


Abstract
The clearance route and the absolute values for hepatic and renal clearance of drugs are important criteria for the selection of drug candidates. Based on pharmacokinetic theory, by assuming that uptake is the rate-determining process for the biliary excretion of drugs, organ intrinsic clearance should be simply estimated by the intrinsic uptake. In this study, to investigate whether organ clearance can be predicted from the in vitro uptake activity, we performed uptake experiments using isolated hepatocytes and kidney slices, integration plot analyses, and in vivo pharmacokinetic studies using 12 barely metabolized drugs in rats. The in vivo hepatic and renal clearance could be approximated by uptake clearance estimated from integration plot analyses, except for the renal clearance of some drugs that was relatively small. The comparison of intrinsic uptake clearance from in vitro experiments and integration plot studies revealed that in vivo hepatic uptake was well explained by uptake into isolated hepatocytes, whereas in kidney, in vivo uptake clearance was 10 to 100 times that in kidney slices and a scaling factor is required for its prediction from in vitro experiments. The organ clearance and the fraction excreted into urine could be predicted from in vitro studies except for drugs whose renal clearance was relatively small. This study suggests that the uptake process is the determining factor for organ clearance of minimally metabolized drugs, and uptake assays using isolated hepatocytes and kidney slices are useful for evaluating the uptake clearance.
To select drug candidates rationally in the early stage of drug development, many characteristics of the drugs should be taken into account. The pharmacokinetic property of drugs determines their systemic exposure and local distribution, and thus it is an important factor for optimizing pharmacological and toxicological effects. It is determined by several intrinsic factors such as metabolism, membrane transport, and protein binding, and various in vitro experimental systems have been established (Roberts, 2001; Balani et al., 2005).
Drugs are cleared mainly from the blood circulation by the liver and kidney, and many kinds of metabolic enzymes and transporters are responsible for their elimination (Ito et al., 2005; Shitara et al., 2006). The relative contribution of the liver and kidney to the overall clearance of drugs is important for considering their pharmacological and toxicological effects. Much evidence has indicated that the function of metabolic enzymes and transporters is modified by several factors such as pathophysiological conditions, genetic polymorphisms, and drug-drug interactions (Shitara et al., 2005, 2006; Ieiri et al., 2006; König et al., 2006; Lynch and Price, 2007). Therefore, to avoid large changes in the pharmacokinetics of drugs in unusual circumstances, clearance from multiple elimination pathways in liver and kidney is thought to be a desirable feature for many drugs. For example, the plasma concentration of enalaprilat was significantly increased by reduced renal function, whereas that of temocaprilat was not significantly changed (Oguchi et al., 1993) because enalaprilat is predominantly excreted from the kidney, whereas temocaprilat is excreted from both the liver and kidney, which can minimize the effect of renal dysfunction because of its alternative elimination route by the liver (Ishizuka et al., 1997, 1998). On the other hand, if the pharmacological target is the liver or kidney, efficient targeting of drugs is needed to maximize the pharmacological effect. Pravastatin, a hydrophilic HMG-CoA reductase inhibitor, is efficiently retained in the enterohepatic circulation, which enables the long-term inhibition of HMG-CoA reductase in liver and the avoidance of its systemic side effects such as myopathy (Kitamura et al., 2008b). Antibiotics excreted mainly into bile and urine are selected for the treatment of bile duct inflammation and infection of the urinary tract, respectively (Tsuji, 2006). Thus, the prediction of the elimination route of drugs is essential for the development of desirable drugs. However, in vitro experimental systems for this prediction have not been established yet.
Apparent intrinsic clearance (CLint, app) consists of 1) uptake clearance from blood to an organ (P1), 2) backflux clearance from the organ to the blood (P2), and 3) metabolism, or biliary or urinary excretion (P3) and can be described as eq. 1 (Shitara et al., 2006): 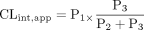
When P3 is much larger than P2, apparent intrinsic clearance can approximate intrinsic uptake clearance (P1). In this case, the uptake clearance is the sole determinant of the overall intrinsic clearance, and therefore the hepatic and renal clearance of drugs can be predicted by uptake clearance.
In the process of the hepatic and renal uptake of organic anions, organic anion transporting polypeptide (Oatp) and organic anion transporter (Oat) family transporters, respectively, which are expressed on the basal membrane, are mainly involved (Shitara et al., 2006). For the characterization of uptake properties of drugs in the liver and kidney, isolated hepatocytes and kidney slices can be used (Shitara et al., 2006). Our group previously demonstrated that tissue uptake clearance of several compounds obtained from a multiple indicator dilution method is well explained by the in vitro uptake clearance into isolated hepatocytes (Miyauchi et al., 1993) in rats. Hasegawa et al. (2003) have shown that rat kidney slices can be useful for predicting the renal uptake clearance of compounds and the relative contribution of Oat1 and Oat3 to their overall uptake. These experimental systems can also be directly applied to humans using cryopreserved human hepatocytes and human kidney slices to predict the hepatic and renal uptake clearance (Hirano et al., 2004; Nozaki et al., 2004).
The purpose of this study was to examine whether hepatic and renal clearance could be predicted simply from in vitro uptake studies using isolated hepatocytes and kidney slices in rats using 12 minimally metabolized anionic drugs from four therapeutic categories (HMG-CoA reductase inhibitors, angiotensin II receptor antagonists, angiotensin-converting enzyme inhibitors, and β-lactam antibiotics), each of which has a different urinary and biliary excreted fraction.
Materials and Methods
Materials. [3H]Pravastatin (44.6 Ci/mmol), [3H]olmesartan (79 Ci/mmol), and [14C]temocaprilat (16.0 mCi/mmol) and unlabeled pravastatin, olmesartan, and temocaprilat were kindly donated by Daiichi-Sankyo Co. (Tokyo, Japan). [3H]Valsartan (81.0 Ci/mmol) and unlabeled valsartan were kindly donated by Novartis Pharma (Basel, Switzerland). [3H]Pitavastatin (16 Ci/mmol) was kindly donated by Kowa Co. (Tokyo, Japan), and [3H]rosuvastatin (79 Ci/mmol) was donated by AstraZeneca (London, UK). Unlabeled pitavastatin was synthesized by Nissan Chemical Industries (Chiba, Japan). Unlabeled rosuvastatin, candesartan, and benazeprilat were purchased from Toronto Research Chemicals (North York, ON, Canada). [3H]Estradiol-17β-glucuronide (E217βG) (53 Ci/mmol), [3H]taurocholate (5.0 Ci/mmol), and [3H]p-aminohippurate (PAH) (4.1 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). [14C]Benzylpenicillin (PCG) (59 mCi/mmol) was purchased from GE Healthcare UK (Little Chalfont, Buckinghamshire, UK). Unlabeled E217βG, taurocholate, PAH, ceftizoxime, and cefmetazole were purchased from Sigma-Aldrich (St. Louis, MO). Unlabeled PCG and enalaprilat were purchased from Wako Pure Chemicals (Osaka, Japan). All other chemicals were of analytical grade and commercially available.
