


Abstract
Cytochrome P450 (P450) protein-protein interactions have been observed with various in vitro systems. It is interesting to note that these interactions seem to be isoform-dependent, with some combinations producing no effect and others producing increased or decreased catalytic activity. With some exceptions, most of the work to date has involved P450s from rabbit, rat, and other animal species, with few studies including human P450s. In the studies presented herein, the interactions of two key drug-metabolizing enzymes, CYP2C9 and CYP2D6, were analyzed in a purified, reconstituted enzyme system for changes in both substrate-binding affinity and rates of catalysis. In addition, an extensive study was conducted as to the “order of mixing” for the reconstituted enzyme system and the impact on the observations. CYP2D6 coincubation inhibited CYP2C9-mediated (S)-flurbiprofen metabolism in a protein concentration-dependent manner. Vmax values were reduced by up to 50%, but no appreciable effect on Km was observed. Spectral binding studies revealed a 20-fold increase in the KS of CYP2C9 toward (S)-flurbiprofen in the presence of CYP2D6. CYP2C9 coincubation had no effect on CYP2D6-mediated dextromethorphan O-demethylation. The order of combination of the proteins (CYP2C9, CYP2D6, and cytochrome P450 reductase) influenced the magnitude of catalysis inhibition as well as the ability of increased cytochrome P450 reductase to attenuate the change in activity. A simple model, congruent with current results and those of others, is proposed to explain oligomer formation. In summary, CYP2C9-CYP2D6 interactions can alter catalytic activity and, thus, influence in vitro-in vivo correlation predictions.
Cytochrome P450s (P450s) are responsible for the metabolism of more than 75% of the drugs on the market (Guengerich, 2006). Multiple subfamilies of cytochrome P450s can activate and metabolize a wide spectrum of substrates by a two-electron transfer catalytic cycle, in which electrons are provided by either NADPH-cytochrome P450 reductase (CPR) or cytochrome b5 (b5), depending on the stage of the cycle (Guengerich, 2001). A number of studies have investigated the role of CPR and b5 and the nature of P450, CPR, and b5 interactions. Although CPR is indispensable for metabolism, cytochrome b5 either has no effect or significantly enhances metabolism (Shimada et al., 1994; Locuson et al., 2006). Yamazaki et al. (1997) demonstrated that b5 stimulated the metabolism of tolbutamide and (S)-warfarin by CYP2C10 but had no effect on bufuralol hydroxylation by CYP1A1 and CYP2D6. In addition, both apo- and holo-b5 enhanced metabolism by CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4, and CYP3A5 in reconstituted systems (Shimada et al., 1994), suggesting that the effect of these protein-protein interactions of the redox partner with P450 may involve enzyme conformational changes or other mechanisms besides just provision of electrons.
In addition to interactions with CPR and b5, P450s can also interact with each other, resulting in changes in metabolism rates (Kaminsky and Guengerich, 1985; Alston et al., 1991; Shimada et al., 1994; Cawley et al., 1995; Tan et al., 1997; Yamazaki et al., 1997; Backes et al., 1998; Backes and Cawley, 1999; Li et al., 1999; Cawley et al., 2001; Hazai and Kupfer, 2005; Kelley et al., 2005, 2006). Reconstituted expressed enzymes [human CYP2C9-CYP2C19 (Hazai and Kupfer, 2005), rabbit CYP1A2-CYP2B4 (Yamazaki et al., 1997), rabbit CYP1A2-CYP2E1 (Kelley et al., 2006), human CYP3A4-CYP1A1 (Yamazaki et al., 1997), human CYP3A4-CYP1A2 (Yamazaki et al., 1997)], microsomes from induced animals (Kaminsky and Guengerich, 1985; Cawley et al., 2001), and triple-expression systems [CYP3A4, CYP2D6, and CPR (Li et al., 1999), CYP2E1, CYP2A6, and CPR (Tan et al., 1997)] have exhibited protein-protein interactions. However, P450-P450 interactions are not universal, because studies with reconstituted enzymes (Kelley et al., 2006) or microsomes from rats (Dutton et al., 1987) did not demonstrate interactions between CYP2E1 and CYP2B4. Initial suggestions that protein-protein interactions occurred because of competition for CPR (West and Lu, 1977) were disproven in subsequent studies using saturating concentrations of CPR (Kaminsky and Guengerich, 1985). CPR activity in the form of cytochrome c was also measured to discount loss of CPR activity as a reason for the observed substrate inhibition.
Understanding and elucidating the mechanisms underlying P450-P450 interactions are important for several reasons. First, they may affect predictions of in vivo clearance from in vitro data. Many times, these predictions are determined using expressed enzyme systems that may not contain the full complement of P450s and, thus, the potential for interactions. Second, a better understanding of P450-P450 interactions may provide insights into the organization of these proteins within the cell and their aggregation characteristics and how these features affect catalytic activity. Third, P450-P450 interactions may provide valuable insights into protein structure-function relationships and how the presence of different proteins may affect individual protein function.
