


Abstract
The objectives of this study were to characterize and compare the reversible inhibition and time-dependent inactivation of cytochromes P450 3A4 and 3A5 (CYP3A4 and CYP3A5) by erythromycin, diltiazem, and nicardipine. In the following experiments, we used cDNA-expressed CYP3A Supersomes and CYP3A-phenotyped human liver microsomes. We estimated the apparent constants for reversible inhibition (Ki(app) and IC50) and the irreversible kinetic constants (KI and kinact) for time-dependent inhibition. Based on an aggregate of Ki(app) and IC50 measurements, all inhibitors showed a greater inhibitory potency for CYP3A4 compared with CYP3A5. In addition, for each inhibitor, the kinact for CYP3A4 was approximately 4-fold higher than that for CYP3A5, indicating a greater propensity for time-dependent loss of CYP3A4 activity than of CYP3A5. Difference spectra experiments revealed an NADPH-dependent peak at ∼455 nm [metabolite-inhibitor (MI) complex] following incubation of all three drugs with CYP3A4. There was no discernable MI complex formation following CYP3A5 incubation with any of the inhibitors. However, when CYP3A4 and CYP3A5 were incubated simultaneously with erythromycin, both enzymes appeared to contribute to the formation of a MI complex. Additional experiments revealed that erythromycin caused a comparable type I spectral change when bound to CYP3A5 and CYP3A4 (Ks = 48 μM and 52 μM, respectively). Moreover, CYP3A5 exhibited only a moderately slower rate for the initial N-demethylation than did CYP3A4 (intrinsic clearance = 41 versus 99 μl/min/nmol, respectively). In conclusion, erythromycin, diltiazem, and nicardipine were weaker inhibitors of CYP3A5 and inactivated the enzyme at a slower rate than their respective effects on CYP3A4. With respect to erythromycin, the failure of CYP3A5 to form a MI complex appears to be the result of slowed or impaired metabolic events downstream from the initial catalytic step, possibly due to a different orientation of the substrate molecule in the active site.
Although CYP3A4 and CYP3A5 share 84% amino acid sequence homology, the two enzymes behave differently with respect to substrate turnover and susceptibility to inhibition. CYP3A5 has generally been considered less important than CYP3A4 because of its polymorphic and relatively low level of hepatic and intestinal expression. However, recent reports indicate that a majority of individuals express some CYP3A5 protein (Jounäidi et al., 1996; Lin et al., 2002) and that it can be found at relatively high levels in some individuals with a heterozygous or homozygous CYP3A5*1 genotype (Kuehl et al., 2001). Moreover, the frequency of polymorphic CYP3A5 expression differs among ethnic groups. For example, CYP3A5 is found at high levels in 10 to 30% of Caucasians and >50% of African-Americans (Lamba et al., 2002; Lin et al., 2002).
The phenotypic significance of the CYP3A5 polymorphism is controversial. For example, Westlind-Johnsson et al. (2003) reported a low CYP3A5*1 allele frequency (4.3%) and a relatively low percentage contribution from CYP3A5 to the total immunodetectable hepatic CYP3A pool (13-27%) in livers carrying the CYP3A5*1 allele. In addition, Floyd et al. (2003) observed no significant difference in midazolam clearance between subjects with a homozygous and heterozygous CYP3A5*3 and those with a homozygous CYP3A5*1 genotype. In contrast, Yamaori et al. (2004) found a much higher CYP3A5*1 allele frequency and a 20 to 60% contribution from CYP3A5 to the total hepatic CYP3A pool for Japanese tissue donors, and Wong et al. (2004) reported a significantly higher midazolam clearance for largely Caucasian cancer patients carrying a CYP3A5*1 allele, compared with those with a homozygous CYP3A5*3 genotype. The reason for these discrepancies is unclear, but recent reproducible, in vivo observations of higher oral clearance of tacrolimus, an excellent CYP3A5 substrate (Bader et al., 2000), in organ transplant patients carrying a CYP3A5*1 allele (Hesselink et al., 2003; Thervet et al., 2003; Haufroid et al., 2004) suggest that the CYP3A5 polymorphism and CYP3A5 expression may play a significant role in inter-individual and interracial variability with respect to metabolic drug elimination.
CYP3A4 and CYP3A5 exhibit similar Km and Vmax values for midazolam 1′-hydroxylation (Gorski et al., 1994a; Gibbs et al., 1999). In addition, both enzymes are efficient at metabolizing nifedipine (Aoyama et al., 1989; Gillam et al., 1995), lidocaine (Bargetzi et al., 1989), tamoxifen (Williams et al., 2002), and dextromethorphan (Gorski et al., 1994b). In contrast, CYP3A5 has been described as a significantly poorer catalyst of testosterone (Wrighton et al., 1990; Waxman et al., 1991), progesterone (Aoyama et al., 1989; Waxman et al., 1991), cyclosporine (Aoyama et al., 1989), clarithromycin (Williams et al., 2002), estradiol (Williams et al., 2002), and valspodar (Fischer et al., 1998) metabolism. However, some quantitative data in the literature are conflicting [erythromycin demethylation, for example (Wrighton et al., 1990; Gillam et al., 1995)], possibly the result of laboratory-specific differences in the enzyme preparation and incubation conditions used.
The few studies comparing the inhibition kinetics for CYP3A4 and CYP3A5 also reported differences in kinetic behavior. For example, both ketoconazole and fluconazole are more potent inhibitors of CYP3A4 than of CYP3A5 (Gibbs et al., 1999). In addition, studies with diltiazem revealed a lower amount of metabolite-inhibitor (MI) complex formation, a higher partition ratio (kcat/kinact), and a lower N-demethylation rate for CYP3A5, compared with CYP3A4 (Jones et al., 1999). Moreover, similar kinetic differences were reported for the suicide inactivation of CYP3A4 and CYP3A5 by mifepristone, with CYP3A5 exhibiting no detectable irreversible inhibition (Khan et al., 2002).
Mechanism-based inactivation is common for CYP3A substrates that contain an amine functional group and undergo N-dealkylation (Pershing and Franklin, 1982; Bensoussan et al., 1995). The process by which substituted amine-containing compounds inactivate CYP3A is presumed to involve consecutive CYP3A-dependent oxidations that eventually generate a nitroso compound that binds tightly to the ferric heme (Montellano, 1995). Erythromycin, diltiazem, and nicardipine have been shown to form human P450 MI complexes in vitro (Lindstrom et al., 1993; Jones et al., 1999; Ma et al., 2000), and thus have the potential ability to inhibit CYP3A in a reversible manner or by mechanism-based inactivation.