Animals. Female Sprague-Dawley (SD) rats (6–7 weeks old) were purchased from Nippon SLC (Shizuoka, Japan). All animals were maintained under standard conditions with a reverse dark-light cycle and were treated humanely. Food and water were available ad libitum. The studies reported in this manuscript were conducted in accordance with the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan).
In Vivo Pharmacokinetic Study. Female SD rats weighing approximately 170 to 200 g were used for these experiments. Under light ether anesthesia, their femoral artery and vein were cannulated with polyethylene catheters (SP-31; Natsume Seisakusyo, Tokyo, Japan), the bile duct was cannulated with a polyethylene catheter (PE-10; Natsume Seisakusyo) for bile collection, and the bladder was cannulated with a polyethylene tube for urine collection. After the surgical procedures, the rats were placed in a restraining cage (Ballman cage; Natsume Seisakusyo) and allowed to recover from the anesthesia. The rats received a bolus intravenous injection of 0.2 mg/kg pitavastatin, 0.5 mg/kg rosuvastatin, 0.5 mg/kg valsartan, 0.08 mg/kg olmesartan, 0.08 mg/kg candesartan, 0.5 mg/kg temocaprilat, 0.5 mg/kg enalaprilat, 0.5 mg/kg benazeprilat, 2 mg/kg PCG, or 1 mg/kg ceftizoxime or constant infusion of pravastatin (76 mg/min/kg after bolus intravenous injection of 0.67 mg/kg) or cefmetazole (0.1 mg/min/kg) into their femoral vein. Blood samples were collected from the femoral artery at designated times. Bile and urine were collected in preweighed test tubes at designated times. Plasma was prepared by centrifugation of the blood samples (15,000g, 5 min, Microfuge; Beckman Coulter, Fullerton, CA). All samples were stored at –20°C until drug concentrations were measured by LC/MS.
Integration Plot Analysis. Female SD rats weighing approximately 170 to 200 g were used for these experiments. Under light ether anesthesia, their femoral artery and vein were cannulated with polyethylene catheters (SP-31; Natsume Seisakusyo). The rats received a bolus intravenous injection of 0.1 mg/kg pravastatin, 0.2 mg/kg pitavastatin, 0.05 mg/kg rosuvastatin, 0.01 mg/kg valsartan, 0.02 mg/kg olmesartan, 0.02 mg/kg candesartan, 0.01 mg/kg temocaprilat, 0.5 mg/kg enalaprilat, 0.5 mg/kg benazeprilat, 1 mg/kg PCG, 1 mg/kg ceftizoxime, or 2 mg/kg cefmetazole. Blood samples were collected from the femoral artery at 15, 30, 45, 60, 90, and 120 s. The rats were sacrificed at 30, 60, or 120 s after dosing, and their livers and kidneys were immediately removed. Plasma was prepared by centrifugation of the blood samples (15,000g, 5 min, Microfuge). Phosphate-buffered saline was added to tissue samples and homogenized to make a 30% homogenate. All samples were stored at –20°C until drug concentrations were measured by LC/MS.
Determination of the Plasma Protein-Unbound Fraction and Blood/Plasma Concentration Ratio. Binding of drugs to plasma proteins was determined by an ultrafiltration method. Plasma was obtained by the centrifugation of blood from female SD rats. Drugs were individually added to the plasma samples, and they were incubated together at 37°C for 5 min. Following the manufacturer's protocol, the specimen was directly applied to Centri-free micropartition devices (Millipore Corporation, Billerica, MA). The concentrations of the drugs in the filtrate and the plasma before filtration were determined by LC/MS. The adsorption of the drugs on the membrane was confirmed to be negligible.
To determine the blood/plasma concentration ratio (RB) values, blood was obtained from female SD rats. Drugs were added to the blood samples individually, and they were incubated together at 37°C for 5 min. Plasma was prepared by centrifugation of the blood samples (15,000g, 5 min, Microfuge). The concentrations of the drugs in the blood and the plasma samples were determined by LC/MS. The protein-unbound fraction in the blood (fB) was calculated by dividing the protein-unbound fraction in plasma (fu) by RB.
Uptake Study Using Rat Freshly Isolated Hepatocytes. Isolation of hepatocytes and an uptake study were conducted as described previously (Yamazaki et al., 1993). Isolated hepatocytes (viability >85%) were suspended in Krebs-Henseleit buffer (118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2, pH 7.4) and stored on ice. Before the uptake study, hepatocytes were preincubated at 37°C for 3 min, and the uptake reaction was started by adding drugs to the hepatocyte suspension. After a designated time, the reaction was terminated by separating the cells from the medium using a centrifugal filtration technique. For this purpose, a 100-μl aliquot of incubation mixture was placed in a 0.4-ml centrifuge tube (Sarstedt, Numbrecht, Germany) containing 50 μl of 2 N sodium hydroxide for radiolabeled compounds or 100 μl of 5 M sodium acetate for unlabeled compounds under a 100-μl layer of an oil mixture (density, 1.05, mixture of silicone oil and mineral oil; Sigma-Aldrich). Samples were then centrifuged for 10 s in a Microfuge. During this process, the hepatocytes pass through the oil layer into the aqueous solution (2 N NaOH or 5 M CH3COONa). In the case of unlabeled compounds, tubes were frozen in liquid nitrogen immediately after centrifugation and stored at –20°C until drug measurement.
The concentrations of pravastatin, pitavastatin, rosuvastatin, valsartan, olmesartan, temocaprilat, PCG, E217βG, and taurocholate were determined by measuring their radioactivity. After overnight incubation at room temperature to dissolve the cells in alkali, the centrifuge tube was cut, and each compartment was transferred to a scintillation vial. The compartment containing dissolved cells was neutralized with 50 μl of 2 N hydrochloric acid and mixed with scintillation cocktail (Clearsol II; Nakalai Tesque, Kyoto, Japan), and the radioactivity was determined in a liquid scintillation counter (LS6000SE; Beckman Coulter). The concentrations of candesartan, benazeprilat, enalaprilat, ceftizoxime, and cefmetazole were determined by LC/MS. Cells in 5 M sodium acetate buffer were taken from the centrifuge tube and sonicated in a new tube to break them down. This sample was used for the measurement of drug concentrations by LC/MS.
Uptake Study Using Rat Kidney Slices. An uptake study using rat kidney slices was performed as described previously (Hasegawa et al., 2002). Kidney slices (300-μm thick) from female SD rats were kept in ice-cold buffer (120 mM NaCl, 16.2 mM KCl, 1 mM CaCl2, and 1.2 mM MgSO4 in 10 mM NaH2PO4-Na2HPO4, pH 7.5). Two slices, each weighing 15 to 25 mg, were randomly selected and then incubated in each well of a 12-well plate with 1 ml of oxygenated buffer after preincubation of slices for 5 min at 37°C. After incubation for designated periods, each slice was rapidly removed from the incubation buffer, washed with ice-cold buffer, blotted on filter paper, and weighed.
