponding change in the exposure to a coadministered drug. The clinical consequences of DDIs range from lack of therapeutic efficacy of a life saving drug to severe adverse drug reactions, including fatalities. Significant drug-drug interactions can lead to termination of development of otherwise promising new therapies, withdrawal of a drug from the market, or severe restrictions/limitations on its use (Wienkers and Heath, 2005). Because of the impact on patient health and safety, DDI was the subject of a position paper in 2003 by scientists from member companies of the Pharmaceutical Research and Manufacturers of America (PhRMA) that focused on the in vitro and in vivo assessment and implications of reversible mechanisms of enzyme inhibition (Bjornsson et al., 2003). The U.S. Food and Drug Administration (FDA) has issued a revised draft guidance for the conduct of in vitro and in vivo drug-drug interaction studies (FDA Guidance for Industry, 2006; http://www.fda.gov/cder/guidance/index.htm). This draft guidance was broadly written with the intention of assisting in the harmonization of approaches and study designs for better comparison between different drugs and data from various laboratories.
Oxidation reactions catalyzed by the cytochrome P450 (P450) enzymes are the most prevalent biotransformations of administered drugs, as well as other xenobiotics and some endogenous compounds (Soars et al., 2007). P450 inhibition has been implicated in the majority of reported clinically relevant drug-drug interactions (Thummel et al., 2000; Bachmann et al., 2003). In addition, the inhibited metabolic pathway could lead to decreased formation of an active metabolite (or formation of a drug from a prodrug) resulting in decreased efficacy. The increase of plasma concentrations caused by DDIs can be substantial, as reported for the interaction between ketoconazole or itraconazole (strong CYP3A4 inhibitors) and triazolam (a CYP3A4 substrate), in which exposure to triazolam increased by 22- or 27-fold after coadministration with ketoconazole or itraconazole, respectively (Varhe et al., 1994). For drugs with narrow therapeutic indices, increases in plasma concentrations can lead to adverse drug reactions.
Inhibition of P450 enzymes can be classified as either reversible or time-dependent inhibition (TDI) (White, 2000). Reversible inhibition involves rapid association and dissociation of drugs and enzymes and may be competitive, noncompetitive, or uncompetitive. In general, TDI results from irreversible covalent binding or quasi-irreversible noncovalent tight binding of a chemically reactive intermediate to the enzyme that catalyzes its formation, resulting in loss of enzyme function. In some cases, TDI could result from reversible inhibition from a metabolite(s) generated in situ. The distinction between the terminology TDI and mechanism-based inactivation (MBI) must be appreciated. A TDI is defined as a compound that demonstrates an increase in the extent of inhibition it causes when it is incubated with the enzyme before addition of the substrate. As such, this is a kinetic definition only. MBI refers to a subset of TDI in which specific biochemical experiments are conducted that show that the enzyme acts upon the substrate to form a chemically reactive metabolite that subsequently inactivates the enzyme (Silverman, 1996). These two terms will be used throughout this document and are not interchangeable.
Metabolic drug-drug interactions resulting from TDI can display a delayed onset due to the time dependence in inhibition and can persist even after the inhibitor has been eliminated because enzymatic activity is only restored by de novo protein synthesis. Time-dependent inhibitors of various human P450 enzymes have been well documented from in vitro studies, and many have been shown to cause DDI (Table 1). Several comprehensive reviews on TDI have been published recently (Zhou et al., 2005, 2007; Venkatakrishnan and Obach, 2007; Johnson, 2008).
Drugs and other compounds known to cause time-dependent inhibition of drug-metabolizing human cytochrome P450 enzymes
Drug metabolism sciences have advanced significantly since the publication of the PhRMA position paper (Bjornsson et al., 2003), the 1997 FDA in vitro drug interactions guidance (Drug Metabolism/Drug Interaction Studies in the Drug Development Process: Studies In Vitro), and even since the release of the updated FDA draft drug interactions guidance in 2006, with reports on drug-drug interactions involving TDI steadily increasing (Ghanbari et al., 2006). Thus, the Drug Metabolism Technical Group of PhRMA assembled a working group of pharmaceutical industry researchers to address the topic of TDI, which builds upon the treatment of reversible inhibition offered in the earlier publication (Bjornsson et al., 2003). This group devised an anonymous survey to collect data on current practices on TDI within pharmaceutical industry research organizations and developed recommendations regarding the conduct of in vitro TDI studies. The outcome of these efforts is presented in this article.
The Science of Time-Dependent Inhibition and Predicting DDI
Bioorganic Chemistry of P450 Inactivation. The P450 catalytic cycle is illustrated in Fig. 1, and the reader is referred to authoritative accounts of the biochemistry of these enzymes (Guengerich, 2001; Ortiz de Montellano and De Voss, 2002). Several steps will be sensitive to inhibition or inactivation including substrate and oxygen binding or electron transfer. The final products of the catalytic cycle can be stable metabolites or reactive intermediates. The reactivity and nature of a formed metabolite will determine further interactions with P450. The biochemical mechanisms by which compounds function as mechanism-based P450 inactivators can be divided into three categories: quasi-irreversible or metabolite-intermediate complex (MIC) formation, heme alkylation, and protein alkylation. In some cases, inactivation can be caused by protein alkylation by heme fragments (He et al., 1998). Comprehensive reviews of the chemistry and biochemistry of these processes have previously been provided and are beyond the scope of the present discussion (Correia and Ortiz de Montellano, 2005; Fontana et al., 2005; Kalgutkar et al., 2007; Hollenberg et al., 2008).
In brief, MICs generally form from molecules possessing amine or methylenedioxyphenyl functional groups. Oxidative bioactivation of these functional groups can lead to intermediate species (e.g., nitroso, carbene, etc.) that form very tight complexes with the heme iron of the P450. The complex probably disrupts oxygen binding and subsequent catalysis, and it produces a characteristic absorption spectrum with a Soret maximum of 448 to 455 nm. Although chemically reversible, MICs are stable under physiological conditions. Several examples of compounds that inactivate P450s by this mechanism include the macrolide antibiotics (Franklin, 1991), calcium channel blockers such as verapamil and diltiazem (Ma et al., 2000), and the selective serotonin reuptake inhibitor paroxetine (Bertelsen et al., 2003).

Cytochrome P450 catalytic cycle.
Another P450 inactivation mechanism involves covalent attachment of a reactive species to either the prosthetic heme of the P450 or to protein itself. Although in some instances heme adducts are not stable, the inactivation is generally considered to be irreversible (Correia and Ortiz de Montellano, 2005). Perhaps one of the best characterized compounds that functions by covalently modifying the heme is the relatively nonselective P450 inactivator 1-aminobenzotriazole (Ortiz de Montellano and Mathews, 1981). Other functional groups that can be activated to MBI by P450 metabolism are listed in Table 2. The reader is referred to reviews of the chemistry of P450 inactivators (Fontana et al., 2005; Kalgutkar et al., 2007; Riley et al., 2007; Hollenberg et al., 2008).
Chemical substituents frequently associated with cytochrome P450 time-dependent inhibition
Enzyme Kinetic Aspects of P450 Inactivation. P450-catalyzed reactions are generally rapid equilibrium interactions between P450 and its substrate, followed by electron transfers (from oxidoreductase/NADPH and cytochrome b5/NADH) and oxygen incorporation (Guengerich, 2001), leading to oxidized product (metabolite) formation. As illustrated in Scheme 1, a P450 enzyme catalyzes the conversion of a mechanism-based inactivator to its reactive form EI* with the rate constant k2. The reactive metabolite can either be released as a stable product (k3) or it can react with the enzyme (k4). The interaction of a reactive metabolite with a P450 enzyme results in inactivation of the enzyme (E-X) through covalent protein binding, heme modification, or formation of MIC (Silverman, 1995; Correia and Ortiz de Montellano, 2005) as discussed above. The rate of E-X formation is related to k2, k3, and k4.
The rate of P450 enzyme inactivation is proportional to inactivator concentration and can be saturated at high concentration (eq. 1): 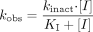 where the kobs is the pseudo first-order rate constant of inactivation at inactivator concentration [I], kinact is the maximum inactivation rate (a theoretical value that cannot be experimentally observed), and KI is the inactivator concentration when the rate of inactivation reaches half of kinact. Further details of the relationships among the micro rate constants and these parameters are beyond the scope of this discussion and can be found elsewhere (Silverman, 1996).
where the kobs is the pseudo first-order rate constant of inactivation at inactivator concentration [I], kinact is the maximum inactivation rate (a theoretical value that cannot be experimentally observed), and KI is the inactivator concentration when the rate of inactivation reaches half of kinact. Further details of the relationships among the micro rate constants and these parameters are beyond the scope of this discussion and can be found elsewhere (Silverman, 1996).