The goals of this study were to 1) compare the effect of erythromycin, diltiazem, and nicardipine on CYP3A4- and CYP3A5-catalyzed midazolam hydroxylation, 2) test whether there is a difference in the CYP3A4 and CYP3A5 inhibition mechanism, and 3) test whether there are qualitative differences between the inhibition kinetic parameters generated from cDNA-expressed enzymes and human liver microsomes that contain predominantly CYP3A4 or CYP3A5. In this regard, direct comparison of metabolic activity with selected CYP3A-phenotyped human liver microsomes might provide more predictive (in vitro to in vivo) experimental data.
Materials and Methods
Reagents. NADPH (nicotinamide adenine dinucleotide phosphate, reduced form), erythromycin, diltiazem, nicardipine, and alkaline phosphatase-conjugated secondary antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Ketoconazole was obtained from Research Diagnostics (Flanders, NJ). Midazolam, 1′-hydroxymidazolam, 4-hydroxymidazolam, and 15N3-midazolam were kindly provided by Hoffman-La Roche (Nutley, NJ). [N-methyl-14C]Erythromycin was a gift from Dr. Paul Watkins (University of North Carolina, Chapel Hill, NC). Acetonitrile and ethyl acetate were purchased from Fisher Scientific Co. (Pittsburgh, PA). N-Methyl-N-(t-butyl-dimethylsilyl) trifluoroacetimide was obtained from Pierce Chemical (Rockford, IL). A CYP3A5-selective antibody (Cat. 458235), and baculovirus-insect cell-expressed CYP3A4 (P207, Lot 14) and CYP3A5 (P235, Lot 10) Supersomes were purchased from BD Gentest (Woburn, MA). The Supersomes (2000 pmol of P450/ml) were coexpressed with cytochrome P450 reductase (2044 and 2808 nmol of cytochrome c reduced/min/ml) but not cytochrome b5. Cytochrome b5 was purchased from PanVera Corp. (Madison, WI). SDS-polyacrylamide gel electrophoresis reagents (37.5:1 bis-acrylamide, ammonium persulfate, and N,N,N′,N′-tetra-methyl-ethylene-diamine) were purchased from Bio-Rad (Hercules, CA). Nitrocellulose was purchased from Schleicher and Schuell (Keene, NH). BCIP/NBT reagents were purchased from Kirkegaard and Perry Laboratories (Gaithersburg, MD). All other chemicals were reagent grade or better.
Tissue Collection and Microsomal Preparation. Human livers were obtained through the Solid Organ Transplant Program at the University of Washington Medical Center and Life Center Northwest (Seattle, WA). Limited descriptive information on the liver donors is provided in Table 1. Liver microsomes were prepared as described elsewhere (Paine et al., 1997) and stored at -80°C. Six livers were selected for time-dependent enzyme inactivation experiments. The respective specific CYP3A4 and CYP3A5 contents were: (CYP3A4 set) HL-114, 86.1 and 3.1 pmol/mg; HL-155, 19.1 and 3.1 pmol/mg; HL-166, 144, and 2.2 pmol/mg; (CYP3A4 + CYP3A5 set) HL-127, 21.1, and 44.8 pmol/mg; HL-154, 5 and 22.2 pmol/mg; HL-167, 19.2 and 39.6 pmol/mg.
Descriptive information for human liver donors
CYP3A4 and CYP3A5 Western Blot Analysis. Human livers used in the following experiments were selected based on the relative abundance of CYP3A4 and CYP3A5. Immunoquantitation of CYP3A4 and CYP3A5 specific content (pmol/mg protein) in the human microsomal preparations have been presented elsewhere (Lin et al., 2002). Purified CYP3A4 and expressed CYP3A5 were used as reference standards to quantitate both enzymes. Integrated optical density measurements were obtained from a Bio-Rad ChemiDoc system and Quantity One Software.
Incubation Systems. The CYP3A4 and CYP3A5 Supersomes utilized for this experiment were both deficient in cytochrome b5. Accordingly, all experiments with the heterologously expressed enzymes were supplemented with a 3:1 molar ratio of cytochrome b5 to CYP3A enzyme. This molar ratio was found, in preliminary experiments, to provide maximum midazolam hydroxylation rates (data not shown). The heterologously expressed CYP3A Supersomes are referred to hereafter as 3A4-Supersomes and 3A5-Supersomes. The CYP3A-phenotyped human liver microsomes were categorized as 3A4-microsomes (weak or no detectable CYP3A5 expression) or 3A4/5-microsomes (expressing relatively high levels of CYP3A5, 22.2-291 pmol CYP3A5/mg protein). For irreversible inhibition experiments, we selected human liver microsomes based on their CYP3A contents. We chose three human liver microsome preparations that contained predominantly CYP3A4 [HL-114 (3.5% CYP3A5), HL-155 (16% CYP3A5), HL-166 (1.5% CYP3A5)], and the three microsomal samples with the highest molar abundance of CYP3A5 [HL-127 (68% CYP3A5), HL-154 (85% CYP3A5), HL-167 (67% CYP3A5)]. These samples are referred to as 3A4-microsomes and 3A5-microsomes.
Protocol for Ki(app) and IC50 Measurements. The effect of erythromycin, diltiazem, and nicardipine on CYP3A4- and CYP3A5-catalyzed midazolam hydroxylation was evaluated under conditions where the substrate and inhibitor were added simultaneously to the reaction mixture. Because the period of product formation was kept brief (4 min), resulting inhibition parameters (Ki(app) and IC50) were considered to reflect primarily the affinity of inhibitor for the CYP3A enzymes. However, the term “apparent” (app) is added to Ki to reflect the possibility of some metabolism-based enzyme inactivation. A previously characterized reversible CYP3A inhibitor, ketoconazole, was also studied for comparison. Incubations with each inhibitor were conducted in solutions containing 100 mM potassium phosphate buffer at pH 7.4 in a final volume of 0.5 ml. All incubations were performed in triplicate. Ketoconazole and erythromycin solutions were prepared in acetone. After addition to the reaction vessel, the solvent (50 μl) was allowed to evaporate before the addition of any additional reagents. Nicardipine was dissolved in dimethyl sulfoxide; the final concentration of solvent never exceeded 1% (v/v). Midazolam and diltiazem were dissolved in buffer.
Substrate, inhibitor, buffer, and CYP3A enzyme [50 μg of protein for microsomes or 10 pmol of Supersomes (∼15 μg of protein)] were preincubated for 5 min at 37°C before the addition of NADPH (1 mM final concentration). The midazolam concentration was varied between 2 and 16 μM (Ki(app) experiments) or was fixed at 4 μM (IC50 experiments). After 4 min, reactions were terminated with the addition of 1 ml of 100 mM Na2CO3, pH ∼11. Internal standards (15N3-1′- and 15N3-4-hydroxymidazolam solution) were added to all tubes, followed by extraction with 5 ml of ethyl acetate. Both 1′- and 4-hydroxymidazolam in the samples and associated standard curves were measured by gas chromatography-negative chemical ionization mass spectrometry as described previously (Schmiedlin-Ren et al., 1997).