The concentrations of pravastatin, pitavastatin, rosuvastatin, valsartan, olmesartan, temocaprilat, PCG, and PAH were determined by measuring their radioactivity. The slice was dissolved in 1 ml of Soluene 350 (PerkinElmer Life and Analytical Sciences). The radioactivity in scintillation cocktail (Hionic-Fluor; PerkinElmer Life and Analytical Sciences) was determined by liquid scintillation counting (LS6000SE; Beckman Coulter). The concentrations of candesartan, benazeprilat, enalaprilat, ceftizoxime, and cefmetazole were determined by LC/MS. PBS (100 μl) was added to the slices followed by sonication to break them down. This sample was used for the measurement of drug concentration by LC/MS.
Quantification of Drug Concentration by LC/MS. Samples were precipitated with 3 volumes of acetonitrile (for in vivo samples and kidney slices) or methanol (for hepatocytes) and centrifuged at 15,000g at 4°C for 10 min. The supernatants were subjected to LC/MS. An LCMS-2010 EV equipped with a Prominence LC system (Shimadzu, Kyoto, Japan), an Alliance HT 2795 separation module with an autosampler (Waters, Milford, MA), and a Micro-mass ZQ mass spectrometer with an electron ion spray interface (Waters) were used. In the measurement using Shimadzu equipment, the interface voltage was 3.5 kV, and the nebulizer gas (N2) flow was 1.5 L/min. The heat block and curved desolvation line temperatures were 200 and 150°C, respectively. In the measurement using Waters equipment, the desolvation temperature was 350°C, and the capillary voltage was 3.8 kV. Detailed conditions for the measurement of each compound are shown in Supplemental Table S1.
Pharmacokinetic Analysis.In vivo study. The area under the plasma concentration-time profile over 120 min (AUC0–120) was calculated using a trapezoidal method. The plasma concentration-time profile was fitted to the two exponential equations using a nonlinear iterative least-squares method using MULTI software (Yamaoka et al., 1981) and AUC0–∞ was estimated by integration of the fitted equation from time 0 to infinity. The plasma clearance (CLtot, p), the biliary clearance based on the drug concentration in plasma (CLbile, p), and the renal clearance based on the drug concentration in plasma (CLrenal, p) were calculated using eqs. 2–4: 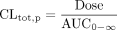

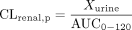 where Xbile represents the cumulative excreted amount in bile over 120 min and Xurine represents the amount in urine over 120 min.
where Xbile represents the cumulative excreted amount in bile over 120 min and Xurine represents the amount in urine over 120 min.
When the drugs were infused intravenously, CLtot, p, CLbile, p, and CLrenal, p were calculated using eqs. 5–7: 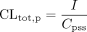
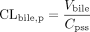
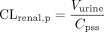 where I represents the infusion rate, Cpss represents the drug concentration in plasma at steady state, Vbile represents the biliary excretion rate at steady state, and Vurine represents the urinary excretion rate at steady state. Cpss was determined as the mean value of the plasma concentration at 30, 60, 90, and 120 min. Vbile and Vurine were determined as the mean value of the renal excretion rate from 30 to 60, 60 to 90, and 90 to 120 min. The clearances based on drug concentrations in whole blood (CLtot, B, CLbile, B, and CLrenal, B) were calculated by dividing CLtot, p, CLbile, p and CLrenal, p by RB, respectively.
where I represents the infusion rate, Cpss represents the drug concentration in plasma at steady state, Vbile represents the biliary excretion rate at steady state, and Vurine represents the urinary excretion rate at steady state. Cpss was determined as the mean value of the plasma concentration at 30, 60, 90, and 120 min. Vbile and Vurine were determined as the mean value of the renal excretion rate from 30 to 60, 60 to 90, and 90 to 120 min. The clearances based on drug concentrations in whole blood (CLtot, B, CLbile, B, and CLrenal, B) were calculated by dividing CLtot, p, CLbile, p and CLrenal, p by RB, respectively.
Integration plot analysis. The AUC0–t was calculated using a trapezoidal method. The tissue uptake amount of drugs per gram of tissue (Xt) normalized by the plasma concentration (Cp) can be described as eq. 8:  where CLuptake, p represents tissue uptake clearance based on the drug concentration in plasma and Xt(0)/Cp(0) represents the initial distribution volume. Based on eq. 8, the AUC0–t/Cp(t) value was plotted against the Xt(t)/Cp(t) value, and each plot was fitted to the straight line using a nonlinear iterative least-squares method. CLuptake, p was obtained as a slope of the fitted line. The uptake clearance based on drug concentration in whole blood (CLuptake, B) was calculated by dividing CLuptake, p by RB. Because renal clearance includes glomerular filtration and tubular secretion in the kidney, the tubular secretion clearance, which corresponds to in vitro uptake clearance in kidney slices, was estimated by subtracting fB × glomerular filtration rate (GFR) (12 ml/min/kg) from CLuptake, B.
where CLuptake, p represents tissue uptake clearance based on the drug concentration in plasma and Xt(0)/Cp(0) represents the initial distribution volume. Based on eq. 8, the AUC0–t/Cp(t) value was plotted against the Xt(t)/Cp(t) value, and each plot was fitted to the straight line using a nonlinear iterative least-squares method. CLuptake, p was obtained as a slope of the fitted line. The uptake clearance based on drug concentration in whole blood (CLuptake, B) was calculated by dividing CLuptake, p by RB. Because renal clearance includes glomerular filtration and tubular secretion in the kidney, the tubular secretion clearance, which corresponds to in vitro uptake clearance in kidney slices, was estimated by subtracting fB × glomerular filtration rate (GFR) (12 ml/min/kg) from CLuptake, B.
In the dispersion model, organ clearance (CLorg) is expressed as a function of the intrinsic clearance (CLint), organ blood flow rate (Q), fB, and dispersion number (DN): 

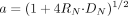
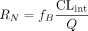
The blood flow rate in the liver was set at 60 ml/min/kg and in the kidney at 40 ml/min/kg (Davies and Morris, 1993), and DN was set at 0.17 (Roberts and Rowland, 1986; Iwatsubo et al., 1996). Because the in vivo intrinsic uptake clearance from blood to tissue (P1vivo) can be regarded as the organ intrinsic clearance (CLint) when the rate-limiting step of the overall clearance is the uptake process, P1vivo (= CLint) was calculated by assigning the CLuptake, B value to the CLorg in eqs. 9 to 12. When P1vivo values were calculated, drugs whose clearance was close to the blood flow (extraction ratio >0.7) were excluded because we cannot obtain the accurate P1vivo value. Detailed procedures for calculation of the parameters for the in vivo uptake clearance of valsartan as an example can be found in the Supplemental Appendix and Supplemental Fig. S1.
In vitro studies. The in vitro intrinsic uptake clearance (P1vitro) was calculated by dividing initial uptake velocity by the drug concentration in the incubation buffer. The initial uptake velocity of the drugs was calculated as a slope of the uptake volume at 0.5 and 1 min in isolated hepatocytes and at 5 min and 10 min in kidney slices. CLorg was calculated based on eqs. 9 to 12, assuming that P1vitro was equal to CLint. In the case of liver, the calculated CLorg is expressed as the predicted hepatic clearance (CLh, predicted). In the case of kidney, the calculated CLorg corresponds to the secretion clearance, and the predicted renal clearance (CLr, predicted) was calculated using eq. 13: 

A simple mathematical model for considering the permeation of drugs in kidney slices. The relationship between the true intrinsic uptake clearance (P1KID, corrected) and the observed uptake clearance in kidney slices (P1KID) was explained theoretically by the simple mathematical model as shown in this figure. The details are described under Materials and Methods.
