


Abstract
The disposition of fexofenadine, a commonly used antihistamine drug, is governed primarily by active transport. Biliary excretion of the parent compound is the major route of systemic clearance. Previous studies demonstrated that fexofenadine hepatic uptake is mediated by organic anion transporting polypeptides. Recently, we showed that in mice fexofenadine is excreted into bile primarily by multidrug resistance-associated protein (Mrp) 2 (Abcc2). In the present study, the roles of Mrp3 (Abcc3) and Mrp4 (Abcc4) in the hepatobiliary disposition of fexofenadine were examined in knockout mice using in situ liver perfusion. Compared with that in wild-type mice, basolateral excretion of fexofenadine was impaired, resulting in a ∼50% decrease in perfusate recovery in Abcc3-/- mice; in contrast, fexofenadine hepatobiliary disposition was unaltered in Abcc4-/- mice. As expected, in Abcc2-/- mice, fexofenadine was redirected from the canalicular to the basolateral membrane for excretion. In Abcc2-/-/Abcc3-/- double-knockout mice, fexofenadine biliary excretion was impaired, but perfusate recovery was similar to that in wild-type mice and more than 2-fold higher than that in Abcc3-/- mice, presumably due to compensatory basolateral transport mechanism(s). These results demonstrate that multiple transport proteins are involved in the hepatobiliary disposition of fexofenadine. In addition to Mrp2 and Mrp3, other transport proteins play an important role in the biliary and hepatic basolateral excretion of this zwitterionic drug.
Fexofenadine is an H1 receptor antagonist commonly used in the treatment of seasonal allergies and chronic urticaria. The intestinal absorption of fexofenadine is mediated by organic anion transporting polypeptides (Oatps), a process that is counteracted by P-glycoprotein-mediated efflux (Cvetkovic et al., 1999). Metabolism accounts for less than 5% of total fexofenadine clearance in humans, and the oral and intravenous exposure of fexofenadine in rats is not altered by the pan-cytochrome P450 mechanism-based inactivator, aminobenzotriazole (Strelevitz et al., 2006). However, specific information regarding fexofenadine metabolism in mice is not available. Biliary excretion of unchanged fexofenadine is the primary route of systemic clearance in mice (Tahara et al., 2005). Although fexofenadine intestinal absorption and blood-brain barrier penetration are limited by P-glycoprotein (Cvetkovic et al., 1999; Tahara et al., 2005), the biliary excretion of fexofenadine is mediated by multiple transport mechanisms (Tian et al., 2008). The unusual dispositional properties of fexofenadine, i.e., low metabolism and extensive transport, have led to its use as a probe for the analysis of both Oatp and P-glycoprotein modulation by various coadministered drugs and herbal products (Banfield et al., 2002; Zhou et al., 2004; Shon et al., 2005; Lemma et al., 2006).
After uptake from sinusoidal blood into the hepatocyte, drugs and derived metabolites may undergo excretion across the canalicular membrane into bile or across the basolateral membrane into blood. For example, carboxydichlorofluorescein is taken up into hepatocytes via basolateral Oatps and is subsequently excreted unchanged into bile by multidrug resistance-associated protein (Mrp) 2 and also undergoes basolateral export into blood by Mrp3 (Zamek-Gliszczynski et al., 2003; Nezasa et al., 2006). Similarly, glucuronide conjugates of acetaminophen and morphine also are subject to bidirectional excretion into bile and blood by Mrp2 and Mrp3, respectively (Xiong et al., 2000, 2002; Zelcer et al., 2005; van de Wetering et al., 2007). Basolateral Mrp3 is similar to canalicular Mrp2 in terms of amino acid sequence, membrane topology, and substrate specificity (König et al., 1999; Ogawa et al., 2000). Mrp3 is highly up-regulated in obstructive cholestasis (e.g., bile duct ligation) and hereditary conjugated hyperbilirubinemia in rats and humans (e.g., Mrp2-deficient animals and patients with Dubin-Johnson syndrome), acting as an overflow pump for hepatic excretion of Mrp2 substrates and conjugated bile acids when biliary excretion is impaired (Hirohashi et al., 1998; König et al., 1999). Although expression levels of Mrp3 protein are relatively high in mice, a 2-fold up-regulation of Mrp3 protein is still observed in Mrp2-knockout mice (Nezasa et al., 2006). On the basis of recent findings that the biliary excretion of fexofenadine is mediated in part by Mrp2 in mice (Tian et al., 2008), we hypothesized that fexofenadine may also undergo hepatic basolateral excretion via Mrp3. In addition to Mrp3, Mrp4 functions as an excretory mechanism on the hepatic basolateral membrane (Rius et al., 2003). Limited evidence of functional overlap between Mrp3 and Mrp4 exists. For example, both Mrp3 and Mrp4 are up-regulated in response to obstructive cholestasis (Hirohashi et al., 1998; Denk et al., 2004). Furthermore, both Mrp3 and Mrp4 mediate the hepatic basolateral excretion of the sulfate metabolites of acetaminophen, 4-methylumbelliferone, and harmol in mice (Zamek-Gliszczynski et al., 2006). Whereas these features suggest that Mrp3 and Mrp4 may be involved in the hepatic basolateral excretion of fexofenadine, the extent to which these pumps function in this capacity has not been investigated.