Experimental conditions to determine these kinetic parameters typically involve an “inactivation” incubation (termed in this article as the “preincubation”) for specific time periods in the presence/absence of a putative inactivator and NADPH followed by P450 activity measurement by dilution into a secondary incubation. The percentage of P450 enzyme activity loss due to inactivation can be calculated using eq. 2 (Obach et al., 2007): 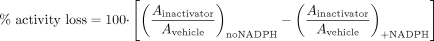 where the A represents measured enzyme activities in the presence and absence of inactivator and NAPDH. (Ainactivator/Avehicle)no NADPH represents the P450 enzyme activity remaining due to the inhibition by the diluted inactivator in the secondary incubation. (Ainactivator/Avehicle)+NADPH represents the enzyme activity remaining due to preincubation loss plus the inhibition by diluted inactivator in the secondary incubation. For multiple time point preincubations, P450 activities from 0 min preincubation with and without inactivator in the presence of NADPH serve as (Ainactivator/Avehicle)no NADPH. Therefore, percentage of enzyme activity loss due to TDI can be expressed as follows (eq. 3):
where the A represents measured enzyme activities in the presence and absence of inactivator and NAPDH. (Ainactivator/Avehicle)no NADPH represents the P450 enzyme activity remaining due to the inhibition by the diluted inactivator in the secondary incubation. (Ainactivator/Avehicle)+NADPH represents the enzyme activity remaining due to preincubation loss plus the inhibition by diluted inactivator in the secondary incubation. For multiple time point preincubations, P450 activities from 0 min preincubation with and without inactivator in the presence of NADPH serve as (Ainactivator/Avehicle)no NADPH. Therefore, percentage of enzyme activity loss due to TDI can be expressed as follows (eq. 3): 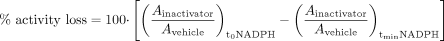
The parameters kinact and KI can be determined by a direct plot of kobs versus inactivator concentration as well as non-least squares fitting using any of a number of commercially available graphing software packages. Historically, a double reciprocal plot of 1/kobs versus 1/[I] was constructed to obtain the inactivation parameters (Kitz and Wilson, 1962). Equations 2 and 3 can be used in abbreviated experiments designed to indicate whether a new molecular entity (NME) is a TDI, in which the number of preincubation time points and number of concentrations of test compound are greatly reduced. Depictions of these two abbreviated experimental designs for detecting TDI are shown in greater detail in Figs. 2 and 3.


A graphical presentation of an abbreviated TDI experiment. In the left panel, the data are shown that would be generated if a complete set of time points were measured. The right panel shows the abbreviated TDI experiment with the same data represented with dashed lines. In this experiment, the test compound is examined at just one concentration (i.e., concentration “C” in this example) and is compared with vehicle control, with preincubations carried out for 30 min in the presence and absence of NADPH. The notations of activity, i.e., “A” are as described in eq. 2. In this case, had a time 0 time point been taken, a noticeable decline in activity of 6% would have been observed in the absence of inactivator and NADPH [i.e., (Avehicle)no NADPH]. In the example of this experimental set-up, the calculation of the percentage activity loss would be as follows:  Thus, even though the inactivator caused an 80% decline in activity, the actual loss attributable to the inactivator at a concentration of C is 60%.
Thus, even though the inactivator caused an 80% decline in activity, the actual loss attributable to the inactivator at a concentration of C is 60%.
Current Status of the Science of Predicting DDI from P450 Inactivation Data. Within the framework of pharmaceutical industrial practices on the assessment of mechanism-based P450 inactivation, a discussion of the utility of generated in vitro parameters for the extrapolation of DDI outcomes must be included. This treatment is not intended to provide a comprehensive review of the literature in this area, but rather to introduce and explain the mathematical models presently in use, the input parameters (in vitro and in vivo) required, and to convey the uncertainty associated with these inputs. A more comprehensive review of in vitro-in vivo extrapolation (IVIVE) with respect to mechanism-based inactivation has been provided by Venkatakrishnan et al. (2007).
Mathematical models for IVIVE of drug interaction magnitude based on reversible P450 inhibition were known for some time to underpredict interactions when the compound producing the DDI was a MBI. A report by Hall and colleagues (Mayhew et al., 2000) described the first viable model that incorporated the kinetic aspects of enzyme inactivation into extrapolation of DDI. The scaling model essentially stated that because intrinsic clearance (CLint) is inversely proportional to exposure [area under the curve (AUC)] and directly proportional to enzyme amount, the fractional change in AUC of a victim drug (substrate) in the presence of an inactivator can be described by the following (eq. 4): 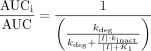 where kdeg is the natural in vivo enzyme degradation rate constant. The model makes excellent conceptual sense, in that the fraction of enzyme remaining at a steady-state (fixed) concentration of inactivator will depend on its natural degradation rate and what amounts to an augmentation of this rate by the rate of inactivation. Several important assumptions must be noted. First, the model assumed that a dose of the affected drug was completely absorbed from the gastrointestinal tract, and that its clearance was hepatic and dependent only on the affected P450. In addition, the model assumed that the inactivator does not alter kdeg or the natural enzyme resynthesis rate (ksyn). Using the model, the authors extrapolated the anticipated drug interaction between CYP3A substrates and fluoxetine, clarithromycin, and N-desmethyldiltiazem and claimed good qualitative agreement with the observed clinical interactions. Predicted values were 1.4, 2.6, and 4.7, respectively, with clinically observed values being 1.26 (Greenblatt et al., 1992), 3.5 to 9.6 (Yeates et al., 1996; Gorski et al., 1998), and 4.8 (Mousa et al., 2000), respectively. The underprediction of the clarithromycin/midazolam interaction can probably be ascribed to the contribution of intestinal metabolism in limiting the bioavailability of midazolam.
where kdeg is the natural in vivo enzyme degradation rate constant. The model makes excellent conceptual sense, in that the fraction of enzyme remaining at a steady-state (fixed) concentration of inactivator will depend on its natural degradation rate and what amounts to an augmentation of this rate by the rate of inactivation. Several important assumptions must be noted. First, the model assumed that a dose of the affected drug was completely absorbed from the gastrointestinal tract, and that its clearance was hepatic and dependent only on the affected P450. In addition, the model assumed that the inactivator does not alter kdeg or the natural enzyme resynthesis rate (ksyn). Using the model, the authors extrapolated the anticipated drug interaction between CYP3A substrates and fluoxetine, clarithromycin, and N-desmethyldiltiazem and claimed good qualitative agreement with the observed clinical interactions. Predicted values were 1.4, 2.6, and 4.7, respectively, with clinically observed values being 1.26 (Greenblatt et al., 1992), 3.5 to 9.6 (Yeates et al., 1996; Gorski et al., 1998), and 4.8 (Mousa et al., 2000), respectively. The underprediction of the clarithromycin/midazolam interaction can probably be ascribed to the contribution of intestinal metabolism in limiting the bioavailability of midazolam.
The model was further refined to include the contribution of the affected P450 to clearance (fm,CYP or fraction metabolized), in the event that the substrate has multiple clearance mechanisms (eq. 5). 

A graphical presentation of an abbreviated TDI experiment. In the left panel, the data are shown that would be generated if a complete experiment was done to determine KI and kinact. The right panel shows the abbreviated TDI experiment with the same data represented with dashed lines. In this experiment, the test compound is examined at just one concentration (i.e., concentration C) and is compared with vehicle control, with preincubations in the presence of NADPH carried out for 30 min. The notations of activity, i.e., A, are as described in eq. 3. In this case, there is a detectable decline in activity even without the inactivator such that 21% of the activity is lost [i.e., (Avehicle)t30,NADPH]. In addition, even without preincubation, the inactivator causes a decline in activity of 13% [i.e., (Ainactivator)t0,NADPH]. These factors need to be accounted for when calculating the percentage decline that occurs:  Thus, even though the inactivator caused an 80% decline in activity, the actual loss attributable to the inactivator at a concentration of C is 62%.
Thus, even though the inactivator caused an 80% decline in activity, the actual loss attributable to the inactivator at a concentration of C is 62%.
In addition, potential contributions of gut metabolism to the drug interaction for CYP3A substrates were considered to yield (Wang et al., 2004; Ernest et al., 2005; Obach et al., 2007) (eq. 6): 
The terms are as described previously: Fg is the fraction of victim drug (substrate) that escapes gut metabolism in the absence of the inactivator; kdeg(CYP3A,gut) is the natural degradation rate constant for CYP3A in the enterocyte; and [I]g is the concentration of inactivator in the gut following oral administration.
For the models discussed, a degree of uncertainty is associated with each input parameter. The inactivation constants KI and kinact are sensitive to the in vitro study design, and there is considerable variability in the experimental designs used (Yang et al., 2005; Ghanbari et al., 2006; Van et al., 2006). The concentration of inactivator at the enzyme active site [I] is not a parameter that can be readily obtained clinically. Several estimates for [I] including total Cmax, unbound Cmax, and estimates of the hepatic portal inlet concentration (unbound and total) have been used. The relationship from which the hepatic portal inlet concentration can be estimated using eq. 7 (Ito et al., 1998; Kanamitsu et al., 2000): 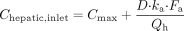 in which D is the oral dose of the inactivator, ka is its absorption rate constant, Fa is the product of the fractions absorbed and escaping gut metabolism, and Qh is the liver blood flow. When corrected for plasma protein binding, this parameter provided reliable predictions for reversible inhibitors of the P450 enzymes (Obach et al., 2006; Einolf, 2007).
in which D is the oral dose of the inactivator, ka is its absorption rate constant, Fa is the product of the fractions absorbed and escaping gut metabolism, and Qh is the liver blood flow. When corrected for plasma protein binding, this parameter provided reliable predictions for reversible inhibitors of the P450 enzymes (Obach et al., 2006; Einolf, 2007).