The detailed calculation procedures of the pharmacokinetic parameters estimated from in vitro assay of valsartan as an example can be found in the Supplemental Appendix and Supplemental Figs. S2 and S3. The predicted value of the fraction excreted into urine (furine, predicted) was calculated using eq. 14:  In the process of in vitro-in vivo scale-up, we used the following parameters: 1.25 × 108 hepatocytes/g of liver, 38.3 g of liver/kg b.wt., and 7.27 g of kidney/kg b.wt.
In the process of in vitro-in vivo scale-up, we used the following parameters: 1.25 × 108 hepatocytes/g of liver, 38.3 g of liver/kg b.wt., and 7.27 g of kidney/kg b.wt.
Simple mathematical model for explaining the relationship between the true intrinsic uptake clearance (P1KID, corrected) and observed uptake clearance inkidney slices (P1KID). One of the possible reasons for the discrepancy between predicted and observed intrinsic uptake clearance in the kidney was that the drug concentration inside the cells of kidney slices was lower than that in the buffer because a kidney slice consists of multiple cell layers and drugs cannot easily penetrate the multilayered cells (see also Results). Therefore, to explain the relationship between the true intrinsic uptake clearance (P1KID, corrected) and the observed uptake clearance in kidney slices (P1KID), we constructed the simple mathematical model as shown in Fig. 1.
We assumed that a kidney slice consists of multiple cell monolayers (number of monolayers = N), and that V, Cn, and Xn represent the volume of extracellular fluid compartment for each layer, the drug concentration in an extracellular fluid compartment at the nth layer, and the amount of drug taken up into an extracellular fluid compartment at the nth layer. In this model, we assumed that the drug is unidirectionally transported from an extracellular fluid compartment at the nth layer to that at the (n + 1)th layer with a clearance of P2 and that the drug is also taken up into an intracellular compartment at each layer from an extracellular fluid compartment at the same layer with a clearance of P1′. We also assumed that the drug concentration in an extracellular fluid compartment at the first layer is regarded as that in the incubation medium (set as A).
In this condition, the mass-balance equation for an extracellular fluid compartment at the nth layer [n = 2 ∼ (N – 1)] can be described as the following equation:  The equation for an extracellular fluid compartment at the Nth layer is as follows:
The equation for an extracellular fluid compartment at the Nth layer is as follows: 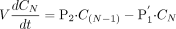 At the steady state, eqs. 15 and 16 can be converted to eqs. 17 and 18:
At the steady state, eqs. 15 and 16 can be converted to eqs. 17 and 18: 
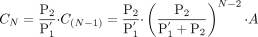
The velocity of drug uptake into an intracellular compartment at the nth layer is as follows (eq. 19): 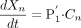 The velocity of drug uptake into the whole kidney slice (v) can be described as the sum of the uptake velocity into an intracellular compartment at each layer.
The velocity of drug uptake into the whole kidney slice (v) can be described as the sum of the uptake velocity into an intracellular compartment at each layer. 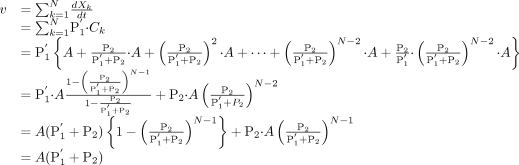 Thus, the uptake clearance in the whole kidney slice (P1KID) is as follows:
Thus, the uptake clearance in the whole kidney slice (P1KID) is as follows: 
P1′ is defined as the intrinsic uptake clearance for each cell monolayer in a kidney slice, whereas P1KID, corrected is defined as the “true” intrinsic uptake clearance for the whole kidney slice.  Thus, when eq. 22 is assigned to eq. 21, the following equation can be obtained:
Thus, when eq. 22 is assigned to eq. 21, the following equation can be obtained: 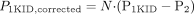
Results
Measurement of in Vivo Organ Clearances in Rats. The pharmacokinetic parameters for 12 drugs after intravenous administration to female SD rats were calculated and are shown in Table 1. Ten drugs, except for pravastatin and cefmetazole, were administered by intravenous bolus injection. It was thought to be difficult to measure the plasma concentration of pravastatin quantitatively because of its rapid elimination from blood circulation after intravenous bolus administration. It was difficult to measure the plasma concentration of cefmetazole after intravenous bolus administration because of its low sensitivity in LC/MS analysis. Therefore, pravastatin and cefmetazole were administered by constant intravenous infusion. At 120 min after bolus administration or at reaching the steady state in the intravenous infusion study (cefmetazole and pravastatin), more than 80% of the dose was excreted into bile and urine in an unchanged form, except for pitavastatin (64%), suggesting that drugs used in this study were mainly eliminated from the body in an unchanged form in female SD rats. We also measured the plasma fu and RB of each drug; the results are shown in Table 2.
Pharmacokinetic parameters of 12 drugs in female SD rats
Female SD rats received a bolus intravenous injection of 0.2 mg/kg pitavastatin, 0.5 mg/kg rosuvastatin, 0.5 mg/kg valsartan, 0.08 mg/kg olmesartan, 0.08 mg/kg candesartan, 0.5 mg/kg temocaprilat, 0.5 mg/kg enalaprilat, 0.5 mg/kg benazeprilat, 2 mg/kg PCG, and 1 mg/kg ceftizoxime or constant infusion of pravastatin (76 mg/min/kg after intravenous administration of 0.67 mg/kg) and cefmetazole (0.1 mg/min/kg). The concentrations of drugs in plasma, bile, and urine were measured, and pharmacokinetic parameters were calculated. Each value is expressed as mean ± S.E. of four to five rats. No. for each compound corresponds to the numerical coding attached to each plot in all the figures.
R B and f u and f B for each compound in female SD rats
fu and RB values were measured and fB was calculated by dividing fu by RB. Each value is expressed as mean ± S.E. of triplicate determinations.
Measurement of the in Vivo Tissue Uptake Clearance Obtained from Integration Plot Analyses. To estimate the in vivo tissue uptake clearance of drugs directly, integration plot analyses were performed. The tissue uptake clearance from blood to organ (liver or kidney) was calculated as the slope of the fitted line of the integration plot as described under Materials and Methods and is shown in Table 3.
Tissue uptake clearance of drugs obtained from integration plot analyses
CLuptake, p (milliliters per minute per gram of tissue) obtained from the slope of an integration plot was converted to CLuptake, B (milliliters per minute per kilogram) using the weight of rat liver (38.3 g of liver/kg b.wt.) and kidney (7.27 g of kidney/kg b.wt.), and RB values. Each value is expressed as mean ± computer-calculated S.D. of three to four rats.