The effect of various concentrations of inhibitor on the formation of 1′- and 4-hydroxymidazolam was evaluated. Ketoconazole concentration was varied between 5 and 250 nM. The concentration of erythromycin was between 10 and 500 μM for human liver microsomes and from 2 to 150 μM for Supersomes. The diltiazem concentration for human liver microsomes and Supersomes was varied from 20 to 1000 μM and 0.5 to 100 μM, respectively. Nicardipine concentration varied from 5 to 1000 nM for Supersomes and from 5 to 10,000 nM for human liver microsomes.
Protocols for kinact and KI Measurements. Experiments were conducted to determine the irreversible effect of erythromycin, diltiazem, and nicardipine on the formation of 1′- and 4-hydroxymidazolam. Incubations were performed in solutions of 100 mM potassium phosphate buffer, pH 7.4, and 1 mM EDTA. To assess time- and concentration-dependent inhibition, 5 × 5 matrices of inhibitor concentration and NADPH-dependent preincubation times were implemented for each inhibitor. Briefly, 180 μl of a solution containing microsomes (200 pmol of 3A4- or 3A5-Supersomes supplemented with 600 pmol of cytochrome b5, or 1 mg of human liver microsomal protein), inhibitor, EDTA, and buffer were preincubated at 37°C for 5 min before the addition of 20 μl of 10 mM NADPH (200-μl total volume). The concentration of erythromycin used for inactivation experiments was between 2 and 100 μM for microsomes, and from 2 to 50 μM in Supersomes. The diltiazem and nicardipine concentrations ranged from 0.5 to 100 μM and from 0.2 to 10 μM, respectively, for both microsomes and Supersomes.
After the addition of NADPH, 25-μl aliquots were transferred to prewarmed tubes containing 475 μl of 1 mM EDTA, 1 mM NADPH, and 8 μM midazolam (final concentrations) at timed intervals of 0, 1, 6, 12, and 25 min. A higher concentration of midazolam (8 μM versus. 4 μM) was used to displace residual inhibitor that was present in the 25-μl transfer volume. After a 4-min incubation with midazolam, the reactions were terminated with 100 mM Na2CO3, pH ∼11. Addition of internal standard and work-up of incubation samples was as described in the previous section.
Determination of Kinetic Parameters. The reversible inhibition mechanism was assigned by graphical analysis using Dixon and Lineweaver-Burk plots. Parameter estimates (Ki(app)) were obtained by fitting to either competitive or noncompetitive models (Segel, 1975) using SAAM II statistical software (SAAM Institute, Inc., Seattle, WA). For all inhibitors coincubated with 4 μM midazolam, the concentration of inhibitor required for half-maximal substrate turnover (IC50) was determined based on a one-enzyme hyperbolic model (KaleidaGraph version 3.5; Abelbeck/Synergy (Reading, PA).
For time-dependent enzyme inactivation, the pseudo first order rate constant for enzyme inactivation (λ) was estimated from eq. 1. 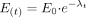
E(t) is the percentage of active enzyme remaining at time t, and E0 was set at 100%. An estimate of λ was obtained from the slope from t = 1 to 12 min (Fig. 1). To determine inactivation kinetic constants (KI and kinact), λ and inhibitor concentration [I] were fitted to eq. 2. 
Estimation of the pseudo first order rate constant for enzyme inactivation (λ) following time-dependent enzyme inactivation of 3A4-microsomes by erythromycin. Microsomes (HL-166 is shown; 1 mg/ml microsomal protein) were incubated with erythromycin (0-100 μM) in the presence of NADPH for 0 to 12 min followed by a 4-min incubation with 8 μM midazolam to assess loss of midazolam 1′-hydroxylase activity. A, time-dependent loss of enzyme activity by different concentrations of erythromycin. B, activity was corrected for loss of activity in control incubations (0 μM ERY).
Under this model, kinact represents the constant for the maximal rate of enzyme inactivation and KI is the concentration of inhibitor required to achieve 50% of the maximum enzyme inactivation (Silverman, 1988). A representation of metabolic inactivation is given in Scheme 1.

In Scheme 1, the initial step describes enzyme (E) and inhibitor (I) binding, with respective on- and off-rate constants k1 and k-1, respectively. The second step describes the catalytic event (k2) that produces the inhibitory metabolite. Once the metabolite is formed, it can be released as product (k3) or can inactivate the enzyme (k4).
MI Complex Formation. MI complex formation was determined for 3A4- and 3A5-Supersomes, as well as for human liver microsomes. MI complex formation was characterized by difference spectroscopy (Cary 9E double beam UV/visible spectrometer, Varian, Inc., Palo Alto, CA) over a wavelength range of 390 to 500 nm. Formation of the MI complex was demonstrated by the appearance of a peak absorbing at approximately λ = 452 nm and was quantitated from absorbance difference spectra (λ452-490 nm). Sample cuvettes contained 200 pmol of CYP3A Supersomes (supplemented with 600 pmol of cytochrome b5) or 1 mg of microsomal protein, NADPH (1 mM final concentration), and inhibitor (50 μM erythromycin, 50 μM diltiazem, or 5 μM nicardipine), whereas reference cuvettes contained identical components except that inhibitor was replaced with the appropriate vehicle solvent. Reactions were initiated by addition of NADPH and were conducted for 30 min at 37°C. For one set of experiments, erythromycin (50 μM) was incubated with 3A4-Supersomes, 3A5-Supersomes, and cytochrome b5, in a molar ratio of 200: 200:1200 pmol, and the resulting difference spectrum was compared with that produced from an incubation of erythromycin (50 μM) and 200 pmol of either 3A4- or 3A5-Supersomes (each supplemented with 600 pmol of cytochrome b5).
CYP3A Binding Spectra. 3A4- or 3A5-Supersomes were suspended in buffer (100 mM potassium phosphate buffer, 1 mM EDTA, pH 7.4) to achieve a final concentration of 200 nM. An equal volume (500 μl) was added to sample and reference cuvettes and a baseline was recorded. Aliquots (1 μl) of erythromycin stock solutions (0-450 μM final concentration) were added to the sample cuvette, whereas the reference cuvette received an equal volume of vehicle (methanol). Difference spectra between 350 and 500 nm were recorded at 25°C using a modernized Aminco DW2 double beam UV/visible spectrometer (Olis Inc., Bogart GA).