However, it was found to overpredict interactions for mechanism-based P450 inactivators, and the free systemic Cmax proved more accurate (Obach et al., 2007). The natural degradation rate constant for the affected P450 (kdeg) is another important parameter for IVIVE that is not presently ethical to directly measure in humans in vivo. A summary of the approaches to estimating kdeg has recently been provided (Yang et al., 2008), with a guidance to avoid “selective use” of one specific value over another a priori. Both fm,CYP, the fraction of systemic clearance associated with a given pathway, and Fg, the fraction escaping gut metabolism (primarily 3A substrates), have high impact on a predicted DDI, based on the models above. Both of these parameters can also be difficult to obtain in vivo. For routinely used probe drugs such as midazolam for CYP3A4, estimates of both can be found in the literature (Obach et al., 2006). However, for new compounds in development, estimates of fm,CYP are typically made from a combination of in vitro reaction phenotyping studies and in vivo information from radiolabeled human absorption, distribution, metabolism, and excretion (ADME) studies (Venkatakrishnan and Obach, 2007). A review of techniques for the estimation of Fg has recently been provided (Yang et al., 2007; Galetin et al., 2008). In brief, in vivo estimates of Fg are rarely available during drug development because they minimally require comparison of intravenous versus oral pharmacokinetic (PK) data. In fact, this technique provides the product of the absorbed fraction (Fa) and Fg rather than Fg alone. Other techniques such as the conduct of pharmacokinetic studies in anhepatic patients (Kolars et al., 1991) and administration of grapefruit juice, assuming selective inhibition of intestinal CYP3A4, have also been used (Ameer and Weintraub, 1997). An in vitro model to approximate Fg has been described previously (Yang et al., 2007) (eq. 8): 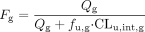 where Qg is the gut (villous) blood flow, fu,g is the unbound drug fraction in the enterocyte, and CLu,int,g is the intestinal free intrinsic clearance.
where Qg is the gut (villous) blood flow, fu,g is the unbound drug fraction in the enterocyte, and CLu,int,g is the intestinal free intrinsic clearance.
The mathematical models described previously are all essentially static models. Although they consider the kinetic aspects of mechanism-based inactivation, the predictions they provide are steady state with regard to both inactivator concentration and enzyme amount (after inactivation). Approaches using physiologically based pharmacokinetic (PBPK) modeling techniques permit the simultaneous dynamic simulation of changes to substrate and inactivator concentration and also enzyme amount, with time. A model described by Yang et al. (2006) examined the potential for inactivation of CYP2D6 by 3,4-methylenedioxymethamphetamine (MDMA) and the potential pharmacokinetic consequences (auto-inactivation). The model demonstrated that CYP2D6 was essentially completely inactivated within 1 h of an MDMA dose, and, therefore, the subsequent administration of additional CYP2D6 inhibitors would not further affect pharmacokinetics.
PBPK models present several theoretical advantages over static approaches due to their ability to consider changes in concentrations of enzyme, substrate, and inactivator with time. For example, a comprehensive comparison of the above-described approaches was recently conducted (Einolf, 2007), in this case also using a commercially available population-based ADME simulator (version 6.4; Simcyp Ltd., Sheffield, UK) (Jamei et al., 2009). The author noted that for mechanism-based inactivators, the mechanistic static models tended to be more predictive than dynamic models used in the simulation software. However, it was also noted that the predictions were improved after the substitution of the kdeg values in the computational dynamic model with the kdeg values used for the static models.
The decision to use any model to predict DDI magnitude will depend on the availability of reliable in vitro data, the development phase of the putative inactivator or substrate, and the need for the prediction in the context of clinical study planning. The static models described above may be more pragmatic than sophisticated PBPK-based approaches, and they have been demonstrated to be of value in early (and perhaps late) drug development for both risk assessment and clinical study planning. With the availability of software tools such as Simcyp, PBPK-based approaches will undoubtedly become of increasing utility and importance.
Results of the PhRMA Survey
General Comments on the Survey. The survey on PhRMA member industry practices regarding time-dependent inhibition of drug-metabolizing enzymes was comprised of 87 questions focused on technical aspects of in vitro experimental study design, the use of the in vitro data generated in making predictions of clinical DDI, and strategic aspects regarding how the data are used in making decisions on progression of NMEs through discovery and development phases. The on-line survey was distributed to 32 research and development organizations of PhRMA member companies, and 17 companies submitted responses (53% response rate) with two thirds coming from companies employing >10,000 staff members (mostly companies with multiple research sites). The survey response period was from March to April 2008, with administration of the survey and tabulation of the data conducted by staff members at PhRMA to maintain anonymity of the respondents. A brief list of points for which the survey revealed concordant and disparate responses is in Table 3.
Similarities and differences in the practice and use of time-dependent inhibition studies in drug discovery and development
Strategic Aspects and Use of TDI Data. The frequency in which companies encounter in vitro DDI due to time-dependent inhibition during the drug discovery-development continuum causes it to be thought of as a relatively common issue across various chemical classes (>5% of compounds tested) for the majority (60%) of the respondents. A smaller percentage of companies felt that it was an uncommon or rare issue (29% and 12% of the respondents, respectively). Structural alerts [i.e., substituents in the NME frequently associated with mechanism-based inactivation of P450 enzymes or bioactivation to reactive electrophiles (Kalgutkar et al., 2007; Riley et al., 2007)] are also considered important, especially if the NME has a similar structure to analogs with known TDI issues (e.g., macrolide antibiotics). The use of structural alerts is particularly prevalent in the drug discovery phase because respondents reported that many of the potentially offending substituents have been replaced in final lead candidate molecules before entering the development phase. Thus, given the propensity of certain chemical features to be mechanistically associated with TDI, slightly greater than half of the surveyed organizations deliberately avoid these features (e.g., alkynes, 3-substituted indoles, methylenedioxy-phenyls, etc.; Table 2) during lead generation. Although not all organizations avoid chemical alerts for TDI, all survey participants (except one) incorporate assays during drug discovery that enable the early detection of TDI. Approximately one third of respondents screen for TDI before lead optimization, and the remainder evaluate TDI during lead optimization. The general strategy is to use an abbreviated experimental format to identify a TDI liability followed by a more rigorous experimental paradigm (later in discovery or early in development) to characterize the magnitude of inhibition. Data from discovery assays seem to be used primarily to flag compounds as high risk, limiting the progression of the compound into development and/or aiding in the design of molecules devoid of the TDI liability. Most respondents (14 of 17) stated that a “cut-off” value is used to flag high-risk compounds in the discovery/research phase. The cut-off most frequently used is based on the loss of enzyme activity relative to a positive control (50% of respondents), predicted clinical AUC change (40% of respondents), and comparison of kinact (20% of respondents) or the ratio of kinact to KI relative to a positive control (20% of respondents) (note some respondents selected multiple measures). During early drug discovery, data from these abbreviated assays are also used to aid lead optimization [structure activity relationships (SAR)] by the majority of respondents (>80%). The general sentiment is that SAR development is possible (75%) with respect to TDI. The remainder of respondents have not looked into SAR or chemical structural constraints for SAR, or felt that SAR around TDI was very challenging.
Survey respondents unanimously indicated that they routinely evaluate compounds for TDI of some human P450 isoforms in the drug development phase. (The specific enzymes most commonly studied are discussed below.) An overwhelming majority of participants stated that during drug development, in vitro TDI parameters are used for the prediction of in vivo DDI and the planning of clinical DDI studies. This indicates that the intent behind gathering these data during the drug development phase is for use in developing a clinical DDI study strategy (e.g., timing of these studies relative to Phases 1–4; which probe substrates to test as objects of DDI; information in product labels, etc.). It is also part of an overall risk assessment made for NMEs during early development.
Responses were varied regarding timing of predictions of clinical DDI and ranged from lead optimization to Phase 1. Several in vitro parameters are considered important at the initiation of a TDI investigation and include knowledge of metabolic stability of a compound in the test system, reversible P450 inhibition, and the P450 isoforms that metabolize the compound.
There was no consensus in the models used to predict the clinical DDI impact of TDI. Respondents used one or more of the following methods to predict clinical DDI potential:
-
simple model to predict change in exposure (AUCi/AUC), assuming that both Fg and fm,CYP = 1 (eq. 4) (Mayhew et al., 2000);
-
models that incorporate experimentally estimated Fg and fm,CYP values (eq. 6) (Obach et al., 2007);
-
models that incorporate the PK parameters of the object and precipitant drugs, such as Simcyp or PK models developed within research organizations.
The majority of survey respondents indicated that the most important input parameter for accurate predictions is the assessment of in vivo inactivator concentration. However, to develop a prediction, a variety of inactivator concentrations are used. Before the acquisition of clinical exposure data, predicted values of inactivator concentration are based on in vitro models, animal pharmacology, or extrapolated human exposures. Various input values for [I] are used by the survey respondents. In most cases, early predictions are refined when the knowledge of the therapeutic concentration of the inactivator becomes available.
Roughly half of the survey respondents have prospectively evaluated the models for prediction accuracy using either literature data alone or a combination of literature and internal data sets. Approximately half of the respondents claimed that the accuracy of the methods used to quantitatively predict the magnitude of DDI from in vitro TDI data are not adequate, with a majority claiming that the magnitudes of DDI are overestimated. Sixty percent of participants responded that there are not yet enough data to provide confidence in the quantitative predictive modeling of TDI outcomes. However, most (90%) claimed that the models can generally categorize clinical TDI potential as weak (<2-fold increase in AUC), moderate (2–5-fold increase in AUC), or strong (>5-fold increase in AUC). In addition to an assessment of in vivo inactivator concentration, the choice of the appropriate P450 degradation rate constant (kdeg) was recognized as very important to the generation of accurate DDI predictions. However, a consensus on the in vivo kdeg values for the P450 isoforms has not been reached. Values that respondents use for P450 half-life values ranged from 11.6 to 140 h, differing slightly from those described in Table 4.
Summary of estimates of turnover half-lives for cytochrome P450 enzymes in humans ( Yang et al., 2008 )
Survey respondents predict the magnitude of a potential DDI from in vitro TDI data to decide on whether an NME can be readily developed, to assist in formulating patient exclusion criteria for early clinical trials, and to determine the necessity of clinical DDI studies. A large majority of survey participants indicated that in vitro TDI data are used to predict the magnitude of clinical DDIs and also influence both the timing and study design of in vivo DDI trials. Positive in vitro TDI data are not used alone to discontinue the progression of an NME. The decision of whether to terminate a compound due to a TDI liability depends on several factors including therapeutic area, therapeutic concentration range, market competitiveness, and probable concomitant medications in the patient population. These criteria are consistent with the model proposed for assessing risk of lead compounds based on bioactivation potential (Evans et al., 2004). Seventy percent of respondents indicated that knowing the mechanism of inactivation affects decision-making. In some cases, this information can help with the selection of alternative potential drug candidates. For example, development of an NME that causes quasi-irreversible MIC formation is viewed by some as a lower risk than an MBI that causes covalent binding.