Studies outlined in this manuscript tested the hypothesis that fexofenadine undergoes basolateral excretion from the liver via Mrp3 and Mrp4. Perfused livers from relevant transporter gene knockout mice were used to examine the role of Mrp2, Mrp3, and Mrp4 in the hepatobiliary disposition of fexofenadine. Furthermore, the combined roles of Mrp2 and Mrp3 in the overall hepatic excretion of this zwitterion were studied in livers from Abcc2-/-/Abcc3-/- double-knockout mice. The data presented herein demonstrate the importance of Mrp2 and Mrp3 in the biliary and hepatic basolateral excretion, respectively, of fexofenadine in mice.
Materials and Methods
Chemicals. Fexofenadine, cetirizine, taurocholate, and Krebs-Henseleit buffer packets were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of reagent grade and were readily available from commercial sources.
Animals. Male C57BL/6 wild-type (WT) (age-matched heterozygotes), Abcc2-/-, Abcc3-/-, Abcc2-/-/Abcc3-/-, and Abcc4-/- mice (23–31g) were generated as described previously (Belinsky et al., 2005, 2007). Abcc2-/- mice have been generated recently by gene-targeting methods in the laboratory of Dr. Gary Kruh (unpublished observations). Abcc2-/-, Abcc3-/-, and Abcc4-/- mice used in this study were derived from mixed 129/C57BL/6 animals and were backcrossed to C57BL/6 for eight generations. Abcc2-/- and Abcc3-/- mice on the C57BL/6 background were used to breed the Abcc2-/-/Abcc3-/- mice. Mice were maintained on a 12-h light/dark cycle with free access to water and rodent chow. Experiments were performed at Fox Chase Cancer Center and approved by their institutional animal care and use committee.
In Situ Liver Perfusion. All experimental procedures were performed under full anesthesia induced with ketamine/xylazine (140/8 mg/kg i.p.). The liver perfusion procedure was modified slightly from the previous report of Tian et al. (2007). Briefly, the common bile duct was ligated above the duodenum to prevent bile from entering the intestine, and the gallbladder was cannulated with PE-10 tubing (BD Biosciences, Parsippany, NJ). The portal vein was cannulated with a 20-gauge catheter (B. Braun Medical Inc., Bethlehem, PA), the abdominal vena cava below the liver was severed immediately by incision, and the inferior vena cava above the liver was cannulated with a 20-gauge catheter. The liver was perfused (5 ml/min with fexofenadine-free continually oxygenated Krebs-Henseleit buffer containing 5 μM taurocholate) for an equilibration period of approximately 15 min. Subsequently, the inferior vena cava was ligated between the liver and kidney to direct all perfusate outflow through the cannulated inferior vena cava above the liver. After the preperfusion period to allow for equilibration of temperature and bile flow, the liver was perfused for 30 min with buffer containing 1.5 μM fexofenadine, followed by a washout with fexofenadine-free buffer for 45 min. Bile was collected in 7-min intervals; outflow perfusate was collected at designated time points in toto. Perfusion pressure and bile flow were monitored to assess liver viability (Chandra et al., 2005).