Several possible mechanisms of altered P450 function resulting from these interactions have been explored. The role of ionic interactions between P450 pairs has been evaluated (Kelley et al., 2005) using high concentrations of buffer salts to disrupt possible ionic interactions of CYP2B4 and CYP1A2 and restore most of the P450 activity. Investigators also have used molecular modeling of protein-protein interactions to evaluate how proteins might interact to form heteromers that may alter activity (Backes et al., 1998; Hazai et al., 2005). However, it remains unclear whether conformational changes, altered catalytic cycle functioning, or other mechanisms are responsible for the observed changes in P450 function.
CYP2C9 comprises approximately 10% of the liver P450, whereas CYP2D6 comprises 2 to 3% but is involved in the metabolism of 30% of all drugs (Shimada et al., 1994). One to 5% of the white population has multiple copies of CYP2D6, leading to greater CYP2D6 proteins, and lower CYP2C9/CYP2D6 ratios in these individuals (Meijerman et al., 2007). In the current study we examined the consequence of CYP2C9-CYP2D6 interactions, and the resulting impact on metabolism rates. In addition, the role of CPR on these interactions was evaluated. Previous work (Gorsky and Coon, 1986) has demonstrated that order of mixing of P450 proteins with redox transfer proteins can alter the rate of metabolism, and thus the order of combination of these enzymes (CYP2C9, CYP2D6, and CPR) in a reconstituted system was also explored to evaluate what role this might play in the degree of interactions observed.
Materials and Methods
Materials. Acetonitrile, dibasic potassium phosphate, methanol, and phosphoric acid were purchased from Thermo Fisher Scientific (Waltham, MA). Dextromethorphan, dilauroylphosphatidylcholine (DLPC), levallorphan (internal standard), NADPH, and triethylamine were purchased from Sigma-Aldrich (St. Louis, MO). Dextrorphan was purchased from Sigma/RBI (Natick, MA). (S)-Flurbiprofen, 4′-hydroxyflurbiprofen, and 2-fluoro-4-biphenyl acetic acid (internal standard) were gifts from the former Pharmacia, Inc. (Kalamazoo, MI). Human CYP2D6.1 and human P450 CPR were purchased from Invitrogen (Carlsbad, CA). CYP2C9.1 was expressed and purified as described previously (Locuson et al., 2006).
Enzyme Reconstitution. Incubations contained P450s, extruded DLPC vesicles (10 μg/incubation), CPR, and substrate. Different mixing schemes (sequential and simultaneous) of CYP2C9, CYP2D6, and CPR were explored. In the sequential mixing schemes, two components were mixed and allowed to equilibrate on ice for 5 min, followed by the addition of the third component and further equilibration on ice for 5 min. In the simultaneous mixing scheme, all three components were mixed consecutively and allowed to equilibrate for 10 min on ice. The order of mixing of the P450s was also investigated, and all experimental mixing conditions are summarized in Table 1. Once all three components had equilibrated on ice, they were was reconstituted in DLPC (extruded through a 200-nm pore size membrane) and allowed to further equilibrate for 5 or 10 min in the case of sequential or simultaneous mixing, respectively. Enzyme mixtures were then added to substrate and buffer and preincubated at 37°C for 8 min before initiation of the reaction.
Order of combination of enzymes in the metabolism of (S)-flurbiprofen by CYP2C9 and the effect of CPR
(S)-Flurbiprofen Metabolism by CYP2C9 in the Presence of CYP2D6. (S)-Flurbiprofen (2, 5, 10, 25, 50, 100, 200, and 300 μM) was incubated with CYP2C9 (5 pmol/incubation), CPR (10 or 20 pmol/incubation for subsaturating or saturating conditions), and CYP2D6 (0–5 pmol/incubation). The ratios of CYP2C9/CYP2D6 ranged from 10:1 to 1:1 in an attempt to mimic in vivo ratios of these enzymes across the range of expression (poor to extensive to ultra-rapid metabolizers). All experiments were performed in 50 mM potassium phosphate buffer (pH 7.4) at 37°C. After initiation of the reaction with NADPH (1 mM final concentration), at 20-min incubation time, the reactions were terminated by adding 200 μl of 180 ng/ml 2-fluoro-4-biphenyl acetic acid (internal standard) in acetonitrile followed by 40 μl of half-strength phosphoric acid to adjust to pH ∼3.0 for HPLC separation. All experiments were performed three times on separate days.
Dextromethorphan Metabolism by CYP2D6 in the Presence of CYP2C9. Dextromethorphan (1, 2, 5, 10, 20, 50, 100, 150, and 200 μM), CYP2D6 (10 pmol/incubation), CYP2C9 (0 or 10 pmol/incubation), and CPR (20 or 40 pmol/incubation) were incubated together in 50 mM potassium phosphate buffer (pH 7.4) at 37°C. Reactions were initiated with NADPH (1 mM final concentration). After 20 min, the reactions were terminated by adding 50 μl of half-strength phosphoric acid followed by 20 μl of 8 μg/ml levallorphan (internal standard) in water. Again, all experiments were performed three times on separate days.
Analysis of 4′-Hydroxyflurbiprofen. 4′-Hydroxyflurbiprofen formation was quantitated exactly as described previously (Tracy et al., 2002).