Most respondents indicated that their research organizations have progressed an NME with TDI properties into clinical development within the last 5 years. There was no consensus regarding cut-off values of TDI parameters that would instigate a clinical DDI study, and reported criteria included predicted -fold change in AUC of a concomitantly administered drug, percent loss of P450 activity observed in vitro, and in vitro TDI parameters. If the test compound inactivates multiple P450 enzymes, a variety of approaches are used to minimize the number of clinical DDI studies. These include “cocktail” studies (i.e., administration of multiple probe substrates), rank-ordering based on either potency (e.g., kinact/KI) or predicted AUC changes, and prioritizing the most clinically important enzyme(s). Of the respondents who advanced NMEs with TDI potential to clinical trials, half observed clinical DDI findings that corresponded to the in vitro data, whereas 42% reported that clinical DDI data were not available at the time of the survey. Although predicted DDI magnitude is frequently used to decide whether to conduct clinical DDI studies, 38% of respondents have received requests from government regulatory agencies to conduct clinical drug-drug interaction studies even when in vitro P450 TDI and competitive inhibition are weak or negative.
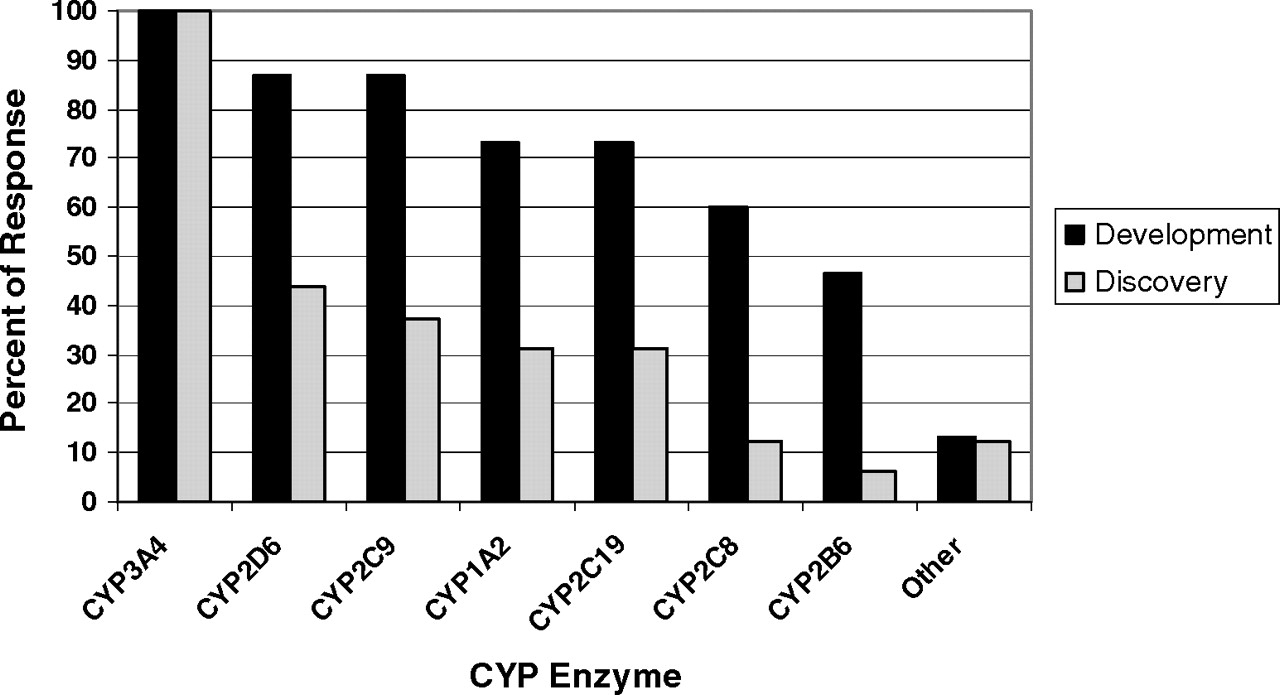
Percentage of respondents who assess new chemical entities for their ability to cause time-dependent inhibition for specific human cytochrome P450 enzymes in drug discovery and development phases.
Mechanistic studies are sometimes pursued when initial findings demonstrate TDI, but most respondents indicated that these studies are seldom performed. These mechanistic studies include measurement of MIC via spectrophotometric methods, determination of partition ratios, reversibility assessments, and detection of P450 adducts using liquid chromatography/mass spectrometry or radiometric detection. When conducted, results from these mechanistic studies can assist in determining the risk due to reactive intermediate formation.
Only 12% of respondents reported performing in vivo DDI studies in animal models. For compounds that display reversible inhibition or induction in addition to TDI, 58% of respondents felt that it is somewhat or very important to use a cell-based assay or an in vivo preclinical DDI study to identify worst-case scenarios.
Experimental Aspects: Drug Development. Overall, there was considerable concordance of responses regarding the technical aspects of conducting TDI studies in the drug development phase. Among the P450 enzymes examined, all respondents replied that compounds are tested as CYP3A inactivators, and the vast majority (>70%) also tested CYP2D6, 2C9, 1A2, and 2C19 (Fig. 4). Approximately half also test CYP2C8 and 2B6, and only two respondents stated that they test other P450 enzymes. All respondents use pooled human liver microsomes (HLMs) as the source of enzyme activity, and a few respondents stated that they also use recombinant enzymes and hepatocytes to make these measurements. When hepatocytes were used to make conclusions regarding TDI, it seems that this may be in conjunction with induction assays, in which inactivation can also be detected. In hepatocyte induction assays both mRNA and enzyme activity endpoints can be measured. If there is discordance between these two outputs such that mRNA is increased or unaffected, while enzyme activity decreases, TDI is possible. This result is especially true if the activity measurement is made after the test compound has been removed from the system. All replied that substrates used for enzyme activity measurement were selective P450 probe substrates.
Survey respondents stated that TDI assays are done in a format wherein the test compound is incubated with enzyme and NADPH (preincubation) followed by dilution into a second mixture containing the P450 selective probe substrate. The dilution factor reported to be used was 6× or greater for all but one respondent. The number of preincubation time points ranged considerably among the survey respondents, with a minimum of four points, but all use six or more test compound concentrations when determining inactivation parameters. Concentrations are selected from preliminary range-finding experiments and reversible inhibition data, with considerations for solubility limitations and anticipated/known relevant clinical concentrations. All run control incubations containing NADPH and no test compound in the preincubation, and a majority also run a control lacking NADPH, but only some normalize the data to control values at each time point. Most respondents do not routinely measure the concentration of their test inactivator throughout the preincubation period. For those who do this, some adjust the incubation time and repeat the experiment while others will report the data as is or with the caveat that inactivation parameters may be underestimated. In the secondary assay to measure P450 activity, probe substrate concentrations used are high, with 80% of respondents using at least five times the KM. In more definitive experiments conducted in drug development, two or three replicate incubations are used (either within-day and some between-day). The log-linear phase of decline is used in data analysis to determine kobs values. Most use nonlinear regression to determine KI and kinact values, although some used simple linearized plots (Kitz-Wilson) or global nonlinear regression of nonlog transformed data. Positive control inactivators at single concentrations are routinely run, and in many cases investigators strive for the positive control data to be within 20% of historical response. Frequently used positive control inactivators include troleandomycin and erythromycin (CYP3A), furafylline (CYP1A2), paroxetine (CYP2D6), and tienilic acid (CYP2C9).
To demonstrate that a compound is not an inactivator, abbreviated experimental designs are used, even in drug development. These approaches use either single test compound concentrations and multiple incubation time points or multiple test compound concentrations at a single incubation time (the “IC50 shift” approach) and are described below. Other abbreviated approaches to determining inactivation are generally not used in the drug development phase; when used, the number of test compound concentrations and time points range from 2 to 5.
Experimental Aspects for Evaluating TDI in Drug Discovery. Sixteen of 17 respondents of this survey screen for TDI during the drug discovery phase. For those that did screen before candidate selection for development, all respondents tested compounds for CYP3A inactivation. Less than half of the companies examined P450 enzymes other than CYP3A4 in the discovery phase. The rank order of the most common P450 enzymes examined after CYP3A were CYP2D6 > CYP2C9 > CYP1A2 = CYP2C19 (Fig. 4). All but one respondent used pooled HLMs to evaluate TDI in drug discovery. Several respondents (6 of 17) also evaluated TDI using recombinant enzymes, and 2 of 17 respondents indicated that they have used human hepatocytes or other cellular models. The majority of respondents used selective P450 probe substrate metabolism with liquid chromatography/tandem mass spectrometry analysis or radiochemical detection to measure the remaining P450 activity after TDI assays in the discovery stage, with only one using fluorogenic substrates.