Analytical Methods. Bile and outflow perfusate samples were analyzed by liquid chromatography with detection by tandem mass spectrometry (Applied Biosystems API 4000 triple quadrupole spectrometer with TurboIonSpray interface; MDS Sciex, Concord, ON, Canada). Fexofenadine and cetirizine (internal standard) were eluted from an Aquasil C18 column (5 μm, 50 mm × 2.1 mm; Thermo Electron, Waltham, MA) using a mobile phase gradient (A: 0.1% formic acid in water; B: 0.1% formic acid in methanol) with 0 to 0.8 min hold at 10% B, 0.8 to 3.5 min linear increase to 85% B, 3.5 to 4.0 min hold at 85% B, 4.0 to 4.5 min return to 10% B, and 4.5 to 5 min hold at 10% B; the flow rate was 0.75 ml/min (Shimazdu solvent delivery system; Shimazdu, Columbia, MD). Fexofenadine and the internal standard, cetirizine, were detected in positive ion mode using multiple reaction monitoring: fexofenadine: 502.3 → 466.4 m/z; and cetirizine: 389.0 → 201.0 m/z. Fexofenadine was quantified with standard curves prepared in the appropriate matrix.
Data Analysis. All data are reported as mean ± S.D. (n = 3–4 per group). Statistical significance was assessed by one-way analysis of variance with Tukey's post hoc test. If the normality test failed, data were log-transformed before statistical analysis. In all cases, p < 0.05 was considered to be statistically significant.
Results
Body Weight, Liver Weight, and Bile Flow. Body weight was comparable between WT and knockout mice (Table 1). Liver weight (normalized for body weight) was significantly increased in Abcc2-/- (∼30%) and in Abcc2-/-/Abcc3-/- (∼46%) relative to that of WT mice. Abcc3-/- and Abcc4-/- mouse liver weights were comparable with those for WT controls (Table 1). The bile flow rate was decreased ∼30% in Abcc2-/- and Abcc2-/-/Abcc3-/- mouse livers relative to that of WT controls but was normal in livers from Abcc3-/- and Abcc4-/- mice (Table 1).
Body weight, liver weight normalized for body weight, and bile flow rate in wild-type and transporter gene knockout mice Values are expressed as mean ± S.D.; n = 3 to 4 per group.
Recovery of Fexofenadine in Outflow Perfusate. Fexofenadine concentrations in outflow perfusate from WT and knockout mouse livers are presented in Fig. 1. For clarity, only mean data are plotted; standard deviations were within ∼50% of the mean data. The total recovery of fexofenadine at the end of the washout period as a percentage of the infused dose was 73 ± 10% in WT, 62 ± 30% in Abcc3-/-, 68 ± 9% in Abcc4-/-, 77 ± 12% in Abcc2-/-, and 71 ± 15% in Abcc2-/-/Abcc3-/- mouse livers. To quantify the impact of Mrp3 and Mrp4 genetic ablation on the basolateral efflux of fexofenadine from the liver, fexofenadine recovery in perfusate during the washout phase was expressed as a percentage of liver fexofenadine content at the end of the infusion (determined as the difference between infused dose and the cumulative mass of fexofenadine excreted in outflow perfusate and bile through 30 min) (Table 2). The fraction of fexofenadine excreted into the outflow perfusate was significantly decreased (∼50%) in livers from Abcc3-/- mice; in contrast, significantly increased perfusate recovery (∼60%) was noted in Abcc2-/- relative to WT mouse livers. Interestingly, perfusate recovery of fexofenadine in livers from Abcc2-/-/Abcc3-/- mice was similar to that in WT control livers. In contrast to livers from Abcc3-/- mice, perfusate recovery of fexofenadine in livers from Abcc4-/- mice was unchanged.
Recovery of fexofenadine in perfusate and bile during the washout phase of mouse liver perfusions and liver concentrations after the washout phase Values are expressed as mean ± S.D. percentage of fexofenadine liver content after the 30-min infusion; n = 3 to 4 per group.