Analysis of Dextrorphan. Dextrorphan was analyzed by HPLC with fluorescence detection. The HPLC system consisted of an Alliance 2695XE pump/autosampler and a 2495 fluorescence detector (Waters, Milford, MA) set at an excitation wavelength of 280 nm and an emission wavelength of 310 nm. The mobile phase consisted of deionized water, methanol, acetonitrile, and triethylamine, pH 3.0 (60:34.3:5.7:0.25, v/v) pumped at 1 ml/min through a 4.6 × 150 mm Cyano Column (Regis Technologies, Inc., Morton Grove, IL). Dextrorphan and internal standard eluted at 6.0 and 7.3 min, respectively.
(S)-Flurbiprofen Spectral Binding. Difference spectra were obtained to evaluate enzyme-substrate affinity based on the alteration in heme iron spin state that occurs when active site water(s) are displaced through (S)-flurbiprofen binding. In brief, various ratios of CYP2C9 (0 and 0.5 μM concentrations) and CYP2D6 (0 and 0.5 μM concentrations) and 0.5 μg of DLPC/pmol of P450 in 50 mM potassium phosphate buffer (pH 7.4) were mixed in the sample and reference cuvettes. Increasing concentrations of (S)-flurbiprofen, dissolved in 50 mM potassium phosphate (pH 7.4) with a small amount of acetonitrile, were added to the sample cuvette (the acetonitrile concentration did not exceed 1%), and phosphate buffer was added to the reference cuvette and a 3-min equilibrium interval was allowed between readings. Spectral binding experiments were conducted on an Aminco DW2000 spectrophotometer with an Olis upgrade (Olis, Inc., Bogart, GA) set to record spectra from 340 to 500 nm wavelength with a slit width of 0.5 mm and 20 scans per datum at 1.0-nm steps. The temperature of the cell was held constant at 30°C with a F12 compact refrigerated circulator with an external ED temperature controller (Julabo USA, Inc., Allentown, PA). The difference in absorbance between high-spin (390 nm) and low-spin (418 nm) components was calculated and plotted against the (S)-flurbiprofen concentration, and the data were fit by nonlinear regression (SigmaPlot 8.0; Systat Software, Inc., San Jose, CA) to estimate the KS (spectral binding constant) from the titration binding curve using the following equation: 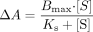

Effect of CYP2D6 on (S)-flurbiprofen metabolism by CYP2C9 at 10 pmol of CPR/incubation. A to E represent SeqAdd1, SeqAdd2, SeqAdd3, Allmix1, and Allmix2, respectively. Data fitting for the plots are shown. For clarity, representative data from a single experiment are presented. However, all experiments were conducted in triplicate over 3 separate days, and the mean values were used to determine degree of inhibition. Single-factor analysis of variance was used for statistical comparisons. OH, hydroxy.
Results
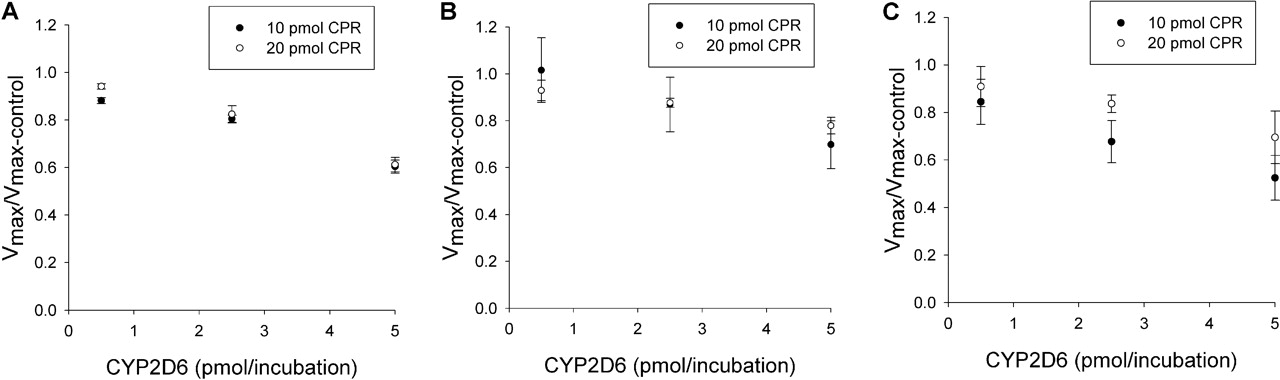
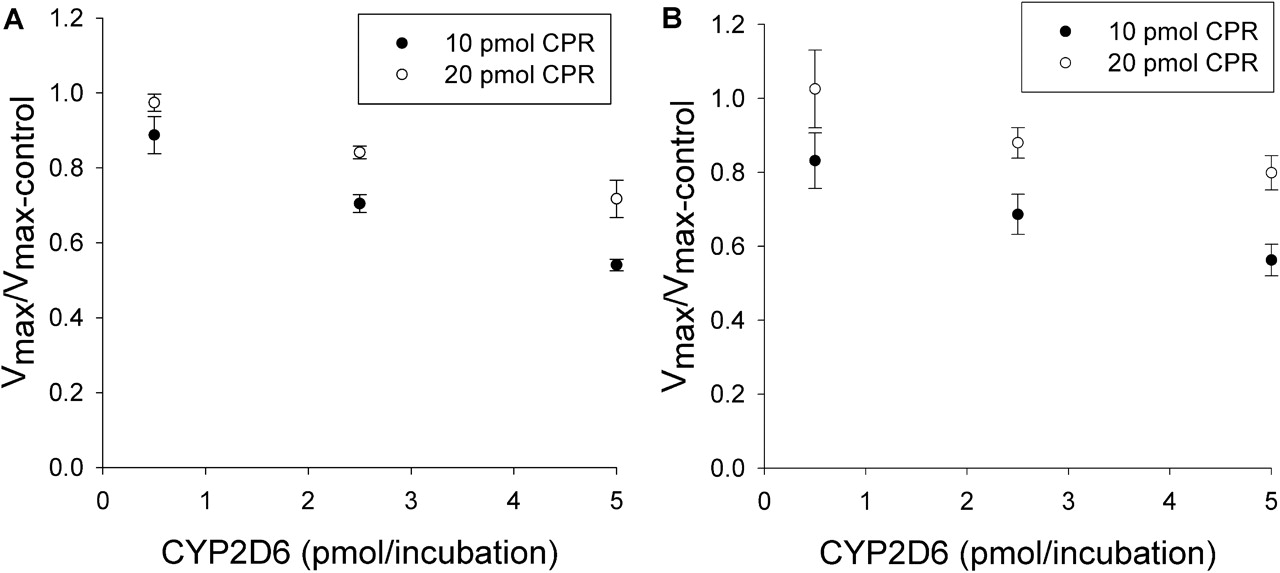