Several companies use an IC50 shift assay as their first TDI assessment (47%), and a lower percentage of companies are measuring a percentage decline or inactivation rate at a single inhibitor concentration (35%). In an IC50 shift experiment, the IC50 is determined for a P450 marker activity before and after the test compound has been incubated with enzyme and NADPH for a set preincubation time (Bertelsen et al., 2003; Obach et al., 2007). Approximately half of respondents perform IC50 shift experiments by conducting the preincubation at a higher concentration and then diluting into the activity assessment incubation (like the inactivation experiments described above), whereas the other half conduct the activity incubation by adding the probe substrate with no dilution step. The majority of respondents stated that the choice of inhibitor concentrations used in the IC50 shift experiment was dependent upon the reversible P450 inhibition IC50 and solubility limits. Interpretation regarding the magnitude of shift observed is not necessarily clear-cut. Some respondents stated that a clear criterion is not established, whereas most cited shift values of 1.2- to 3-fold as indicative of a positive finding, with one respondent citing a 10-fold shift as being clearly positive. A smaller percentage (18%) perform an abbreviated KI-kinact study (defined as abbreviated due to minimal number of inhibitor concentrations and time points). Definitive KI-kinact studies are not performed as a first assessment of TDI by any respondent company. For a couple of companies with different research sites, it was noted that different methods were used within the same company for early screening (e.g., IC50 shifts and percentage decline in activity).
Unresolved Scientific Issues. Virtually all participants in the survey agree that the purpose of preclinical in vitro TDI studies is to use the data to predict in vivo drug-drug interactions and to propose a clinical DDI study strategy. However, as mentioned above, many respondents expressed the opinion that there are not enough clinical data to have confidence in predictive modeling of TDI outcomes and maintain that the various methods used to predict the magnitude of DDI from in vitro TDI data are not accurate enough and often overestimate the DDI outcomes.
Several factors may contribute to the lack of quantitative prediction from in vitro TDI data and as such represent issues in need of resolution with further investigation. These include the following: 1) models used to predict the clinical DDI impact of TDI may not be satisfactory, partly due to the limited knowledge and uncertainty of parameters used for the prediction (described in more detail below); 2) P450 enzymes, such as CYP3A4, demonstrate complicated biochemical behaviors that are not fully understood at a molecular level; and 3) in vivo drug disposition involves numerous complex physiological processes. Furthermore, P450 enzymes, although important, may not be the only biochemical entities determining the outcome of clinical DDI because other factors such as active tissue uptake mediated by drug transport proteins could affect the magnitude of DDI. Deriving a prediction of an in vivo outcome from a simple in vitro experiment requires knowledge of numerous in vivo processes, many of which may be unknown for a given drug. These complicating factors are not restricted to compounds that are TDI because they apply to reversible inhibitors as well.
Although all of these factors could contribute to the difficulty of quantitative DDI prediction, it is apparent that accurate information and knowledge to make the prediction better are not always possessed. The general equations used to calculate the potential for a TDI to cause an increase in AUC as a result of inactivation of hepatic P450 (cited above) contain five important terms: [I]in vivo, KI, kinact, kdeg, and fm,CYP. For CYP3A4, one may also add a term for the effect on CYP3A in the intestine. For each of these five parameters, the question of what values are the most appropriate can be posed, because the use of different reported values will give different predicted AUC increases.
[I]in vivo. The majority of survey respondents indicated that an important parameter for accurate predictions is the assessment of in vivo inactivator concentration, [I]in vivo. However, as important as it is, there is no consensus as to what [I]in vivo value should be used for the prediction of clinical DDI. Although unbound maximum plasma concentration (Cmax,u) is the most commonly used value, other parameters, such as Cmax,total, unbound hepatic inlet Cmax, average total or unbound plasma concentration, and enzyme site concentration from PK model predictions, have also been used. However, all values obtained from systemic concentrations are only surrogates for those at the enzyme active site. In addition, the best [I]in vivo value can be dependent on the compound (e.g., potentially compensating for active hepatic uptake). Thus, there are considerable uncertainties on the [I]in vivo values used for DDI prediction.
KI andkinact. In well designed experiments, KI and kinact can be accurately determined. However, it is well known that these values can vary depending on the enzyme source (HLM, human hepatocytes, and recombinant human P450). The variability can be due to differences in the availability of the inhibitor or substrate to the P450 in each system (nonspecific microsomal or hepatocyte binding, excessive metabolism, etc.). In addition, the well accepted biochemical method in which inactivation parameters are generated (i.e., preincubation with inactivator and enzyme followed by dilution into substrate) may not reflect the in vivo situation in which inactivator and substrate are simultaneously present (i.e., the presence of the substrate could slow the rate of inactivation by competitive inhibition).
kdeg. The choice of the appropriate in vivo P450 degradation rate constant (kdeg), derived from P450 half-life values, is very important to the generation of accurate DDI predictions. However, the survey revealed no consensus on the in vivo kdeg values for human P450 isoforms. Both in vitro and in vivo systems, using various approaches, have been used to estimate the turnover half-lives of human hepatic P450 enzymes. Reported half-lives for all human P450 enzymes range from 23 to 140 h (Table 4). In some reports, large variability in half-life can be found in the same experiment for a single P450. For example, individual CYP3A4 half-life estimates range from 2 to 158 h in one experiment and 85 to 806 h in another experiment (Yang et al., 2008). The influence of such a large difference in enzyme half-life on the recovery of enzyme levels from de novo biosynthesis can be enormous.
fm,CYP. The fraction of metabolism contributed by a specific P450 to the total clearance (fm,CYP) is another important parameter used for the prediction of clinical DDI. The calculated AUC increase can be very sensitive to fm,CYP, particularly when the value approaches unity. There are few reports in the literature describing experimental determinations of fm,CYP, despite the importance of this parameter in DDI prediction. These can sometimes be derived from genetic polymorphism studies, or they can be estimated by combining data from human ADME studies in which metabolic pathways are determined and data from in vitro reaction phenotyping experiments. Sometimes, an fm,CYP of 1 is assumed as a worst-case scenario, but this assumption can contribute to overestimations of predicted magnitude of clinical DDI.
It is very clear from the current knowledge on P450 biochemistry, physiological aspects of drug metabolism and distribution, and the models available for DDI prediction, that uncertainties exist in several critical areas. More research is needed to improve the models. Despite these limitations, the majority of survey respondents felt comfortable that the current models can generally categorize clinical DDI potential as weak, moderate, or strong from in vitro TDI data.
Experimental Approaches for TDI Studies: Recommendations
The detection of mechanism-based inactivators needs to satisfy a set of criteria that have evolved for characterization of this type of inhibition (Silverman, 1995). This, together with the difficulty in quantitatively predicting the mechanism-based inactivation potential of NME from chemical structure, is consistent with the U.S. FDA recommendation of minimally examining NME for time-dependent inhibition after a 30-min preincubation before assessment of enzyme activity (FDA Draft Guidance for Industry on Drug Interaction Studies, 2006). Hence, TDI experiments have evolved as a surrogate method for detection of mechanism-based inactivators. The published literature on mechanism-based inactivation studies shows significant variation in experimental design, which can result in considerable differences in the estimated inactivation kinetic parameters (Ghanbari et al., 2006). This limited guidance together with the numerous published methods for detecting mechanism-based inactivation of P450 enzymes prompted the PhRMA Drug Metabolism Technical Group to sponsor development of consensus recommendations on the best practices for in vitro assessment of mechanism-based inactivation of P450 enzymes. In addition, a result from the survey indicated that approximately 80% of respondents would welcome a consensus set of TDI assays that are acceptable to the FDA. Therefore, a standardized approach would help to minimize interlaboratory variability for detection and assessment of potency of mechanism-based inactivators.
Examination of published mechanism-based inactivators of P450 enzymes identified functional groups on compounds responsible for mechanism-based inactivation via MIC formation and covalent binding to heme and/or apoprotein (Franklin, 1991; Murray, 1997; Fontana et al., 2005; Kalgutkar et al., 2007; Riley et al., 2007; Hollenberg et al., 2008). However, not all compounds with such functional groups are mechanism-based inactivators, but nonetheless such a structural alert knowledge base can be useful for triggering screening of compounds for mechanism-based inactivation of P450 enzymes. Time-dependent inhibition of P450 enzymes is often used in the pharmaceutical industry to distinguish between reversible inhibition and mechanism-based inactivation and to screen out compounds with this type of P450 enzyme inactivation property. Hence, the methods for detecting time-dependent inhibition of P450 enzymes, if not properly designed, can result in false positive results due to reversible inhibition from a metabolite(s) generated in situ and false negative results from inadequate resolution of the method to separate potent reversible inhibitors from potent time-dependent inhibitors. The recommended methods to identify a time-dependent inhibitor include a standard dilution method and an IC50 shift method. Once the NME tests positive for TDI of P450 enzymes, further experiments would be required before it can be concluded to be a mechanism-based inactivator. These follow-up mechanistic experiments include determination of the partition ratio, demonstration of irreversibility of inhibition, protection by substrate or competitive inhibitor, and demonstration of inactivation of the catalytic site before release of reactive intermediates. However, these mechanistic follow-up experiments are not needed for making predictions of DDI and are therefore not required. The experimental approaches and recommendations for all of these methods will be discussed below.
Standard Dilution Method and Determination ofKI andkinact. The most commonly used method to evaluate the time-dependent inhibition potential is the dilution method as described elsewhere (Waley, 1985; Silverman, 1995, 1996; Guengerich, 1999; Tucker et al., 2001). Although HLMs, hepatocytes, purified P450 enzymes, and cDNA-expressed recombinant P450 enzymes have been used for time-dependent inhibition studies in different laboratories, pooled human liver microsomes seem to be the preferred source of enzyme activity for time-dependent inhibition studies. In brief, human liver microsomes (1–2 mg/ml or lower, if possible) are preincubated at 37°C with various concentrations of NME in buffer containing NADPH. (In some cases, addition of EDTA can reduce the baseline level of activity decline that can occur when enzymes are preincubated in the presence of NADPH and in the absence of NME.) At various time points, aliquots of the preincubation mixtures are diluted (≥10-fold) with fresh assay buffer containing NADPH and probe substrate at a saturating concentration (≥4-fold KM) to monitor the remaining enzyme activities. The 10-fold or greater dilutions and the use of a saturating probe substrate concentration minimize the contribution of reversible inhibition from test compound and/or its metabolite to residual enzyme activities.