CYP3A-Catalyzed Erythromycin N-Demethylation. 3A4- and 3A5-Supersomes were supplemented with cytochrome b5 (1:3 P450/b5 molar ratio) and suspended in 100 mM potassium phosphate buffer, 1 mM EDTA, pH 7.4, to achieve a final P450 concentration of 100 nM (100 pmol in 1 ml). [N-methyl-14C]Erythromycin was added to achieve a final concentration of 1.25 to 235 μM. After a 5-min preincubation at 37°C, the reaction was initiated with the addition of NADPH (1 mM final concentration) and stopped after 15 min. 14C-labeled formaldehyde was extracted with hexane and quantified as described by Zhang et al. (1996). Rate data were best fit by a single-enzyme hyperbolic equation. Vmax and Km were estimated using nonlinear regression (WinNonlin; Pharsight, Mountain View, CA). The intrinsic formation clearance was calculated as the ratio Vmax/Km.
Results
For three of the four inhibitors studied, the Ki(app) determined under reversible inhibition conditions (no preincubation of inhibitor and 4-min incubation with NADPH) for midazolam 1′-hydroxylation was less for 3A4-Supersomes than for 3A5-Supersomes (Table 2). Erythromycin, however, exhibited a comparable inhibitory effect on CYP3A5 and CYP3A4 activity (Ki(app) 29.4 versus 33.2 μM, respectively). The mechanism of CYP3A4 and CYP3A5 inhibition appeared to be competitive for erythromycin, nicardipine, and diltiazem and noncompetitive for ketoconazole. The ratio of Ki(app) (CYP3A5/CYP3A4) was 7.9, 0.9, 3.1, and 10.8 for ketoconazole, erythromycin, diltiazem, and nicardipine, respectively.
Model-fitted Ki(app), Km, and Vmax parameters for inhibition of 3A4- and 3A5-Supersomes
Experiments were conducted in triplicate. Values reported are mean ± S.D. [of parameter estimate from competitive (C) inhibition model fit, except for ketoconazole, which was best fit by a noncompetitive (N) model]. A Ki(app) for the inhibition of CYP3A5 midazolam 4-hydroxylation by erythromycin and nicardipine could not be estimated.
Ki(app) values were also calculated for the inhibition of midazolam 4-hydroxylation. With respect to CYP3A4, the parameter estimates for ketoconazole-, erythromycin-, and diltiazem-mediated inhibition of midazolam 4-hydroxylation were generally slightly higher (0.8- to 3.3-fold) than those obtained for inhibition of the 1′-hydroxylation pathway. Again, the mechanism of inhibition of CYP3A4- and CYP3A5-catalyzed midazolam 4-hydroxylation was competitive for erythromycin, nicardipine, and diltiazem, and noncompetitive for ketoconazole. A Ki(app) for CYP3A5 in the presence of erythromycin and nicardipine could not be estimated because of a failure to successfully fit any of the models to the 4-hydroxymidazolam experimental data. For the two inhibitors with a complete data set (ketoconazole and diltiazem), the corresponding CYP3A5/CYP3A4 Ki(app) ratio for the 4-hydroxylation pathway was 4.5 and 3.4, respectively.
For all four inhibitors studied, estimates of the Km for CYP3A4-catalyzed midazolam 4-hydroxylation (Table 2) were uniformly higher (9.1-41.8 μM) than those obtained for 1′-hydroxylation (2.5-4.3 μM). This is consistent with the multiple binding site model for many CYP3A4-catalyzed reactions, including midazolam hydroxylation (Shou et al., 1994; Ueng et al., 1997). With respect to CYP3A5-catalyzed 4-hydroxylation, the estimated Km obtained from incubations with ketoconazole and diltiazem (30.8 and 21.2 μM, respectively) were also higher than that obtained for the 1′-hydroxylation pathway (4.7 and 3.9 μM, respectively).
The effect of ketoconazole, diltiazem, erythromycin, and nicardipine on human liver microsomal midazolam hydroxylation was evaluated using a full range of inhibitor concentrations determined from Ki(app) experiments with Supersomes. Apparent IC50 values were estimated for 32 different preparations of human liver microsomes that were classified as 3A4-microsomes or 3A4/5-microsomes (Table 1). The mean IC50 values are shown in Table 3. Ketoconazole was the most potent inhibitor of liver microsomal midazolam 1′-hydroxylation, followed by nicardipine, erythromycin, and diltiazem. The mean IC50 value was significantly higher (p < 0.05) for 3A4/5-microsomes compared with 3A4-microsomes for all inhibitors except diltiazem, where no statistical difference was observed. In addition, the estimated IC50 for nicardipine, erythromycin, and ketoconazole were moderately correlated to CYP3A5 as a percentage of total CYP3A content (data not shown). Using midazolam 4-hydroxylation as a CYP3A marker reaction gave similar results for the IC50 experiments. For brevity, only data for 1′-hydroxylation are reported in the following sections since midazolam 4-hydroxylation provided no additional inhibitory insight.
IC50 values for inhibition of different CYP3A microsomal preparations
Experiments were conducted in triplicate with 4 μM midazolam using midazolam 1′-hydroxylation as a marker of CYP3A activity. Values reported are mean ± S.D. for human liver microsomal incubations, and mean ± S.E. of the model fit for CYP3A Supersomes. No statistical tests were performed for comparison of Supersomes.
IC50 experiments were duplicated using 3A4- and 3A5-Supersomes supplemented with a 3:1 molar ratio of cytochrome b5. Compared with CYP3A4, the IC50 for CYP3A5 inhibition was higher for ketoconazole (5.8-fold), erythromycin (1.6-fold), diltiazem (1.9-fold), and nicardipine (1.3-fold). Interestingly, the apparent IC50 values for nicardipine in both the 3A4- and 3A5-Supersomes were approximately 15-fold lower than the values generated for liver microsomes (e.g., 24.5 versus 349 nM for CYP3A4 and 33.0 versus. 519 nM for CYP3A5).
We examined the irreversible inhibition of CYP3A4 and CYP3A5 using Supersomes and selected human liver microsomes (three 3A4-microsomes and three 3A5-microsomes). Samples were preincubated with erythromycin, diltiazem, and nicardipine in the presence of NADPH for varying lengths of time. Equations 1 and 2 were fit to the data to determine inactivation kinetic parameters. Representative results for the time-dependent loss of midazolam 1′-hydroxylation activity following erythromycin preincubation (Fig. 1) and the replot for KI and kinact determination (Fig. 2) are presented. Mean parameters obtained from the inactivation experiments are summarized in Tables 4 and 5. For erythromycin, there was no difference between the model-fitted KI value obtained from 3A4-microsomes compared with 3A5-microsomes. With respect to kinact, the value generated from incubations of erythromycin with 3A5-microsomes was approximately one-third that obtained from 3A4-microsomes. There was insufficient time-dependent inactivation resulting from incubations of diltiazem and nicardipine with 3A5-microsomes to permit calculation of KI and kinact.