Comparison between Organ Clearance (CLbile, B and CLrenal, B)and Tissue Uptake Clearance (CLuptake, B, liver and CLuptake, B, kidney)in Vivo. To show the importance of uptake clearance as a determinant factor for organ clearance, the organ clearance (CLbile, B and CLrenal, B) was compared with the respective tissue uptake clearance (CLuptake, B, liver and CLuptake, B, kidney) (Fig. 2). In the liver, CLbile, B values for 6 and 9 of 12 compounds were correlated with CLuptake, B, liver values within a 2- and 3-fold range of difference, respectively (Fig. 2A). On the other hand, in the kidney, CLrenal, B values for 6 and 8 of 12 compounds were correlated with CLuptake, B, kidney values within a 2- and 3-fold range of difference, respectively, but CLrenal, B values for pitavastatin, valsartan, olmesartan, and candesartan, whose CLrenal, B values were relatively small among test compounds, were much smaller than CLuptake, B, kidney values (Fig. 2B).
Estimation of the Uptake Clearance from in Vitro Uptake Studies Using Isolated Hepatocytes and Kidney Slices. Time-dependent uptake of drugs was measured in the presence of tracer and excess amount of compounds (for hepatocytes: 0.1 and 100 μM for [3H]pravastatin, [3H]pitavastatin, [3H]rosuvastatin, [3H]valsartan, and [3H]olmesartan; 6.3 and 300 μM for [14C]temocaprilat; 1.7 μM and 1 mM for [14C]PCG; and 10 μM and 1 mM for enalaprilat, benazeprilat, ceftizoxime, and cefmetazole; for kidney slices: 0.1 μM and 1 mM for [3H]pravastatin; 0.1 and 100 μM for [3H]pitavastatin, [3H]rosuvastatin, [3H]valsartan, and [3H]olmesartan; 6.3 and 300 μM for [14C]temocaprilat; 1.7 μM and 1 mM for [14C]PCG; 10 μM and 1 mM for enalaprilat, benazeprilat, and cefmetazole; and 10 μM and 10 mM for ceftizoxime). All of the drugs were taken up linearly into isolated rat hepatocytes within 1 or 2 min and into kidney slices within 15 min under tracer conditions (data not shown). In all cases, the uptake clearance into isolated hepatocytes or kidney slices was decreased in the presence of an excess of compounds, suggesting that their uptake is saturable and mediated by transporters. The uptake clearance of all drugs in isolated hepatocytes and kidney slices is shown in Tables 4 and 5.
Uptake clearance of drugs in rat isolated hepatocytes
Time-dependent uptake of drugs in rat isolated hepatocytes was measured in the presence of tracer amount of drugs in the incubation buffer (0.1 μM for [3H]pravastatin, [3H]pitavastatin, [3H]rosuvastatin, [3H]valsartan, and [3H]olmesartan; 6.3 μM for [14C]temocaprilat; 1.7 μM for [14C]PCG; 10 μM for enalaprilat, benazeprilat, ceftizoxime, and cefmetazole). The in vitro intrinsic uptake clearance (P1 hep) was calculated by dividing initial uptake velocity by the drug concentration in the incubation buffer. Each value is expressed as mean ± S.E. of triplicate determinations.
Uptake clearance of drugs in rat kidney slices
Time-dependent uptake of drugs into rat kidney slices was measured in the presence of tracer amount of drugs in the incubation buffer (0.1 μM for [3H]pravastatin, [3H]pitavastatin, [3H]rosuvastatin, [3H]valsartan, and [3H]olmesartan; 6.3 μM for [14C]temocaprilat; 1.7 μM for [14C]PCG; 10 μM for enalaprilat, benazeprilat, cefmetazole, and ceftizoxime). The in vitro intrinsic uptake clearance (P1 kid) was calculated by dividing initial uptake velocity by the drug concentration in the incubation buffer. Each value is expressed as mean ± S.E. of triplicate determinations.
Comparison of the Intrinsic Uptake Clearance Obtained from Integration Plot Analyses with That from in Vitro Studies. To examine whether the in vivo intrinsic uptake clearance (P1vivo) obtained from integration plot analyses could be predicted from an in vitro uptake assay (P1HEP for isolated hepatocytes and P1KID for kidney slices), the P1vivo values were compared with the P1HEP and P1KID values (Fig. 3). For this analysis, drugs whose clearance was close to the blood flow (extraction ratio >0.7) were excluded because the absolute value of blood flow rate sensitively affects the calculated P1vivo values for these drugs, and we cannot obtain the accurate ones. Thus, we did not use the data of five (pravastatin, pitavastatin, rosuvastatin, temocaprilat, and PCG) and five (pravastatin, temocaprilat, benazeprilat, cefmetazole and PCG) compounds in the plots for liver and kidney, respectively. In the liver, P1vivo values could be well predicted from in vitro P1HEP values (Fig. 3A). On the other hand, for kidney, the P1vivo value for each compound was approximately 10 to 100 times larger than the in vitro P1KID value (Fig. 3B). One of the possible reasons for the discrepancy is that the drug concentration inside the cells of kidney slices was lower than that in the buffer because a kidney slice consists of multiple cell layers and drugs cannot easily penetrate the multilayered cells, whereas in the physiological condition, drugs can access every cell through blood perfusion. Therefore, the uptake clearance calculated by the uptake amount per unit weight of kidney slice normalized by the drug concentration in the medium might underestimate the true intrinsic uptake clearance. To mimic this situation, we constructed the simple model to explain the relationship between in vitro P1KID and in vivo P1vivo values and estimate the true intrinsic uptake clearance (P1KID, corrected) based on the results of an in vitro uptake assay using kidney slices (see Materials and Methods). Because of analyses using this model, the relationship between P1KID and P1KID, corrected could be described as the following equation by fitting the in vitro P1KID and in vivo P1vivo values for each compound to eq. 23: 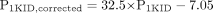 Thus, the P1KID, corrected values were well explained by the P1vivo values and used for the further analyses as an intrinsic uptake clearance in kidney.
Thus, the P1KID, corrected values were well explained by the P1vivo values and used for the further analyses as an intrinsic uptake clearance in kidney.
Prediction of the Organ Clearance from in Vitro Uptake Clearance. Based on the intrinsic uptake clearance obtained from in vitro studies (P1HEP and P1KID), the hepatic and renal clearances of all test compounds were calculated by the simple scale-up method and dispersion model. In the case of kidney, P1KID, corrected values instead of P1KID values were used for intrinsic uptake clearance. The predicted organ clearances were compared with the in vivo observed values (Fig. 4). As a result, the hepatic and renal clearance could be predicted from the results of the in vitro uptake assay using isolated hepatocytes and kidney slices, except for the renal clearances of pitavastatin, valsartan, olmesartan, and candesartan, which were relatively small.
Prediction of the Elimination Routes of Drugs from the in Vitro Uptake Assay. Using the predicted hepatic and renal clearances (CLh, predicted and CLr, predicted), the fraction excreted into urine (furine) was calculated by eq. 14 and compared with the observed values. As shown in Fig. 5, furine values for nine compounds could be reasonably predicted from the results of the in vitro assay, but in the case of pitavastatin, valsartan, olmesartan, and candesartan, the predicted furine values overestimate the real furine values because of the underestimation of the renal clearances predicted from the in vitro assay.