Fexofenadine concentrations in outflow perfusate from wild-type C57BL/6 (•), Abcc3-/- (▵), Abcc4-/- (▿), Abcc2-/- (□), and Abcc2-/-/Abcc3-/- (⋄) mouse livers (mean data are plotted; n = 3–4 per group). Livers were perfused (5 ml/min) with fexofenadine for 30 min followed by a 45-min washout phase.
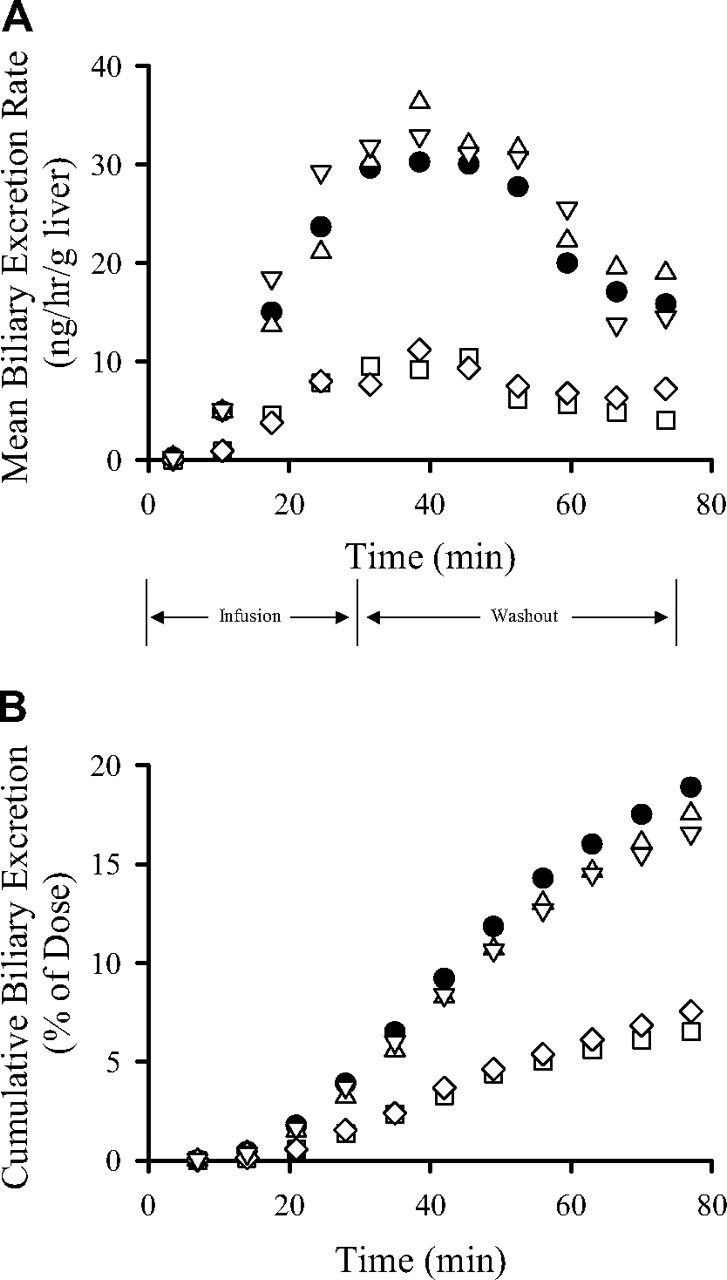
Recovery of Fexofenadine in Bile. Fexofenadine biliary excretion rates and cumulative biliary excretion in livers from WT and knockout mice are presented in Fig. 2, A and B, respectively; standard deviations were within ∼50% of the mean data and are not included in the figures for clarity. The biliary recovery of fexofenadine was not significantly different in Abcc3-/- and Abcc4-/- mouse livers relative to that in WT controls (Fig. 2B; Table 2). Fexofenadine biliary excretion rates and cumulative biliary excretion were significantly decreased in livers from Abcc2-/- and Abcc2-/-/Abcc3-/- mice (Fig. 2). The biliary recovery of fexofenadine during the washout phase, normalized for liver content of fexofenadine at the end of the infusion, was ∼61% and ∼53% lower in Abcc2-/- and Abcc2-/-/Abcc3-/- relative to that in WT mouse livers, respectively (Table 2). Interestingly, liver fexofenadine concentrations at the end of the washout period were not altered in Abcc3-/-, Abcc4-/- and Abcc2-/-/Abcc3-/- mice (Table 2). However, consistent with our previous report (Tian et al., 2008), liver fexofenadine concentrations decreased ∼2-fold in Abcc2-/- mice after the washout phase (Table 2).