Effect of CYP2D6 on CYP2C9 Metabolism of (S)-Flurbiprofen. The effects of various amounts of coincubated CYP2D6 and CPR on the metabolism of (S)-flurbiprofen are presented in Figs. 1 and 2. Figure 1 shows the metabolism of (S)-flurbiprofen when three concentrations of CYP2D6 and 10 pmol of CPR were used in each incubation, and Fig. 2 shows the same when 20 pmol of CPR was used in each incubation. In all cases, a CYP2D6 concentration-dependent inhibition was observed, resulting in a decrease in Vmax but less change in Km. When 2.5 or 5 pmol of CYP2D6 was preincubated with CYP2C9, significant inhibition (p < 0.05) was observed. Figures 3 and 4 summarize the effect of CYP2D6 on the Vmax of (S)-flurbiprofen metabolism by CYP2C9 when the order of combination of the enzymes was varied. When CYP2C9 was combined with CYP2D6 followed by the addition of CPR (SeqAdd1), a reduction in Vmax was observed (Fig. 3A). Increasing the levels of CPR did not restore the CYP2C9 activity. A similar trend was observed when CYP2C9 and CPR were initially combined and then CYP2D6 was added (SeqAdd2) (Fig. 3B). However, when CYP2D6 and CPR were initially combined, followed by the addition of CYP2C9, addition of more CPR reduced the amount of inhibition (SeqAdd3) (Fig. 3C), demonstrating that order of mixing can affect the nature of inhibition. Figure 4 depicts the inhibition of CYP2C9-mediated metabolism of (S)-flurbiprofen when CYP2C9, CYP2D6, and CPR were mixed together simultaneously and preincubated before the addition of DLPC. Figure 4A depicts the data obtained when the order of mixing was CYP2C9, then CPR, followed by CYP2D6 (Allmix1), and Fig. 4B shows data when the order of mixing was CYP2C9, then CYP2D6, followed by CPR (Allmix2). Inhibition was observed at all levels of CYP2D6, and this inhibition was significantly reduced (p < 0.05) by the addition of more CPR. There was no difference observed between Allmix1 and Allmix2, wherein the order of addition of CYP2D6 and CPR was reversed. In all cases, as the amount of CYP2D6 was increased, the degree of inhibition of (S)-flurbiprofen metabolism was also increased. However, at low CYP2D6/CYP2C9 ratios (0.2:1), less than 10 to 20% inhibition was observed. At the highest tested CYP2D6/CYP2C9 ratio of 1:1, the ratios of Vmax + 2D6/Vmax-control ranged from 50 to 80%. Hence, a halving of maximal velocity was observed at the highest ratio of CYP2D6/CYP2C9. The effect of saturating levels of CPR on the amount of inhibition in various mixing schemes is summarized in Table 2. Control incubations of CYP2D6 with (S)-flurbiprofen (no CYP2C9 present) demonstrated that CYP2D6 was unable to metabolize (S)-flurbiprofen to the 4′-hydroxy metabolite, and no additional peaks were noted in the chromatograms (data not shown).
Effect of CPR on 1:1 CYP2C9/CYP2D6-mediated (S)-flurbiprofen metabolism for various mixing schemes
Addition of excess (20 pmol) CPR significantly (p < 0.05) increased the Vmax ratios in Allmix1 and Allmix2 but not in SeqAdd1 and SeqAdd2. In SeqAdd3, a significant (p < 0.05) increase in Vmax ratios was seen at 2.5 pmol of CYP2D6 only between the two CPR levels. No significant changes were observed in the Km ratios upon addition of excess CPR in all systems. Single-factor analysis of variance was used for statistical comparisons. Data are the mean ± S.D.