A simplified two time-point screening approach is frequently used by pharmaceutical companies to support drug discovery efforts. First, the percentage of activity remaining in the presence of one or two concentrations of the test compound (e.g., 10 and 50 μM) before or after 30-min preincubation is determined at a saturating probe substrate concentration after dilution. The enzyme activity of a sample containing vehicle without preincubation can serve as control activity. Because this is an initial experiment to assess whether TDI could possibly occur, the high concentration is recommended to avoid false negatives. To assess TDI potential of a test compound, the percentage of activity loss is calculated using eq. 9: 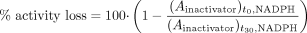 It should be noted that this equation represents a degenerate version of eq. 3, in which the level of decline in activity of the vehicle control is assumed to be negligible. If there is a considerable decrease in activity in the vehicle control, then correction for this factor is advocated.
It should be noted that this equation represents a degenerate version of eq. 3, in which the level of decline in activity of the vehicle control is assumed to be negligible. If there is a considerable decrease in activity in the vehicle control, then correction for this factor is advocated.
Alternately, preincubation of test compounds with and without NADPH for 30 min followed by assessment of residual enzyme activity after dilution into an incubation mixture containing standard probe substrate at a saturating concentration is another simplified method frequently used to evaluate time-dependent inhibition. Recent improved methods can reliably detect time-dependent inhibition of P450 enzymes (Atkinson et al., 2005; Lim et al., 2005; Obach et al., 2007). By increasing the single concentration for testing from 10 to 50 μM (Riley et al., 2007), the evaluation of TDI at a concentration corresponding to 10 × IC25 (Obach et al., 2007) or titration with multiple concentrations (Lim et al., 2005) improved detection of time-dependent inhibitors including weak time-dependent inhibitors. However, using a single concentration (10 or 50 μM) has the advantage of higher throughput and is ideally suited for evaluation of time-dependent inhibition during the drug discovery stage (Wang et al., 2002).
Ultimately, the use of kinetic parameters of inactivation, kinact and KI, is recommended for predicting the human drug interaction potential of time-dependent inhibitors because it is well established (Mayhew et al., 2000; Wang et al., 2004; Obach et al., 2007). Further determination of the inactivation kinetic parameters is recommended when an NME causes more than 20 to 25% loss of activity after 30-min preincubation in a simplified experiment because it has been shown that inactivators causing less than this degree of change are not associated with DDI. At least five preincubation time points should be used to establish pseudo first-order rate constants for inactivation (kobs), which are calculated from the negative slopes of the lines using linear regression analysis of the natural logarithm of the remaining activity as a function of time. (Note that if a biphasic decline in activity is observed, then only the data points representing the initial rate should be used.) To determine kinact and KI values, at least five inhibitor concentrations (plus vehicle control) are recommended. The kinact and KI values can be derived by nonlinear regression analysis using eq. 1. Good enzyme kinetic practice dictates that the kinetic parameters should only be reported if the inhibitor concentrations adequately encompass KI. Ideally, the selected concentration range for testing permits the time-dependent inhibition process to reach saturation. These two parameters can also be generated by the double reciprocal Kitz-Wilson plot (1/kobs versus 1/[I]), where kinact is estimated from the reciprocal of the y-intercept and KI from the negative reciprocal of the x-intercept; however, it should be noted that modern methods of nonlinear regression provide better estimates of kinetic parameters than those generated after linearization of the data. In some cases, limitations on the aqueous solubility of inhibitor will limit the upper concentrations that can be tested, and in these cases, the use of the linearized Kitz-Wilson plot will be necessary to estimate the kinetic parameters.
The following are recommended general aspects of experimental approaches:
-
The preferred enzyme source is pooled human liver microsomes at 1 to 2 mg/ml during preincubation (or lower concentrations if possible based on limits of detection of analytical methods). Non-specific microsomal protein binding may be an issue for certain NME, and correction may be needed for better estimation of KI.
-
At least a 10-fold dilution after preincubation is necessary to quench any further inactivation during the incubation step and minimize further inhibition of probe substrate by the NME and its metabolites. If the NME is also a potent reversible inhibitor, more than 20-fold dilution and a longer preincubation time would improve the method for the evaluation of a time-dependent inhibitor.
-
CYP3A4 is the most frequently affected P450 enzyme for TDI and should always be assessed. The recommended probe substrates and positive control TDI for CYP3A4 together with other P450 isoforms are listed in Table 5.
-
Use of a concentration of the probe substrate that approaches saturation (≥4-fold KM) is recommended to minimize further inhibition of probe substrate by test compound and its metabolites.
-
Vehicle and minus NADPH control samples should be included.
-
Low concentrations of organic solvent vehicle (final solvent concentration in the preincubation ≤1%) should be used to minimize inhibition of P450 activities (Iwase et al., 2006).
Recommended in vitro probe substrates and inactivators for evaluation of TDI (including MBI) of P450 enzymes
IC50 Shift Method. The time-dependent IC50 shift approach (Fig. 5) is another method used in the discovery stage for rigorous comparison of several lead candidates for nomination of a compound into development. The data obtained from this method can also be useful for setting up the concentration range for determination of inactivation kinetic constants. The IC50 shift method has the capability for simultaneous detection of both reversible inhibitors and time-dependent inhibitors, plus the added benefit for potentially predicting drug-drug interactions (Obach et al., 2007). In this assay, human liver microsomes (0.25 to 1 mg/ml) are preincubated for 30 min with various concentrations of NME that surround 10-fold (or greater) of their reversible inhibition IC50 value in the presence and absence of NADPH. The preincubation samples are then diluted 10-fold (or greater) into a solution containing probe substrate and NADPH for determination of the remaining enzyme activity. Probe P450 substrate concentrations around their KM values are used. A left shift of the IC50 curve from samples preincubated with NADPH compared with those without NADPH suggests a potential for enzyme inactivation (Fig. 5). However, this left shift of the IC50 curve could also be theoretically due to generation of metabolites during the first incubation that are highly potent reversible inhibitors of the enzyme. Furthermore, the lack of a time-dependent shift of the IC50 curves cannot exclude the possibility that a weak irreversible inhibition may be masked by potent reversible inhibition from test compound. In addition, the resolution of the IC50 shift method in detection of compound that is both a potent reversible inhibitor and a potent mechanism-based inactivator can be improved by using a saturating probe substrate concentration (≥4-fold KM) and longer preincubation time to generate more of the reactive intermediate. The shifted IC50 instead of the degree of shift of IC50 was found to correlate well with the ratio of kinact/KI obtained from the standard dilution assay (Obach et al., 2007). Compounds that are known to be TDI showed IC50 shifts of ≥1.5× with a 30-min preincubation (Berry and Zhao, 2008), suggesting the use of such values as a cutoff for identifying a compound as a TDI.
The potential contribution of potent reversible inhibition by metabolites generated in situ to shifts in IC50 can be assessed by generation of a parallel set of preincubation samples in the IC50 shift experiment, extraction of these by acetonitrile precipitation, evaporation of the supernatant, and reconstitution in incubation buffer (Li et al., 2005). Then, fresh microsomes along with probe substrate and cofactors are added to measure reversible inhibition. The maximum degree of separation derived from the difference due to loss of enzyme activity in the first pair of IC50 curves is attributed to enzyme inactivation plus reversible inhibition by parent compound and its metabolites. In comparison, the maximum difference between the second pair of IC50 curves is only due to different extents of reversible inhibition of the enzyme by parent compound versus its metabolites (with an assumption of negligible inactivation during activity measurement). The net enzyme inactivation is calculated from the difference between the differences of these two pairs of IC50 curves. The potency of time-dependent inhibition can then be defined as a ratio of the percentage of maximum net loss of activity over the inhibitor concentration that resulted in percentage of maximum net loss of activity. A good correlation of this ratio with the ratio of kinact/KI obtained in the dilution assay was observed for nine marketed drugs (Li et al., 2005).
Other Experiments That Can Yield Mechanistic Insight. Upon observation of TDI and generation of the parameters needed to make predictions of DDI, there are other follow-up experiments that can be done to permit a greater understanding of the underlying mechanism for TDI. It is not proposed that such experiments are required. Rather, these are described here to provide the reader with a greater understanding of other experiments that can yield mechanistic insight into the TDI caused by any given NME or chemical series to which it belongs.
Partition Ratio. The partition ratio (r) is defined as the number of moles of inactivator metabolized to product for each mole of inactivated enzyme (Silverman, 1995). Although the partition ratio itself is not useful for predicting DDI, it has been used as a measure of the biochemical efficiency of the mechanism-based inactivation process and can be used to compare potency of NME in early drug research (Lim et al., 2005). The kinetic definition of the partition ratio is as follows (eq. 10): 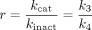 where the kinetic constants k3 and k4 have been previously defined in Scheme 1.
where the kinetic constants k3 and k4 have been previously defined in Scheme 1.
Factors like reactivity of the reactive intermediate and the rate of diffusion of the reactive intermediate out of the active site of the P450 enzyme can influence the magnitude of the partition ratio of a compound. A highly efficient mechanism-based inactivator will have a low partition ratio because relatively few of the reactive intermediate molecules formed will be able to leave the active site without binding covalently to the enzyme or complexing with the heme iron. On the other hand, a higher proportion of the less reactive intermediate molecules will be metabolized to product and leave the active site, resulting in a high partition ratio for weak mechanism-based inactivators.

Illustration of IC50 shift data and change in inhibition at a single concentration (IC25).