Discussion
Previous studies using transporter gene knockout mice identified Mrp2 as the predominant protein responsible for fexofenadine biliary excretion (Tian et al., 2008). In contrast, fexofenadine biliary excretion was not impaired in mice lacking the breast cancer resistance protein or P-glycoprotein (Tahara et al., 2005). In agreement with previous work, Mrp2 was responsible for ∼60% of fexofenadine biliary excretion in the current study. However, the present data also establish the importance of Mrp3 in the hepatic basolateral excretion of fexofenadine. Approximately 50% of fexofenadine basolateral excretion may be attributed to Mrp3-mediated transport when outflow perfusate concentrations are in the range of 300 ng/ml based on the 2-fold decrease in perfusate recovery of fexofenadine in livers from Abcc3-/- mice. These data highlight the functional interplay between Mrp2 and Mrp3 in fexofenadine hepatobiliary disposition. However, the basolateral excretion of fexofenadine in Abcc2-/-/Abcc3-/- mice was not impaired. Fexofenadine liver concentrations in Abcc3-/- and Abcc2-/-/Abcc3-/- mouse livers were similar to those in WT mouse livers. Mechanism(s) responsible for the Mrp2- and Mrp3-independent component of fexofenadine hepatobiliary clearance remain to be elucidated.

Biliary excretion rate (A) and cumulative biliary excretion (B) of fexofenadine in perfused livers from wild-type C57BL/6 (•), Abcc3-/- (▵), Abcc4-/- (▿), Abcc2-/- (□), and Abcc2-/-/Abcc3-/- (⋄) mice (mean data are plotted; n = 3–4 per group). Livers were perfused (5 ml/min) with fexofenadine for 30 min followed by a 45-min washout phase.
Mrp2 plays a critical role as an organic anion excretory system in liver detoxification. Unlike human MRP3, MRP2 protein is highly expressed in humans under normal conditions (Stöckel et al., 2000). Protein expression of Mrp3, the“Mrp2 backup system,” is increased markedly in rats and humans that are deficient in Mrp2 (Hirohashi et al., 1998; König et al., 1999; Xiong et al., 2002). Mrp3 constitutive expression levels in mice are already high, but in Mrp2-knockout mice, Mrp3 is further up-regulated (Nezasa et al., 2006). In contrast, this functional compensation is not reciprocal in mouse liver; the absence of Mrp3 does not result in a notable up-regulation of Mrp2 protein or function in mouse liver, as demonstrated by this and other studies (Belinsky et al., 2005; Zamek-Gliszczynski et al., 2006). This finding suggests that hepatic protection mechanisms other than Mrp2-mediated biliary excretion exist in mice when Mrp3 function is ablated. Mrp4 is another efflux transport protein located on the hepatic basolateral membrane (Rius et al., 2003). Limited evidence of functional overlap between Mrp3 and Mrp4 (Hirohashi et al., 1999; Denk et al., 2004; Zamek-Gliszczynski et al., 2006) prompted the exploration of fexofenadine disposition in livers from Abcc4-/- mice. Interestingly, fexofenadine basolateral excretion was not affected by the absence of Mrp4. The impact of genetic ablation of Mrp4 has been associated with impaired sulfotransferase 2a1 expression and function (Assem et al., 2004), but an appreciable effect on the function or expression of other hepatic transport proteins has not been observed (Assem et al., 2004; Mennone et al., 2006; Zamek-Gliszczynski et al., 2006). Whether Mrp4 serves as the backup system for Mrp3-mediated fexofenadine basolateral excretion requires further investigation.