Comparison between in vivo organ clearance (CLbile, B and CLrenal, B) and organ uptake clearance (CLuptake, B, liver and CLuptake, B, kidney) in liver and kidney. CLuptake, B values for the liver and kidney were obtained from integration plot analyses. CLbile, B and CLuptake, B, liver (for the liver) are plotted in A, and CLrenal, B and CLuptake, B, kidney (for the kidney) are plotted in B. An enlarged view of B (range of x-axis and y-axis: 1–100 ml/min/kg; plots inside the circle are shown in C. Plots represent the following: 1, pravastatin; 2, pitavastatin; 3, rosuvastatin; 4, valsartan; 5, olmesartan; 6, candesartan; 7, temocaprilat; 8, enalaprilat; 9, benazeprilat; 10, PCG; 11, ceftizoxime; and 12, cefmetazole. The solid line represents the line of unity, and the dashed lines represent the lines of 1:2 and 2:1 correlations.
Discussion
In this study, to demonstrate whether the hepatic and renal clearances of hardly metabolized drugs can be predicted from the results of an in vitro uptake assay using isolated hepatocytes and kidney slices, assuming that the intrinsic uptake clearance approximates the organ intrinsic clearance, we investigated the relationship between the predicted organ clearance calculated from in vivo integration plot analyses and in vitro uptake assays and the observed organ clearance for 12 drugs in rats. These drugs are anionic at neutral pH and are excreted mainly into bile or urine in an unchanged form with minimal metabolism. In addition, these drugs are known to be substrates of drug transporters such as Oatps and Oats.
To examine the importance of the tissue uptake process in the overall clearance of test drugs, the organ clearance (CLbile, B or CLrenal, B) was compared with the respective uptake clearance measured by integration plot analyses (CLuptake, B, liver or CLuptake, B, kidney) in rats. CLbile, B was comparable to CLuptake, B, liver, suggesting that the hepatic uptake of compounds was thought to be a determinant factor for overall hepatic clearance (Fig. 2A), whereas CLrenal, B was comparable to CLuptake, B, kidney for eight drugs, but not for pitavastatin, valsartan, olmesartan, or candesartan (Fig. 2B). The renal clearance of these exceptional drugs was relatively small and underestimates their CLuptake, B, kidney values, suggesting that the uptake process was not a rate-limiting step in their overall renal clearance. Thus, it is possible that backflux from kidney to blood is larger than elimination from kidney to urine or that significant reabsorption from urine to blood is involved in their renal excretion.

Comparison between in vivo intrinsic uptake clearance (P1vivo) and in vitro intrinsic uptake clearance (P1HEP and P1KID) in liver and kidney. P1vivo values were calculated from CLuptake, B values obtained from integration plot analyses. P1vivo, liver and P1HEP values (for the liver) are plotted in A, and P1vivo, kidney and P1KID values (for the kidney) are plotted in B. A, the solid line represents the line of unity and the dashed lines represent the lines of 1:2 and 2:1 correlations. B, the solid curve represents the theoretical value calculated from eq. 24 derived from the simple mathematical model (see Materials and Methods). The solid line represents the line of unity, and the dashed lines represent the lines of 1:10 and 1:100 correlations as indicated. Plots represent the following: 1, pravastatin; 2, pitavastatin; 3, rosuvastatin; 4, valsartan; 5, olmesartan; 6, candesartan; 7, temocaprilat; 8, enalaprilat; 9, benazeprilat; 10, PCG; 11, ceftizoxime; and 12, cefmetazole. Please note that 1, 2, 3, 7, and 10 and 1, 7, 9, and 10 were not plotted in Fig. 3, A and B, respectively, because we cannot obtain the accurate P1vivo values for drugs with a high extraction ratio (>0.7). The details are described under Results.
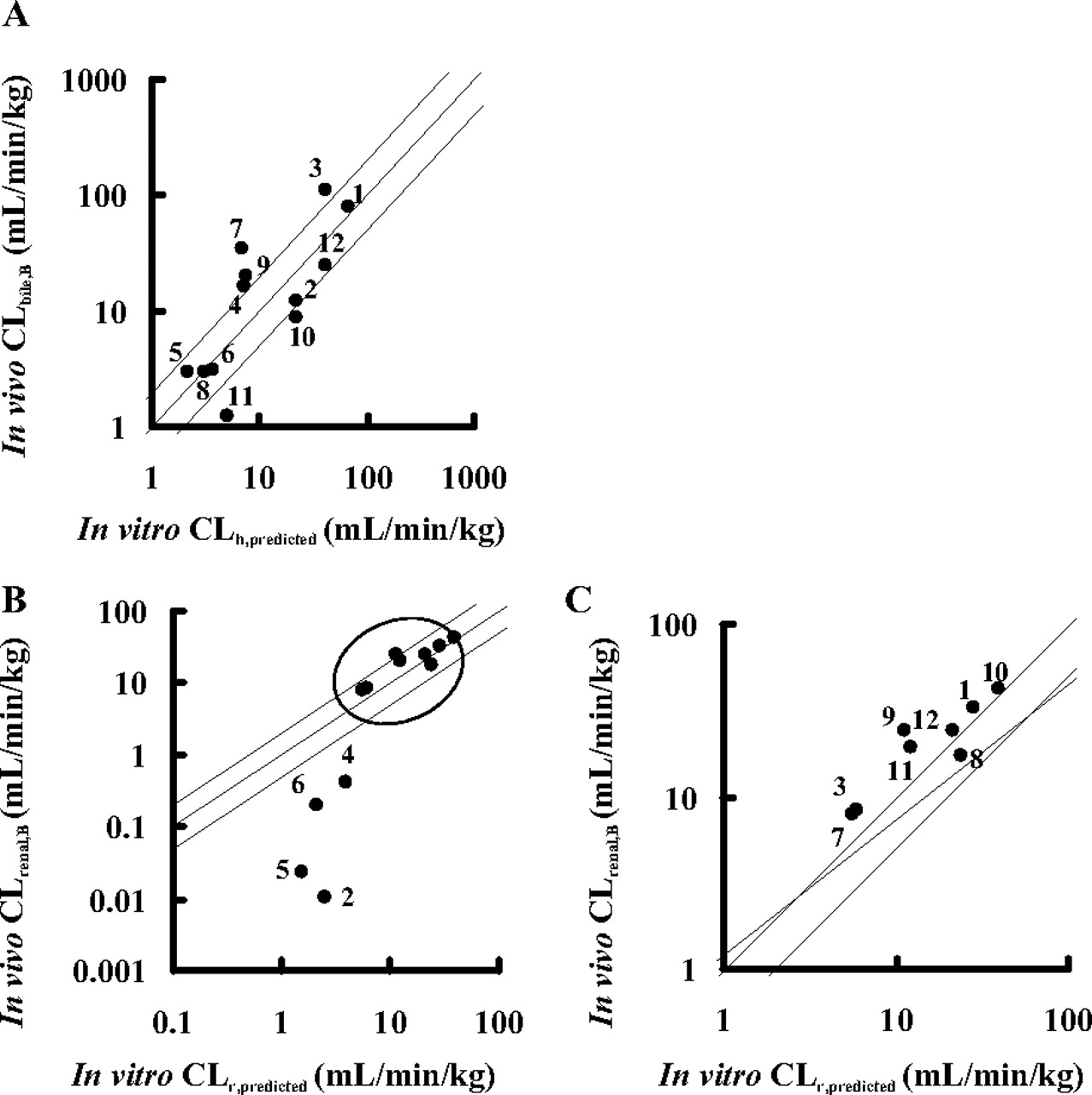
Comparison between the observed (CLbile, B and CLrenal, B) and the predicted organ clearance (CLh, predicted and CLr, predicted) in liver and kidney. CLh, predicted and CLr, predicted were calculated based on eqs. 9 to 12, assuming that uptake clearance obtained from rat-isolated hepatocytes and kidney slices was equal to the intrinsic clearance in each organ (CLint). For calculation of CLr, predicted, the true intrinsic uptake clearance (P1KID, corrected), corrected based on the simple model analysis to explain the relationship between in vitro P1KID and in vivo P1vivo values, was used. The details are described under Materials and Methods. CLbile, B and CLh, predicted (for the liver) are plotted in A, and CLrenal, B and CLr, predicted (for the kidney) are plotted in B. The enlarged view of B (range of x-axis and y-axis: 1–100 ml/min/kg; plots inside the circle) is shown in C. Plots represent the following: 1, pravastatin; 2, pitavastatin; 3, rosuvastatin; 4, valsartan; 5, olmesartan; 6, candesartan; 7, temocaprilat; 8, enalaprilat; 9, benazeprilat; 10, PCG; 11, ceftizoxime; and 12, cefmetazole. The solid line and the dashed lines represent the line of unity and the lines of 1:2 and 2:1 correlations, respectively.