Determination of irreversible inhibition constants for erythromycin and 3A4-microsomes. Microsomes (HL-166 is shown; 1 mg/ml microsomal protein) were incubated with erythromycin (0-100 μM) in the presence of NADPH for differing incubation times. The pseudo first order rate constant for enzyme inactivation (λ) was plotted versus erythromycin concentration to estimate KI and kinact (14.4 μM and 0.045 min-1, respectively). The curve represents the line of best fit.
K1 estimates for different CYP3A preparations obtained from inactivation experiments
Calculation of K1 was based on the loss of midazolam 1′-hydroxylation activity. Experiments were conducted with at least five inhibitor concentrations, preincubation times varying from 1 to 25 min, and with 8 μM midazolam. Human liver microsomal preparations contained predominantly CYP3A4 or CYP3A5, as described under Materials and Methods. Data for microsomes are reported as mean ± S.D. (except for erythromycin and CYP3A5, where data from only two livers could be fit to the inactivation model). Values for Supersomes are mean ± S.E. for the model fit. No statistical tests were performed for comparison of Supersomes.
kinact (min−1) estimates for the different CYP3A preparations obtained from inactivation experiments
Calculation of kinact was based on the loss of midazolam 1′-hydroxylation activity. See Table 4 for additional details.
No such complications were observed for time-dependent inhibition of CYP3A Supersomes. Erythromycin and nicardipine KI values for 3A4- and 3A5-Supersomes were comparable (Table 4). In contrast, the KI for diltiazem and 3A4-Supersomes was appreciably lower than that obtained for 3A5-Supersomes (1.23 versus. 