Methods reported for determination of partition ratios of mechanism-based inactivators of P450 enzymes included using kcat/kinact ratio (Jones et al., 1999), from substrate depletion (Kunze and Trager, 1993) and from titration of enzyme with inactivator (Kent et al., 1998). Purified P450 enzymes, cDNA-expressed recombinant P450 enzymes, and human liver microsomes have been used for determination of partition ratio by these three methods. Note that there may be differences in the apparent partition ratio generated using human liver microsomes compared with the intrinsic partition ratio determined using recombinant P450 enzyme due to depletion of the test compound caused by metabolism by the target P450 or other P450 enzymes. The titration method is commonly used for estimation of partition ratio of mechanism-based inactivators of P450 enzymes based on a literature survey (Ghanbari et al., 2006), which indicated that the titration method used recombinant P450 enzymes twice as frequently compared with use of human liver microsomes. A detailed description of the methods used to determine partition ratios is beyond the scope of this article and can be found in other references (Kent et al., 1998; Atkinson et al., 2005; Lim et al., 2005; Hollenberg et al., 2008).
Dialysis and Microsomal Washing. After demonstration that a compound is a time-dependent inhibitor, the nature of the inhibition can be assessed by dialysis (Ma et al., 2000) or microsomal washing (Schlezinger et al., 1999) experiments to investigate whether the decrease in activity is due to tight reversible or covalent binding of metabolites to the heme and/or protein or due to formation of relatively more unbound and/or more potent metabolites. Small volumes (e.g., 1 ml) of microsomal samples preincubated with vehicle control, test compound, and positive control for the specific P450 of interest (known MBI, e.g., mibefradil for CYP3A) can be dialyzed against a large volume (e.g., 1 L) of buffer at 4°C with stirring overnight. In general, the samples will not show significant volume shift because of the low microsomal protein concentration used (e.g., 0.5 mg/ml). However, it is recommended to determine the protein concentration after dialysis. Activities after dialysis are compared with the dialyzed vehicle control sample to determine whether there is recovery of activity in the test compound sample.
Repeated microsomal washing is another method to test whether enzyme activity can be recovered after treatment with an inactivator. Preincubated samples are spun in an ultracentrifuge at 100,000g for ≥30 min, and the protein pellet is resuspended with a dounce to provide a homogeneous microsomal suspension. This procedure is usually repeated 2 to 3 times, and each time aliquots of samples are set aside. After this washing procedure, activities are measured along with the protein concentrations. The protein-normalized enzyme activities are compared with the preincubation vehicle control. If the washed microsomal samples of the NME and negative control show recovery of activities while the positive control does not, the NME can be classified as a time-dependent inhibitor that is not a mechanism-based inactivator.
Ferricyanide Treatment of MIC. The quasi-irreversible MIC generated by coordinate bond formation between the reactive intermediate and the ferrous iron of the heme can be readily identified from the absorbance maximum at the Soret peak at approximately 448 to 455 nm by difference spectra scanning (Franklin, 1991). Thus, the formation of MIC can be measured in the samples containing one or two concentrations of test compound (e.g., 50 or 100 μM) by scanning from 380 to 500 nm using a dual-beam spectrophotometer to monitor the formation of an absorbance maximum at 455 nm. Human liver microsomes or recombinant P450 are incubated with the test compound in the sample cuvette and vehicle in the reference cuvette. The reaction is initiated by the addition of NADPH.
The MIC can be dissociated, as indicated by a reduction in the Soret absorbance maximum, upon oxidation with ≥50 mM potassium ferricyanide (Buening and Franklin, 1976). Alternatively, the MIC formed during preincubation can be dissociated from further incubation with potassium ferricyanide before another 5-fold or greater dilution for residual activity assessment using ≥4-fold KM probe substrate concentration in the presence of a minimum 50 mM potassium ferricyanide (Lim et al., 2005; Watanabe et al., 2007). Oxidation with potassium ferricyanide only dissociates nitrogen-based MICs (Franklin, 1991); therefore, oxidation with ferricyanide is not recommended for detection of other MIC-forming compounds.
Protection by Substrate or Competitive Inhibitor. Another criterion for a mechanism-based inactivator is the demonstration of decrease in the observed rate of inactivation when a substrate or competitive inhibitor is included during preincubation (Silverman, 1996). The observed rate of inactivation in the presence of substrate or a competitive inhibitor is compared with the rate in the absence of substrate or a competitive inhibitor to determine whether protection from inactivation is afforded. It should be noted that multiple substrate binding sites have been reported for CYP2C9 (Kumar et al., 2006) and CYP3A4 (Kenworthy et al., 1999); therefore, the fulfillment of this criterion may not always be possible for some inactivators of these two P450 enzymes.
Inactivation of Catalytic Site Before Release of Reactive Intermediates. Another criterion of a mechanism-based inactivator is that inactivation of the enzyme occurs before release of the reactive intermediates from the active site of the enzyme (Silverman, 1996). It is informative to differentiate between enzyme inactivation due to nonspecific covalent modification outside the active site versus that which occurs at the active site. This type of inactivation will also increase in a time-dependent manner with increasing nonspecific covalent modification of the enzyme from buildup of concentration of the reactive species in solution. This can be addressed by determining whether inactivation is reduced when electrophile scavenging reagents such as reduced glutathione (1–5 mM) or reactive oxygen species scavengers like catalase and superoxide dismutase are included (Kunze and Trager, 1993; Regal et al., 2000).
Recommendations. A decision flow chart is presented in Fig. 6. An abbreviated version of the standard dilution approach, such as the measurement of a change in the amount of inhibition observed when the NME (at 1–2 concentrations) is preincubated with enzyme and cofactor (Figs. 2 and 3), is recommended. The concentration tested should be sufficiently high (e.g., 10–50 μM) to ensure that weak TDI are identified (Riley et al., 2007), with the potential limitation of solubility taken into consideration for some compounds. False positive results, i.e., inactivation observed at a high concentration that turns out to be irrelevant for DDI in vivo will be captured when KI and kinact are measured in follow-up studies. Alternately, the IC50 shift method (Fig. 5) can be used for initial testing of NME for time-dependent inhibition. Human liver microsomes are the preferred source of enzyme activity. The selection of the specific P450 enzymes to be tested can be based on reaction phenotyping information (i.e., the individual enzymes that are responsible for the turnover of the NME) and/or the isoforms that are competitively inhibited by the NME. However, there can be exceptions to using only these criteria for selection of P450 enzymes to test. For example, some sponsor companies have observed TDI in the absence of significant compound turnover. As such, it is generally good practice to evaluate TDI potential for all of the major human P450 enzymes important in the clearance of drugs during the development phase, especially CYP3A4 (Table 5). Whereas many known P450 TDI act upon CYP3A, there are several drugs known to inactivate other important human P450 enzymes (Table 1) and cause DDI. If the results indicate no observable time-dependent inhibition, then this result should obviate any further investigation. Identification of the threshold that defines whether an NME is a TDI is important and should be established for the experimental paradigm being used, using known positive control inactivators (Table 5). The reason for this is that comparison to different types of control incubations [i.e., vehicle control with preincubation done in the presence of cofactor (Fig. 3) or a control in which NME is included but the preincubation is done in the absence of cofactor (Fig. 2)] could yield different conclusions. Furthermore, the typical limits on intra- and interassay precision (i.e., 10–15%) need to be appreciated when defining what degree of time-dependent inhibition is detectable versus simple experimental variability. In assays in which single concentrations of NME are tested, a decrease of 20 to 25% activity over 30-min preincubation (corrected for vehicle control) represents a reasonable cutoff to identify those compounds that are TDI meriting further study. For the IC50 shift approach, a decrease of 1.5- to 2-fold for IC50 with a 30-min preincubation represents a significant enough change to categorize an NME as a TDI.

Schematic flow chart for identification and characterization of P450 TDI. (Refer to the text for specifics around decision points and threshold criteria.)
For those NME demonstrating TDI using an abbreviated experimental method, further follow-up experiments using a standard dilution method with multiple time points and multiple inhibitor concentrations to determine the kinetic parameters for inactivation (KI and kinact) are recommended for verification that the NME is a time-dependent inhibitor. These parameters are also essential input values needed for the prediction of human DDI potential from in vitro data. For those NME in which KI and kinact values, along with other input parameters (e.g., [I]in vivo, fm,CYP), suggest the likelihood of a point estimate DDI ≥2-fold, verification with a clinical DDI study using an appropriate probe substrate (Bjornsson et al., 2003) should be considered. Confidence in such predictions of DDI magnitude is dependent on the confidence of individual input parameters (e.g., [I]in vivo, kdeg) and the known performance of the algorithm used at scaling in vitro TDI data to the magnitude of DDI caused by known P450 inactivators. Because there is no consensus on kdeg values to use in the prediction of DDI and no method to definitively determine such values in humans, individual laboratories should take an empirical approach to determine specific kdeg values that accurately scale DDI for positive control compounds using inactivation kinetic parameters generated in their own laboratories. As an alternate shorter approach, determination of kobs at a single high [I] can provide an estimate of kinact and when used in the prediction equations will yield an upper estimate of the magnitude of DDI. The selection of the single high value for [I] will need to be scientifically defensible.
Certainly the science around P450 inactivation will continue to develop. Algorithms in which in vitro data are used to predict in vivo DDI will probably improve. Methodologies used to generate the in vitro data will probably become more automated and streamlined. In addition, as medicinal chemists venture into unknown chemical space, it will probably be the case that new structural motifs will be identified as P450 inactivators. Other drug-metabolizing enzymes may also increase in importance, and any concerns for inactivation of these will become relevant to the discovery and development of new drugs. The basic concepts underlying the recommendations described in this article for P450 enzymes should be applicable to other enzymes, although experimental details will differ. As these advances are made, revisiting these recommendations will be needed and adaptations made.
Acknowledgments
We thank the PhRMA Drug Metabolism Technical Group for their sponsorship of this effort. We also thank Michael Garvin, Vail Miller, and Paul Larsen (PhRMA) and Robin Keeney (AstraZeneca) for assistance in executing the survey.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.026716.