The existence of multiple transport and backup transport systems on both the basolateral and canalicular membranes complicates the extrapolation of transport studies from single-knockout to double-knockout animals (van de Wetering et al., 2007). The biliary and basolateral excretion of the Mrp2 and Mrp3 substrate, morphine-3-glucuronide, in Abcc2-/-/Abcc3-/- mice after morphine administration was severely impaired, resulting in considerable hepatic accumulation of the glucuronide metabolite. However, residual plasma concentrations of morphine-3-glucuronide in Abcc2-/-/Abcc3-/- mice were substantial, and the concentrations of morphine-3-glucuronide were similar between Abcc2-/-/Abcc3-/- and WT mice 24 h after morphine administration, which resulted in near-normal urinary excretion of morphine-3-glucuronide, suggesting that a low-capacity backup transport system was present for the basolateral excretion of morphine-3-glucuronide into plasma in Abcc2-/-/Abcc3-/- mice (van de Wetering et al., 2007). In the present study, fexofenadine biliary excretion in livers from Abcc2-/-/Abcc3-/- mice was impaired to the same extent as in mice lacking only Mrp2; fexofenadine basolateral excretion in livers from these double-knockout mice was similar to that in WT mice and was more than 2-fold higher than that in Mrp3 single-knockout mice. The unchanged liver concentrations of fexofenadine in Abcc3-/- mice and considerable basolateral excretion of fexofenadine in the livers of Abcc2-/-/Abcc3-/- mice are consistent with the hypothesis that additional clearance mechanisms compensate for the loss of Mrp3 function. Basolateral Mrps (other than Mrp3), the bidirectional organic anion transport proteins, or other basolateral export proteins may contribute to increased basolateral clearance of fexofenadine in Abcc2-/-/Abcc3-/- mouse livers. The possibility of altered fexofenadine metabolism in Abcc3-/- and Abcc2-/-/Abcc3-/- mouse livers cannot be ruled out because no detailed information on fexofenadine metabolism in mouse livers has been published.
In summary, fexofenadine is taken up into the hepatocyte and undergoes biliary as well as basolateral excretion. Using perfused livers from relevant transporter gene knockout mice, Mrp2 and Mrp3 were identified as important transport proteins mediating fexofenadine biliary and hepatic basolateral excretion, respectively. Mrp4 did not appear to contribute to hepatic basolateral excretion of this zwitterionic drug in the intact mouse liver in the presence of Mrp3 and/or Mrp2 proteins. Mechanism(s) responsible for the residual, Mrp2- and Mrp3-independent, biliary and basolateral excretion of fexofenadine remain to be elucidated. Once again, this study exemplified the importance of using physiologically relevant systems such as the intact liver to evaluate the contributions of hepatic transport proteins in drug pharmacokinetic studies.
Acknowledgments
The authors thank Arlene S. Bridges, Ph.D., for analytical support and Peijin Zhang, Ph.D., for helpful scientific discussions.
Footnotes
-
This work was supported by the National Institutes of Health (GM41935 to K.L.R.B. and CA73728 to G.D.K.) and by the National Cancer Institute (Core Grant CA06927 to Fox Chase Cancer Center).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019273.
-
ABBREVIATIONS: Oatp, organic anion transporting polypeptide; Mrp/MRP, multidrug resistance-associated protein; Abcc2-/-, Mrp2 gene knockout; Abcc3-/-, Mrp3 gene knockout; Abcc2-/-/Abcc3-/-, Mrp2 and Mrp3 double-knockout; WT, wild-type C57BL/6; Abcc4-/-, Mrp4 gene knockout.
-
↵1 Current affiliation: Wyeth, Discovery Pharmacokinetics, Andover, MA.
-
↵2 Current affiliation: Eli Lilly and Company, Drug Disposition, Indianapolis, IN.
-
↵3 Current affiliation: Department of Medicine, University of Illinois at Chicago, Chicago, IL.
- Received October 15, 2007.
- Accepted February 12, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}