8.70 μM, respectively). For all three inhibitors, there were significant differences in the maximal rate of CYP3A4 and CYP3A5 inactivation, with CYP3A5 exhibiting a kinact that was approximately one-fourth that of CYP3A4 (Table 4). Comparing KI and kinact results from incubations with CYP3A Supersomes and human liver microsomes revealed one notable difference (Tables 4 and 5). With respect to CYP3A4, there was a 15-fold higher KI and a 50% lower kinact for diltiazem incubated with liver microsomes versus Supersomes.
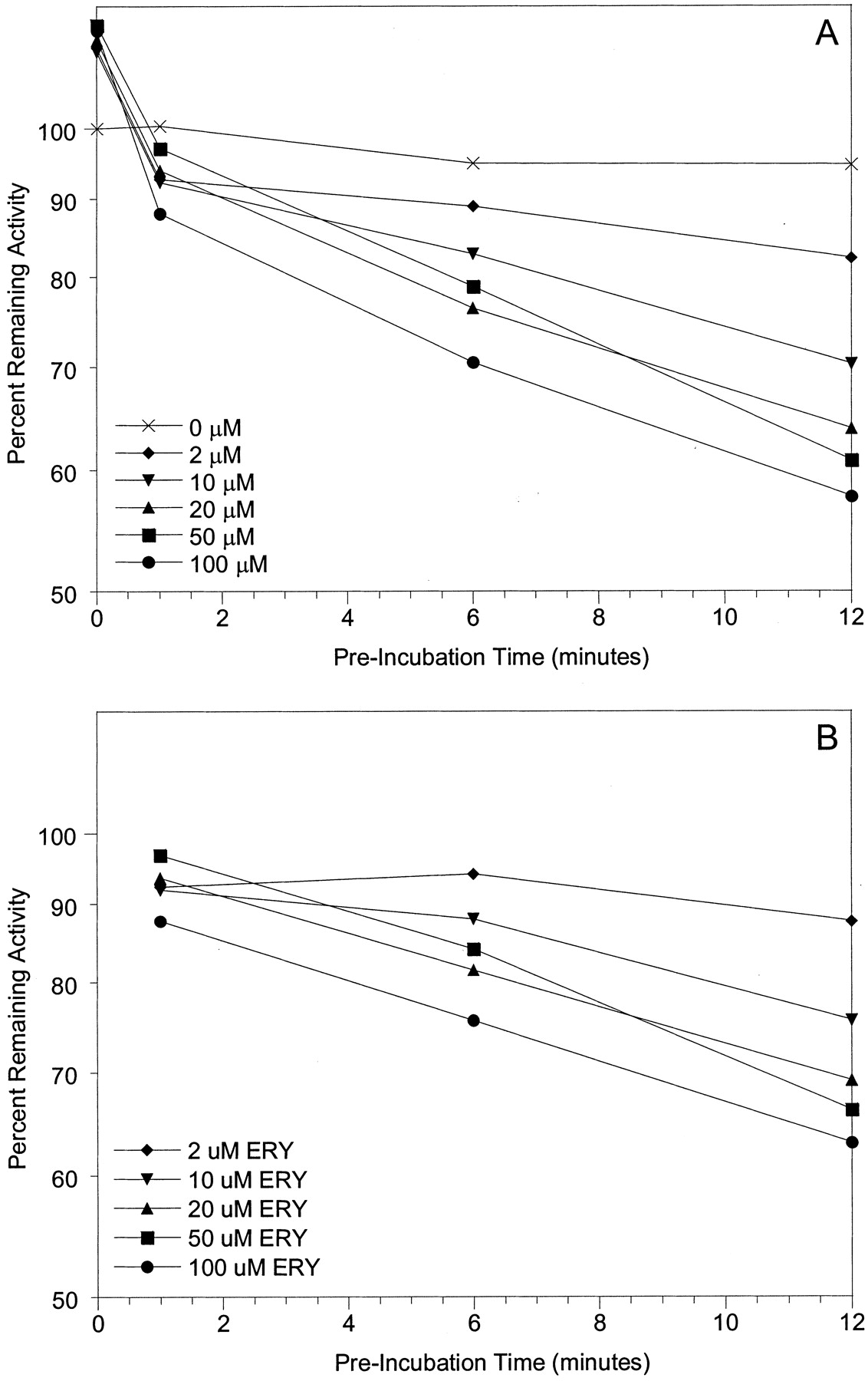
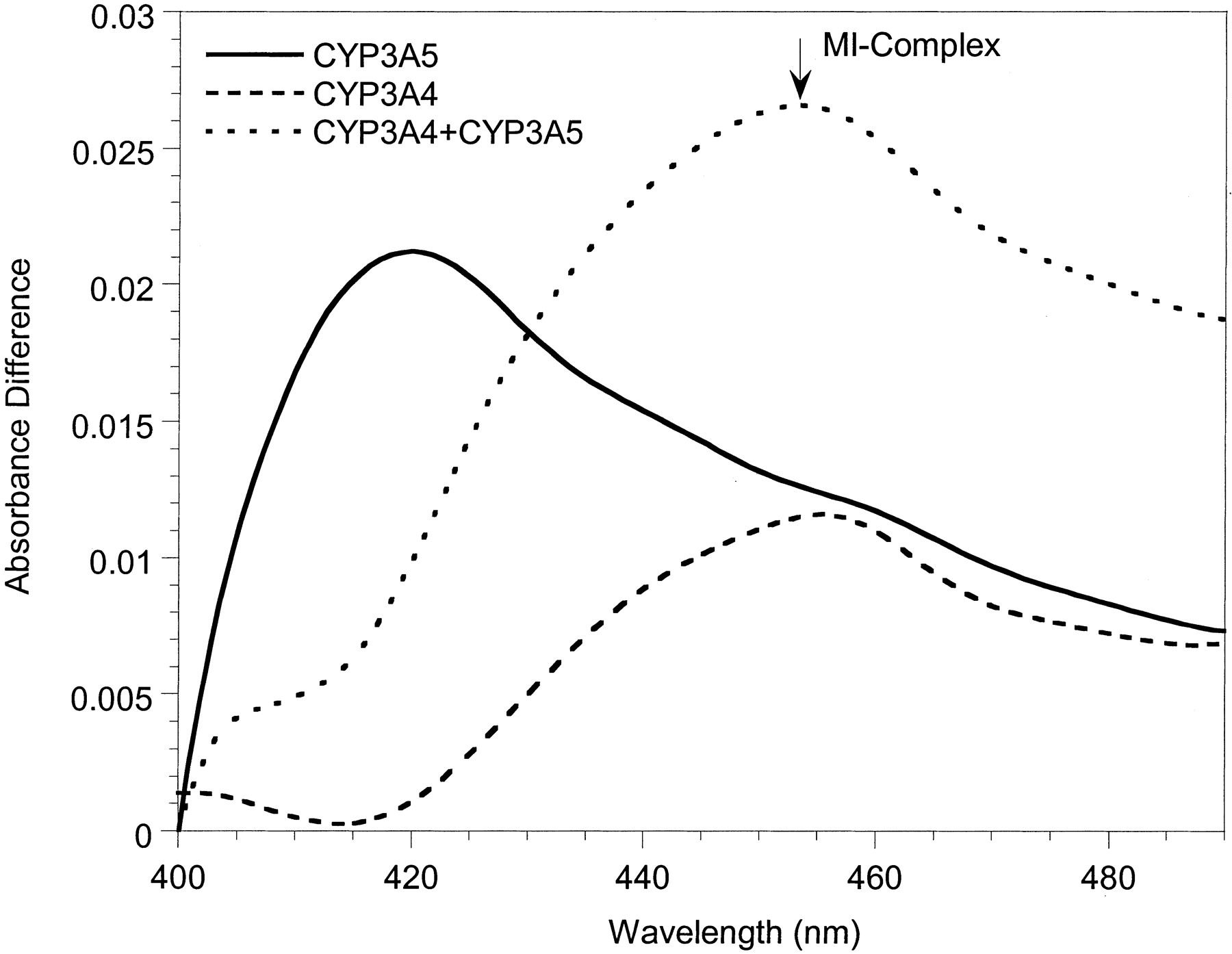
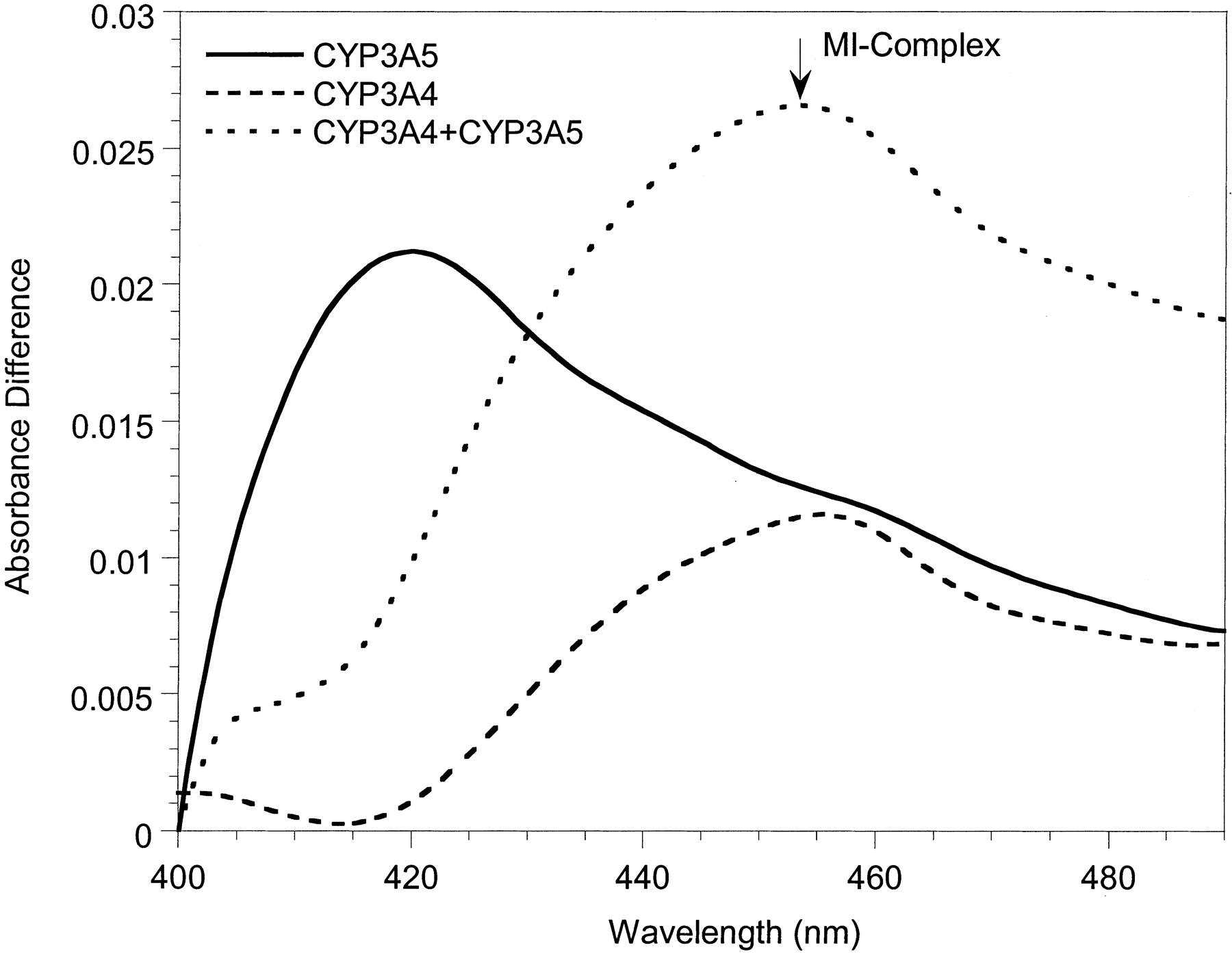
To investigate possible mechanistic differences between the inactivation of CYP3A4 and CYP3A5, spectral analyses were performed with Supersomes and human liver microsomes. MI complex formation (absorbance change between 452 and 455 nm) was used as the surrogate marker for enzyme inactivation. Representative spectra for 3A4-Supersomes and all three inhibitors showed clear absorbance at wavelengths typical of a MI complex (Fig. 3A). Similar data were obtained with 3A4-microsomes (not shown). In contrast, for erythromycin and diltiazem, we were unable to detect MI complex formation with either 3A5-Supersomes (Fig. 3B) or 3A5-microsomes (not shown). Only nicardipine gave a small 452- to 455-nm absorbance change for CYP3A5-containing samples, and it was minimal compared with CYP3A4. The failure to observe a MI complex for erythromycin and CYP3A5 was a surprise given that there was time-dependent loss of catalytic activity for both 3A5-Supersomes and two of the three 3A5-microsomes studied.