Next, to examine whether the in vivo uptake clearance could be predicted from in vitro uptake assays, the intrinsic uptake clearance obtained from integration plot analyses (P1vivo) was compared with the in vitro uptake clearance (P1HEP or P1KID) (Fig. 3). P1HEP was almost comparable to P1vivo, liver (Fig. 3A), indicating that hepatic intrinsic uptake clearance can be predicted from in vitro uptake studies. Previous reports have also demonstrated that the in vivo uptake clearance collected using a multiple indicator dilution method and integration plot analysis could be predicted from the in vitro uptake clearance using isolated hepatocytes by simple scale-up calculations (Miyauchi et al., 1993; Kato et al., 1999). On the other hand, because it is difficult to isolate the renal tubular cells, kidney slices are proposed as a good in vitro tool for the characterization of renal uptake. Hasegawa et al. (2003) characterized active uptake of drugs and established a methodology for examining the contribution of Oat1 and Oat3 to the overall renal uptake of drugs. However, it remains to be clarified whether the in vivo renal clearance can be predicted using in vitro uptake assays. As a result, P1vivo, kidney was 10 to 100 times larger than P1KID (Fig. 3B). We hypothesized that this apparent discrepancy was caused by the decreased exposure of drugs to the inner cell layer of kidney slices. Then, to determine the theoretical relationship between the apparent uptake clearance per unit weight of kidney slices and true intrinsic clearance, we constructed the simple mathematical model and derived eq. 24 (see Materials and Methods). The slope of this equation (32.5) represents half of the number of cell layers in a kidney slice, assuming that the drug-containing buffer can access both sides of the kidney slice equally. The thickness of the kidney slice was approximately 300 μm, and the size of cells is generally 3 to 10 μm, suggesting that a kidney slice consists of 30 to 100 cell layers, which is comparable to the estimated number of cell layers. Thus, we corrected the uptake clearance in kidney slices to the true intrinsic uptake clearance (P1KID, corrected) using eq. 24.
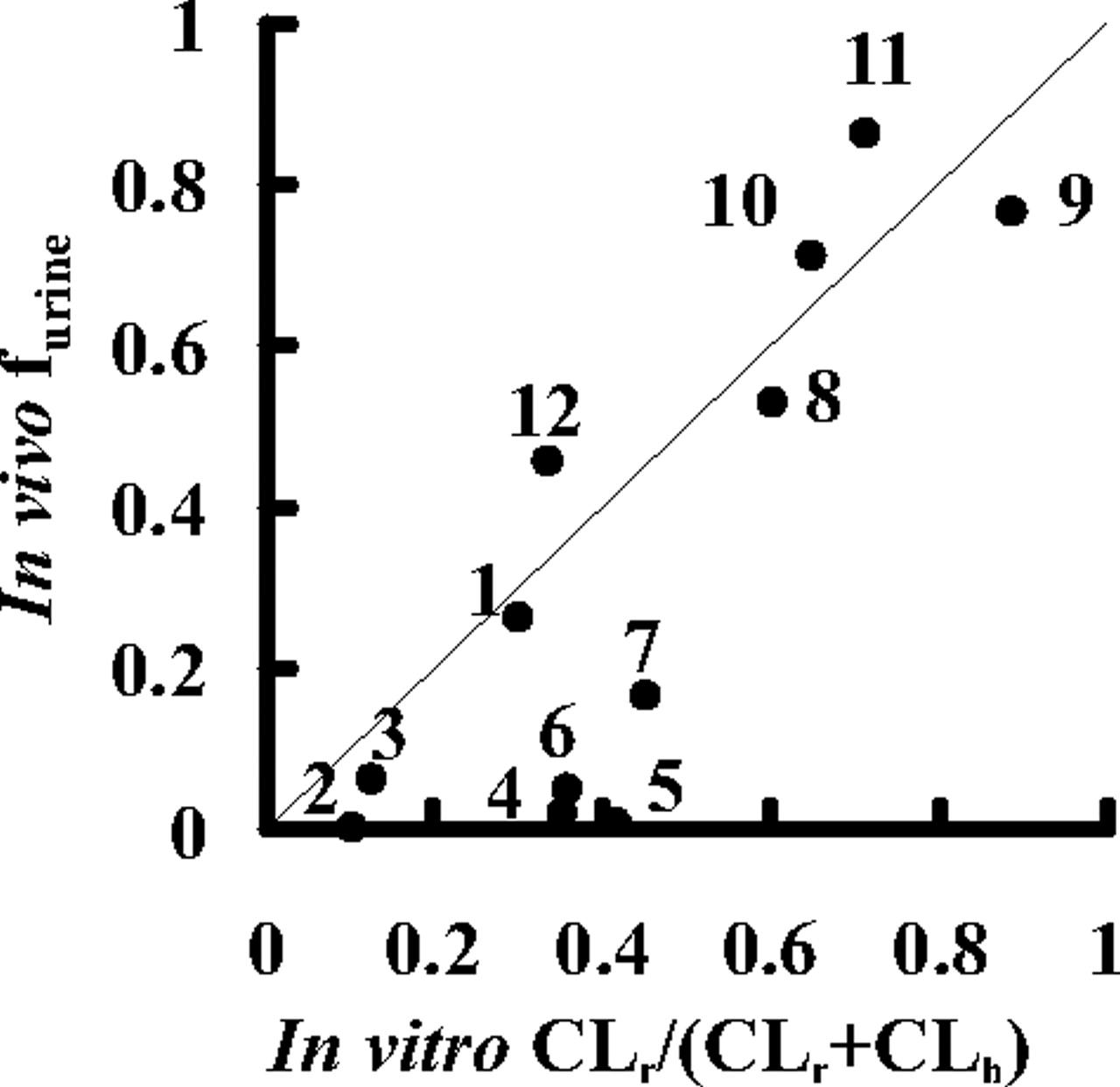
Comparison between the observed and the predicted fraction excreted into urine (furine) of 12 compounds in female SD rats. The fraction excreted into urine (furine) was predicted by eq. 14 using CLh, predicted and CLr, predicted values. The predicted and observed fractions excreted into urine are plotted. Plots represent: 1, pravastatin; 2, pitavastatin; 3, rosuvastatin; 4, valsartan; 5, olmesartan; 6, candesartan; 7, temocaprilat; 8, enalaprilat; 9, benazeprilat; 10, PCG; 11, ceftizoxime; and 12, cefmetazole. The solid line represents the line of unity.