Effects of CYP2C9 on Dextromethorphan Metabolism by CYP2D6. The presence of equimolar ratios of CYP2C9 had no effect on the metabolism of dextromethorphan by CYP2D6 (Fig. 5). Furthermore, control incubations confirmed that no detectable dextrorphan was formed when dextromethorphan was incubated alone with CYP2C9. The Vmax values for dextromethorphan O-demethylation by CYP2D6 ranged from 0.2 to 0.4 pmol/min/pmol of CYP2D6, whereas Km was between 4 and 8 μM.
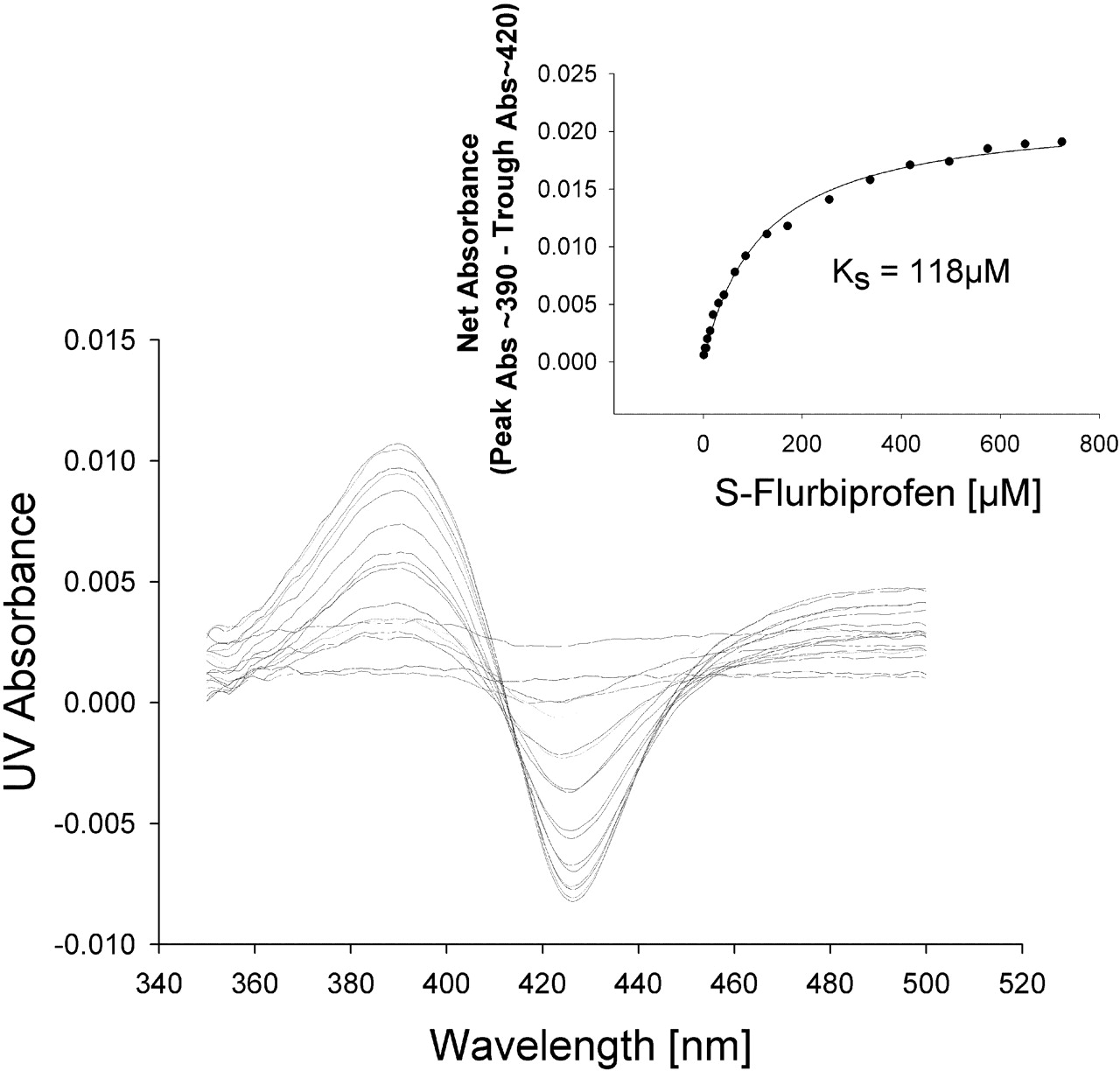
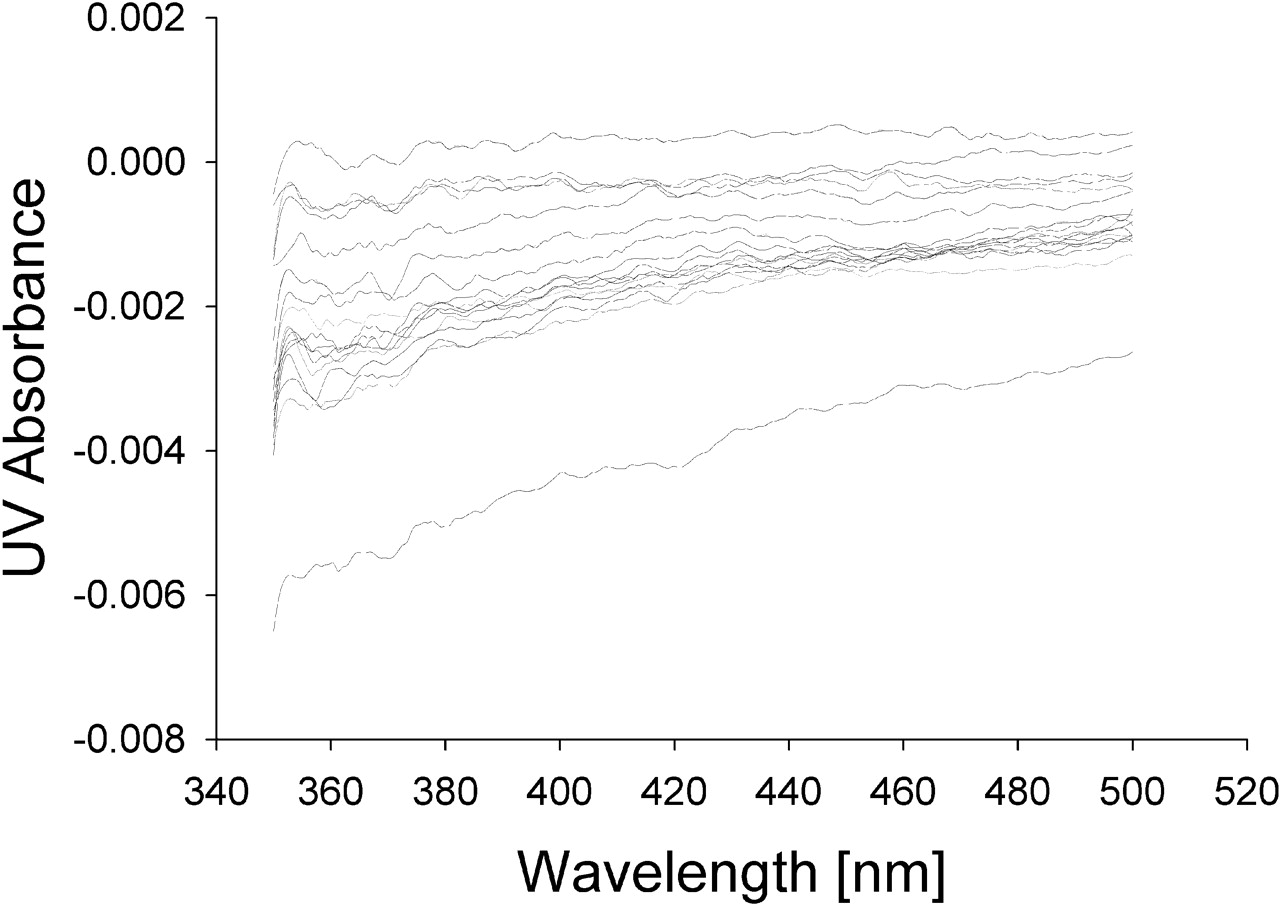
Spectral Binding Studies. To determine whether the protein-protein interactions between CYP2C9 and CYP2D6 resulted in an alteration in binding affinity for the substrate, P450 spectral binding titration studies were performed. In the presence of CYP2C9 alone (control) the spectrally determined binding affinity (KS) for (S)-flurbiprofen was 6 μM (Fig. 6). The addition of equimolar CYP2D6 shifted the KS for (S)-flurbiprofen to 118 μM (Fig. 7), resulting in a decrease in binding affinity. A control binding experiment with (S)-flurbiprofen and CYP2D6 alone confirmed that (S)-flurbiprofen was not causing a spin state change in CYP2D6, suggesting that (S)-flurbiprofen did not bind to CYP2D6 (Fig. 8). To further confirm that (S)-flurbiprofen does not bind to the CYP2D6 active site, the metabolism of dextromethorphan by CYP2D6 in the presence of (S)-flurbiprofen was evaluated, and (S)-flurbiprofen had no effect on dextromethorphan metabolism by CYP2D6 (data not shown). This absence of competitive inhibition, along with the spectral binding data presented above, further reinforces that (S)-flurbiprofen does not enter the CYP2D6 active site, ruling out the possibility that (S)-flurbiprofen binding to the CYP2D6 heme contributed to the alterations in the observed binding affinity of (S)-flurbiprofen for CYP2C9. The 20-fold increase in KS toward (S)-flurbiprofen in the presence of CYP2D6 indicates an increased dissociation of the flurbiprofen-CYP2C9 complex when CYP2D6 is present and is consistent with the inhibitory results from the kinetic studies in the reconstituted systems.