Metabolite-intermediate complex formation by erythromycin (50 μM), diltiazem (50 μM), troleandomycin (20 μM), and nicardipine (10 μM) with 200 pmol of 3A4-Supersomes (A) and 3A5-Supersomes (B) (supplemented with 600 pmol of cytochrome b5). Absorbance differences were monitored from 0 to 40 min; t = 40 min scans are shown.
To confirm the binding of erythromycin to CYP3A5, a spectral binding constant (Ks) was determined under experimental conditions similar to that used to detect MI complex formation. A different instrument was employed to achieve greater sensitivity. Type I spectral changes were observed upon addition of erythromycin to 3A4- and 3A5-supersomes (Fig. 4). Ks estimates for erythromycin and 3A4-supersomes (52 ± 4.2 μM) and 3A5-supersomes (48 ± 5.9 μM) were comparable. In addition, incubations performed with erythromycin and NADPH revealed only a moderately higher Km and lower intrinsic formation clearance for the initial N-demethylation reaction catalyzed by CYP3A5 (31 μM and 41 μl/min/nmol), in comparison to CYP3A4 (15 μM and 99 μl/min/nmol).

CYP3A spectral changes induced by erythromycin. Difference spectra were obtained following the addition of substrate (0-450 μM) to 3A4-Supersomes (200 pmol/ml) (solid line) and 3A5-Supersomes (200 pmol/ml) (dashed line) supplemented with cytochrome b5 (600 pmol/ml).
To test the hypothesis that CYP3A5 is deficient at generating downstream (2°, 3°, etc.) oxidative erythromycin metabolites but capable of forming the ultimate MI complex, we conducted an experiment in which 50 μM erythromycin was coincubated with Supersomes: CYP3A5 + CYP3A4 + cytochrome b5 (200:200:1200 pmol), CYP3A5 + cytochrome b5, and CYP3A4 + cytochrome b5. Although erythromycin-dependent MI complex was nondetectable with CYP3A5 alone, the absorbance difference (λ452-490 nm) produced from the CYP3A5 + CYP3A4 coincubation was 61% greater than that observed from an incubation of inhibitor with CYP3A4 alone (Fig. 5).
Increased MI complex formation during coincubation of CYP3A5 + CYP3A4 + cytochrome b5 (200:200:1200 pmol/ml) with 50 μM erythromycin compared with each CYP3A enzyme separately.
Discussion
In comparison to CYP3A4, the catalytic activity of CYP3A5, as reflected by midazolam 1′-hydroxylation, was less susceptible to time-dependent (erythromycin, diltiazem, and nicardipine) and reversible (ketoconazole) inhibition. For all inhibitors studied, except erythromycin, the reversible Ki(app) for 3A5-Supersomes was more than 2-fold higher than the corresponding Ki(app) for 3A4-Supersomes. For all inhibitors except diltiazem, the estimated IC50 for CYP3A5 was significantly higher than that obtained for CYP3A4 when compared using human liver microsomes.
A similar picture emerged from time-dependent experiments to determine kinact using Supersomes. With respect to erythromycin, diltiazem, and nicardipine, CYP3A5 had a slower (∼1/4) maximum rate of enzyme inactivation (kinact) than did CYP3A4. Moreover, results from spectroscopic experiments strongly suggest that CYP3A5 is not as efficient as CYP3A4 at forming a MI complex from erythromycin, diltiazem or nicardipine. Thus, it is possible that CYP3A5 is deficient at forming intermediate metabolites, unable to form the terminal N-oxide metabolite responsible for inactivation, or that the terminal metabolite could not coordinate effectively with the CYP3A5 heme. Furthermore, the pharmacophore for CYP3A5 inhibition is smaller than that for CYP3A4 (Ekins et al., 2003). The consequences of the small pharmacophore would be less energetic feasibility for inhibition by large-molecule inhibitors and a lower likelihood of secondary or tertiary metabolism.
Additional erythromycin binding experiments in the absence of NADPH revealed comparable Ks values for CYP3A4 and CYP3A5, indicative of similar binding affinity of the parent molecule. In the presence of NADPH, CYP3A5-catalyzed erythromycin N-demethylation proceeded with an intrinsic clearance that was one-half that of CYP3A4. Although it was a slower catalyst for the reaction, CYP3A5 was not completely inactive. This result suggests a more profound deficiency with one or more downstream oxidation events. To address the question of CYP3A5 competency for MI complex formation, equal amounts of CYP3A4 and CYP3A5 were incubated with erythromycin and the MI complex was measured. The increased absorbance observed with CYP3A5 + CYP3A4 coincubation confirms that CYP3A5 can complex with a terminal N-oxide metabolite but, again, suggests that it is inefficient at sequential formation of the precursor metabolites. Interestingly, the data also suggest that if there is sufficient CYP3A4 present in vivo, the release of intermediary erythromycin metabolites may cause efficient inactivation of both CYP3A4 and CYP3A5. However, the time course and maximal inhibitory effect might still be different from that observed when only CYP3A4 is expressed in vivo.