Finally, in vivo organ clearance was estimated from the results of an in vitro uptake study (Fig. 4). When calculating the organ clearance from intrinsic clearance, we used a dispersion model with DN of 0.17 because a previous report suggested that it is the best prediction method compared with other models regardless of the magnitude of the clearance (Roberts and Rowland, 1986). The predicted hepatic clearance (CLh, predicted) was almost comparable to the observed one (CLbile, B) (Fig. 4 A), suggesting that the hepatic clearance of compounds could be predicted from the results of the uptake assay using isolated hepatocytes. The renal clearance was calculated in the same way as that of liver using the corrected intrinsic uptake clearance (P1KID, corrected). The observed renal clearance (CLrenal, B) was correlated with the predicted clearance (CLr, predicted) for each drug within a 2-fold range of difference, except for pitavastatin, valsartan, olmesartan, and candesartan, whose renal clearances were not well predicted from their uptake clearances (Fig. 4B). Moreover, the fraction excreted into urine (furine) calculated by the predicted organ clearance (CLh, predicted and CLr, predicted) was almost comparable to the observed value with the exception of these four compounds (Fig. 5). These results indicated that the absolute values of the organ clearances and the contribution of the liver and kidney to the overall elimination of 8 of 12 substrates could be well predicted from the in vitro uptake studies using isolated hepatocytes and kidney slices. Therefore, the uptake is the rate-limiting step for organ clearance in most cases, and the backflux clearance from organ to blood should be much smaller than the excretion clearance from organ to bile or urine.
Regarding the backflux in the liver, multidrug resistance-associated proteins 3 and 4 on the sinusoidal membrane of hepatocytes are involved in the sinusoidal efflux of several glucuronide and sulfate conjugates, and parent compounds such as fexofenadine and methotrexate (Kitamura et al., 2008a; Matsushima et al., 2008; Tian et al., 2008). However, the significance of these transporters on the sinusoidal efflux in humans has not been clarified. Sandwich-cultured hepatocytes enable us to evaluate the sinusoidal efflux and biliary excretion separately, so sandwich-cultured human hepatocytes might provide us with information regarding the relative contribution of backflux and sequestration of drugs (Bi et al., 2006). For reabsorption in the kidney, candidate transporters on the brush border membrane in proximal renal tubular cells have been identified. In rats, Oatp1a1 is reported to be involved in the reabsorption of organic anions such as estradiol-17β-glucuronide and dibromosulfophthalein, and their renal clearance was smaller in male rats than in female rats because of its gender-specific expression (Gotoh et al., 2002; Kato et al., 2002). However, in this study, the possible involvement of reabsorption was observed even in female rats, implying the existence of other mechanisms. There is no information regarding the backflux in the kidney to date. Future studies will be needed to clarify the molecular mechanisms of backflux and reabsorption in the kidney. More recently, it has been demonstrated that vectorial transport of drugs could be observed across the monolayer of epithelial cells from the proximal renal tubule seeded onto the culture insert, and the expression of several transporters was also confirmed (Lash et al., 2006, 2008). This kind of experimental system may help us investigate bidirectional drug transport in the kidney.
The Oatp family transporters and Oat1 and Oat3 are mainly involved in the uptake of organic anions in the liver and kidney, respectively. The substrate specificity of Oatp transporters overlaps that of Oat3 such as pravastatin, olmesartan, temocaprilat, and PCG. Thus, the fraction excreted into bile and urine of organic anions may be determined by the relative transport activity of Oatp transporters and Oat3. By comparing the transport activity of transporter-specific ligands or relative expression level in organ samples and transporter expression systems, the major elimination route of drugs can be estimated from the relative transport activity in each expression system. In this study, we selected minimally metabolized compounds for analysis. Recent reports have suggested the importance of uptake transporters in the hepatic clearance of some drugs such as repaglinide, bosentan, and atorvastatin, which are extensively metabolized by cytochrome P450 enzymes (Shitara et al., 2006). Even in the case of extensively metabolized drugs, if the uptake process is mediated by transporters, this prediction method may be applied for these substrates because the hepatic clearance may be solely determined by the uptake clearance.
To predict the human pharmacokinetics, human cryopreserved hepatocytes and kidney slices are now available and have been used in the characterization of the contribution of uptake transporters to the overall uptake of drugs and drug-drug interactions (Hirano et al., 2004; Nozaki et al., 2004). Using the same approach, we will be able to predict the elimination route of drugs in humans using in vitro uptake assays.
In summary, absolute values of hepatic and renal clearances and the fraction excreted in urine can be predicted for minimally metabolized drugs from in vitro uptake studies using isolated hepatocytes and kidney slices, except for drugs whose renal clearance was relatively small. Although further studies are needed for more accurate prediction, this model will be applicable to drug screening for the prediction of organ clearance and the distribution of compounds in the liver and kidney.
Acknowledgments
We express our great appreciation to the following: Daiichi-Sankyo Co. (Tokyo, Japan) for providing the [3H]pravastatin, [3H]olmesartan, [14C]temocaprilat, and unlabeled pravastatin, olmesartan, and temocaprilat; Novartis Pharma (Basel, Switzerland) for providing the 3H-labeled and unlabeled valsartan; Kowa Co. (Tokyo, Japan) for providing the [3H]labeled and unlabeled pitavastatin; and AstraZeneca (London, UK) for providing the 3H-labeled and unlabeled rosuvastatin. We thank Dr. Junko Iida and Futoshi Kurotobi (Shimadzu, Kyoto, Japan) for technical support of the LC/MS system.
Footnotes
-
This study was supported in part by the New Energy and Industrial Technology Development Organization of Japan (Development of Technology to Create Research Model Cells); and the Ministry of Education, Culture, Sports, Science and Technology [Grant 19890119].
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.026062.
-
ABBREVIATIONS: Oatp, organic anion transporting polypeptide; Oat, organic anion transporter; E217βG, estradiol-17β-glucuronide; PAH, p-aminohippurate; PCG, benzylpenicillin; SD, Sprague-Dawley; LC, liquid chromatography; MS, mass spectrometry; AUC, area under the plasma concentration-time profile.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Accepted April 9, 2009.
- Received December 8, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}