-
ABBREVIATIONS: DDI, drug-drug interactions; PhRMA, Pharmaceutical Research and Manufacturers of America; FDA, Food and Drug Administration; P450, cytochrome P450; TDI, time-dependent inhibition; MBI; mechanism-based inactivation; MIC, metabolite-intermediate complex; NME, new molecular entity; IVIVE, in vitro-in vivo extrapolation; AUC, area under the curve; ADME, absorption, distribution, metabolism, and excretion; PK, pharmacokinetic; PBPK, physiologically based pharmacokinetics; MDMA, 3,4-methylenedioxymethamphetamine; SAR, structure activity relationships; HLM, human liver microsome.
- Accepted April 7, 2009.
- Received January 13, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




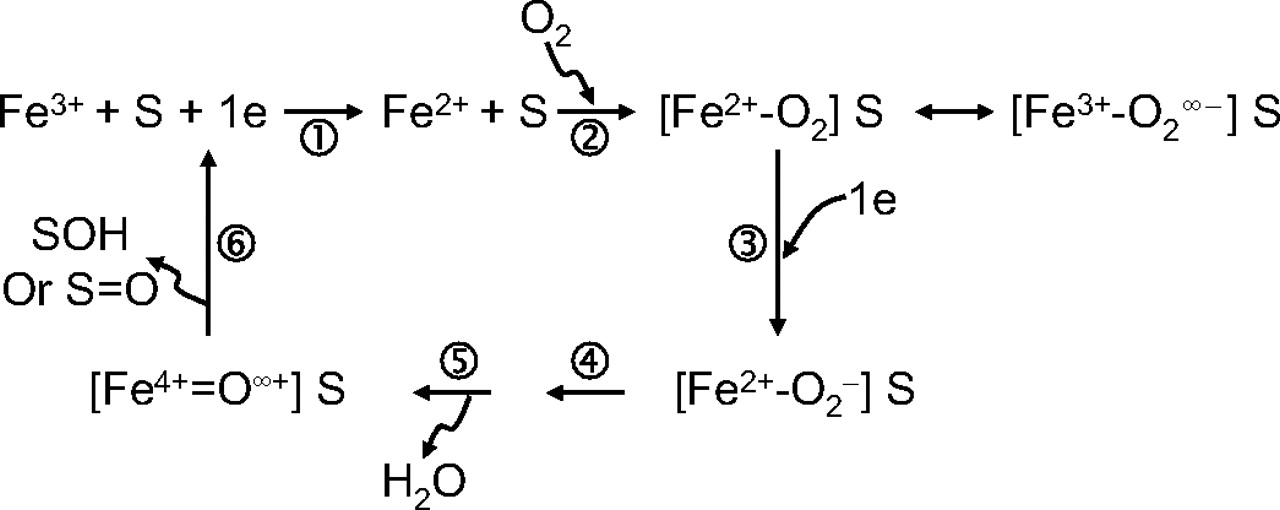{kind=link}
{kind=link}
{kind=link}
{kind=link}
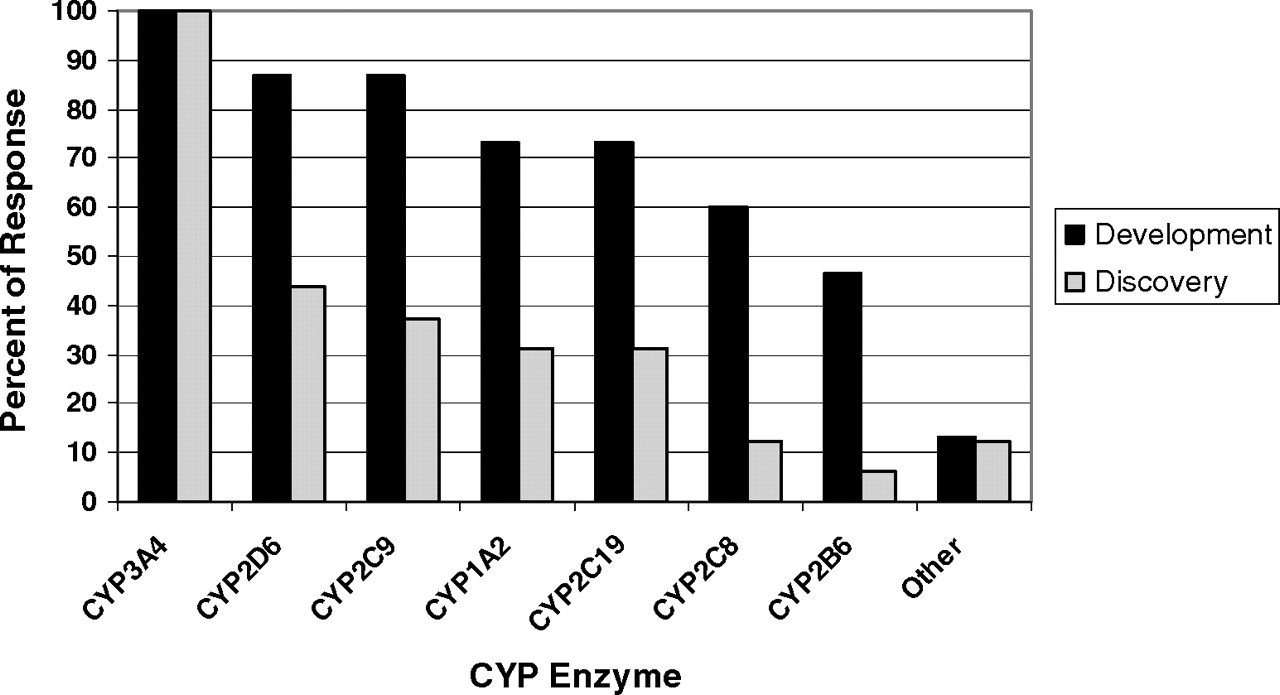{kind=link}
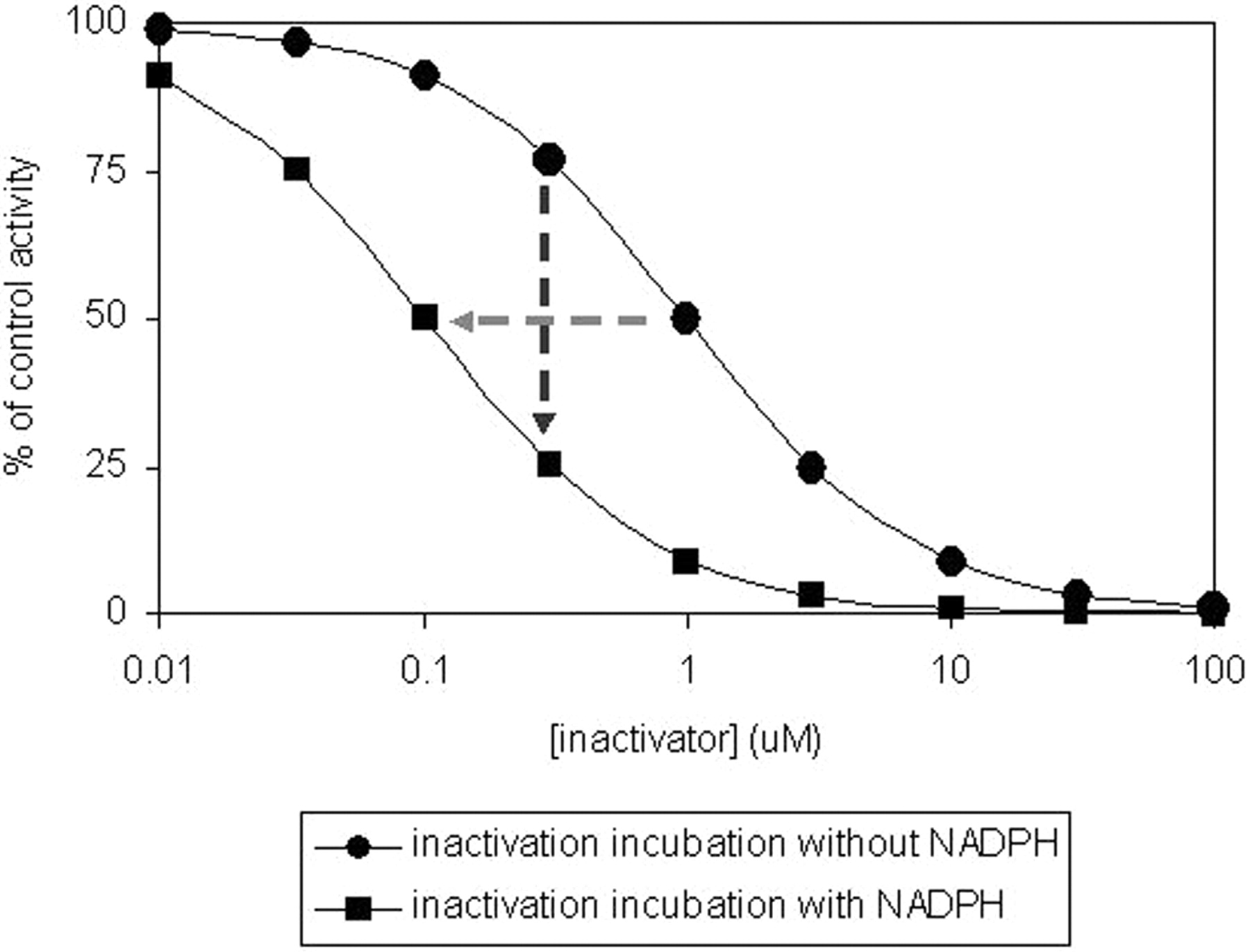{kind=link}
{kind=link}