Although KI could be estimated from CYP3A4 and CYP3A5 Supersome incubations with all three mechanism-based inhibitors, erythromycin and nicardipine showed no quantitative difference in the inhibitor concentration needed to achieve one-half the maximal CYP3A4 and CYP3A5 inactivation rate. In contrast, diltiazem gave a lower KI for 3A4-Supersomes compared with 3A5-Supersomes. Given that the KI was not different for erythromycin and nicardipine, we can conclude that the ratio of CYP3A5/CYP3A4 kinact (∼1/4) will determine the reduced rate of CYP3A5 inactivation at all inhibitor concentrations.
The CYP3A4 and CYP3A5 KI estimates were greater than the corresponding Ki(app) estimates for erythromycin and diltiazem, but much lower for nicardipine. Theoretically, the Ki(app) obtained from a reversible inhibition experiment should approximate the KI obtained from an enzyme inactivation experiment, if the rate constant for irreversible formation of the first product (leading to inactivated enzyme) is smaller than the rate constant for dissociation of the ES complex. The observation that the Ki(app) and KI were different might be the result of experimental error or indicative of a more complex reaction leading to the MI complex. A Dixon plot of the nicardipine data for 3A4-Supersomes (Fig. 6) indicated an excellent convergence at the Ki(app) (8.5 nM), although the regression lines deviated from the observed data points at the highest nicardipine concentration studied (1 μM). In addition, although there was noise in the nicardipine-3A4 inactivation data (Fig. 7) used to estimate KI (590 nM), it seems unlikely to fully explain the discrepancy with Ki(app).

Dixon plot for the inhibition of 3A4-Supersomes by nicardipine. Inhibition of midazolam 1′-hydroxylation was evaluated under conditions where the inhibitor and substrate were added simultaneously to the enzyme and NADPH without preincubation of the inhibitor with NADPH. The main plot focuses on the intersection of regression lines for estimating KI. The inset shows inhibited rates for all nicardipine concentrations tested (0.005-1 μM).

Determination of irreversible inhibition constants for nicardipine and 3A4-supersomes. Nicardipine (0.02-2 μM) was preincubated with 200 pmol 3A4-supersomes, 600 pmol cytochrome b5, 1 mM EDTA, 1 mM NADPH and 100 mM potassium phosphate buffer, for up to 12 min. At various times after the addition of NADPH, an aliquot was removed and added to 8 μM midazolam solution to assess remaining CYP3A4 catalytic activity.
The data we obtained for time-dependent inactivation of CYP3A4 are in general agreement with what has been published previously, although reported kinact values vary widely. Kanamitsu et al. (2000) determined erythromycin kinact values of 0.062 min-1 and 0.097 min-1 in human liver microsomes and expressed CYP3A4, respectively. The erythromycin values from this study are only slightly lower (0.046 min-1 and 0.042 min-1, respectively). With regard to diltiazem, Jones et al. (1999) obtained a kinact of 0.17 min-1 with expressed CYP3A4, compared with 0.030 min-1 from this study. Ma et al. (2000) obtained a kinact for diltiazem (0.01 min-1) closer to the estimate in this report, but they also reported a result for nicardipine (2 min-1) much higher than we observed. It is possible that the slightly discrepant kinact findings are due to differences in the estimation of the pseudo first order rate constant (λ).
A secondary objective of the studies described was a direct comparison of the time-dependent inactivation kinetics generated from CYP3A Supersomes and human liver microsomes that contain predominantly CYP3A4 or CYP3A5. Both in vitro enzyme systems are commonly used to evaluate in vivo drug-drug interaction potential, although differences in enzyme “environment” could affect sequential inhibitor catalysis and kinetic results. The data we report provide some evidence for such phenomena, although they are incomplete. The most complete results were obtained from incubations with CYP3A4. From this data set, KI parameters for Supersomes and microsomes differed appreciably (15-fold) for diltiazem, but not erythromycin and nicardipine. Similarly, the 3A4-Supersome-microsome difference in kinact was greater for diltiazem (2-fold) than for the other metabolites. Without evidence of confounding factors (i.e., nonspecific binding or inhibitor depletion), one might assume that the liver microsomal data best approximate what would occur in the in vivo state.
To evaluate the validity of our liver microsomal inactivation parameter estimates, we calculated the effect of erythromycin on in vivo CYP3A4-dependent metabolism using a formula presented by Mayhew et al. (2000). The formula estimates the effect of a mechanism-based inactivator on steady-state CYP3A4 levels. Based on our liver microsomal estimates of KI and kinact for erythromycin (10.9 μM and 0.046 min-1, respectively) and an expected unbound plasma concentration of 0.66 μM, we estimate that steady-state CYP3A4 levels would drop approximately 75% during multiple-dose erythromycin therapy. Results from an in vivo study with erythromycin (500 mg, three times a day, for 7 days) and midazolam (0.05 mg/kg, intravenously) indicated a 54% reduction in midazolam clearance following erythromycin treatment (Olkkola et al., 1993). Thus, the in vitro prediction matches qualitatively the observed inhibitory effect.
To summarize, CYP3A4 and CYP3A5 are affected differently in vitro by clinically important drugs, both in terms of enzyme affinity and in the potential for mechanism-based enzyme inactivation. For the time-dependent mechanism, the principal difference between CYP3A4 and CYP3A5 appears to be a reduced ability of CYP3A5 to catalyze sequential oxidative reactions necessary for MI complex formation. In the case of erythromycin, CYP3A5 is capable of generating the first demethylated metabolite and binding the terminal nitroso metabolite to form a MI complex, but it is inefficient at sustaining the intervening sequential oxidative steps. Thus, the magnitude of a CYP3A-dependent drug-drug interaction observed in vivo may depend on the relative substrate turnover in vivo by CYP3A5 compared with CYP3A4 and the duration of inhibitor administration.
Footnotes
-
↵1 Current address: Centre of Excellence for Drug Discovery, Department of Drug Metabolism and Pharmacokinetics, GlaxoSmithKline, Research Triangle Park, NC 27709.
-
↵2 Current address: Department of Pharmaceutical Sciences, St. Jude Children's Research Hospital, Memphis, TN 38103.
-
Supported in part by U.S. Pubic Health Service Grants GM07750, GM32165, GM63666, and ES07033.
-
ABBREVIATIONS:Ki(app), apparent inhibition constant derived from simultaneous incubation of inhibitor with substrate and NADPH; KI, inactivation constant or concentration that produces one-half the maximal irreversible enzyme inactivation rate; kinact, the maximal irreversible enzyme inactivation rate; IC50, inhibitor concentration that produces a 50% reduction in product formation rate at a defined substrate-enzyme concentration.
- Received February 12, 2004.
- Accepted June 23, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}