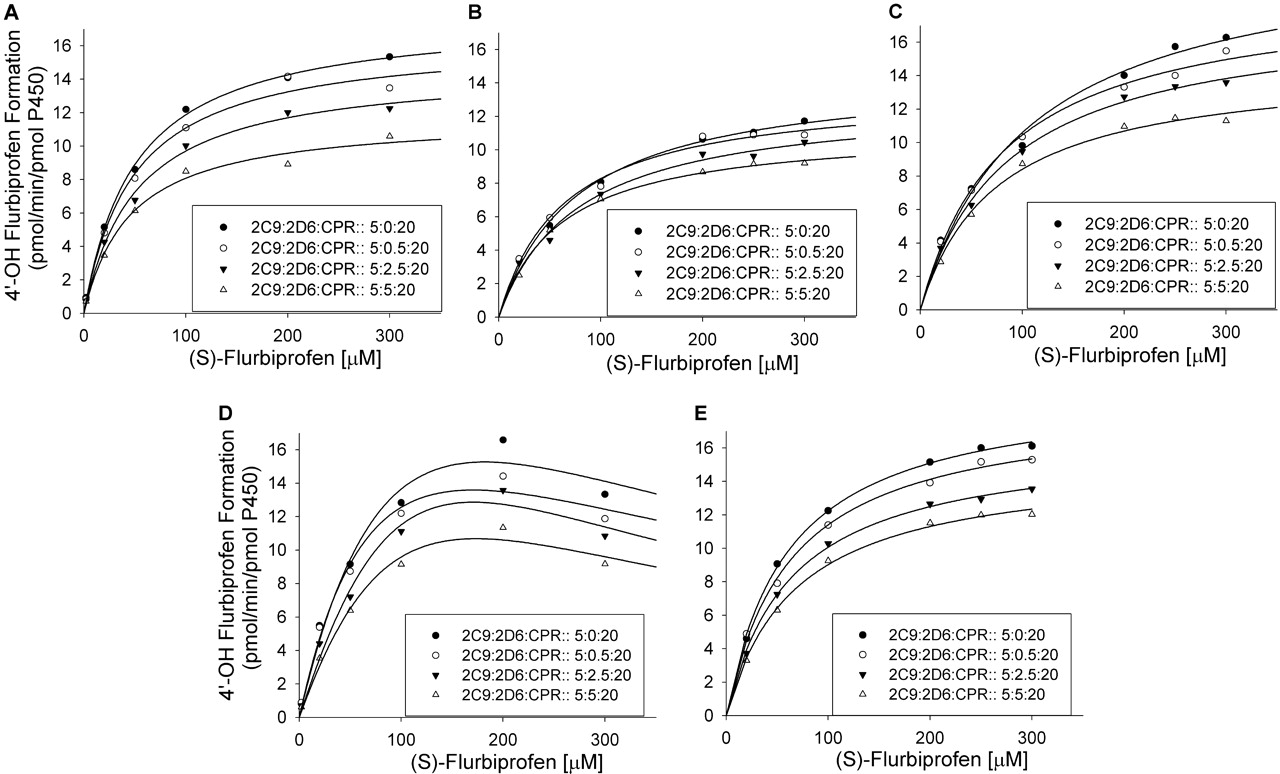
Effect of CYP2D6 on (S)-flurbiprofen metabolism by CYP2C9 at 20 pmol of CPR/incubation. A to E represent SeqAdd1, SeqAdd2, SeqAdd3, Allmix1, and Allmix2, respectively. Data fitting for the plots are shown. For clarity, representative data from a single experiment are presented. However, all experiments were conducted in triplicate over 3 separate days, and the mean values were used for inhibition degree determination. Single-factor analysis of variance was used for statistical comparisons. OH, hydroxy.

Effect of CYP2D6 on CYP2C9-mediated (S)-flurbiprofen metabolism for three levels of CYP2D6 and two levels of CPR. A to C depict the effect of CPR on the amount of inhibition for SeqAdd1, SeqAdd2, and SeqAdd3, respectively. The ratios of Vmax are significantly different (p < 0.05) from unity when the CYP2D6 levels are 2.5 and 5 pmol/incubation. In A and B, increasing CPR (20 pmol versus 10 pmol) did not affect the degree of inhibition. In C (SeqAdd3), a significant (p < 0.05) decrease in degree of inhibition was observed when CPR levels were 20 pmol and CYP2D6 was at 2.5 pmol/incubation, compared with 10 pmol of CPR. Single-factor analysis of variance was used for statistical comparisons.

Inhibition of CYP2C9-mediated metabolism of (S)-flurbiprofen when CYP2C9, CYP2D6, and CPR were mixed together simultaneously. A and B show the effect of CPR on the amount of inhibition for Allmix1 and Allmix2, respectively. Vmax ratios were significantly different (p < 0.05) from unity when the CYP2D6 levels were 2.5 and 5 pmol/incubation. In all cases, the degree of inhibition was reduced significantly (p < 0.05) when 20 pmol of CPR/incubation was used, compared with 10 pmol of CPR/incubation. Single-factor analysis of variance was used for statistical comparisons.

Effect of CYP2C9 on dextromethorphan metabolism by CYP2D6 at 20 and 40 pmol/incubation of CPR. No CYP2C9 (control) and 1:1 CYP2C9:CYP2D6 ratios were tested.
Discussion
Protein-protein interactions involving cytochrome P450 proteins can affect catalytic turnover in either a positive or negative manner and consequently affect in vitro-in vivo correlations of drug disposition. Several previous studies have demonstrated either inhibition or stimulation of P450-mediated metabolism due to protein-protein interactions (Cawley et al., 1995, 2001; Yamazaki et al., 1997; Hazai and Kupfer, 2005), but others have not observed an effect (Kelley et al., 2006), suggesting that the effects may be isoform- and/or substrate-dependent in a manner that affects the rate of metabolism. In the present work, it was demonstrated that CYP2D6 coincubation with CYP2C9 results in a gene dose-dependent inhibition of CYP2C9-mediated flurbiprofen hydroxylation with no effect on Km. This effect was not completely reversed by addition of more CPR. Furthermore, the order of mixing of the constituents had a measurable effect on the degree of inhibition noted. It is interesting to note that CYP2C9 had no effect on CYP2D6 catalytic activity. CYP2D6 expression is polymorphic, resulting in substantially different levels of expression of active protein among individuals, which may affect CYP2C9 activity in individuals and complicate in vitro-in vivo correlations.
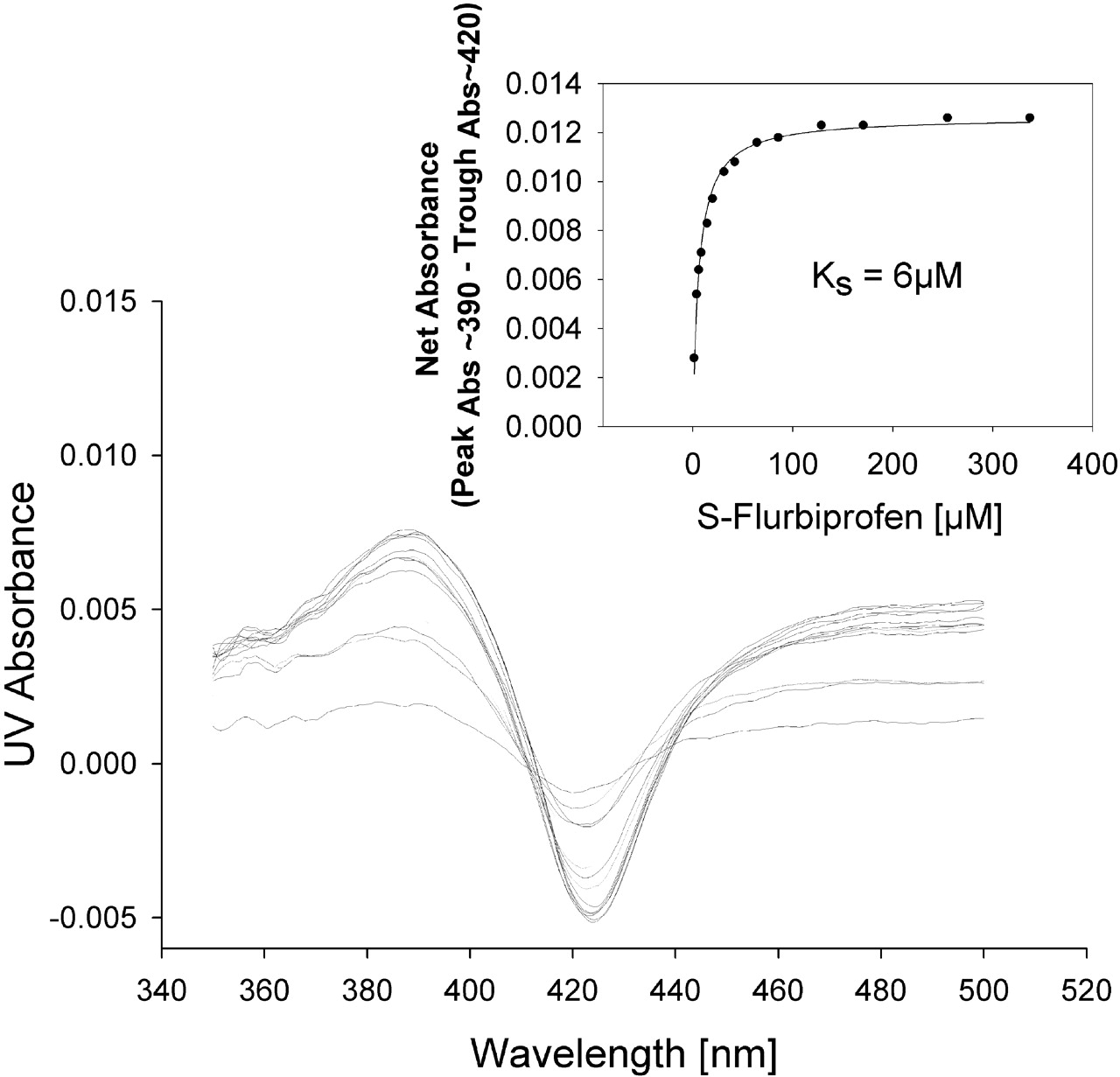
Spectral binding studies of (S)-flurbiprofen with CYP2C9. The inset shows the absorbance (Abs) difference as a function of (S)-flurbiprofen concentration used to calculate KS.
Although some of the parameters affecting protein-protein interactions have been explored, other factors such as the role of interactions on KS (affinity) of an enzyme and the role of order of addition of interacting enzymes have not been evaluated in a systematic manner. When equimolar amounts of CYP2D6 were added to CYP2C9, the spectral binding constant KS increased 20-fold, but the Km remained unchanged. This apparent paradox can be rationalized by examining the factors that make up KS and Km as shown in the following kinetic model: 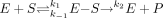 KS is the ratio of k–1/k1, where k–1 is the dissociation of the enzyme (E)-substrate (S) complex, k1 is the formation of the E-S complex, and k2 is the rate of product formation. Because Km is a sum of KS and the fraction k2/k1, for Km to remain the same, while KS increases 20-fold would require that k1 remains unchanged while k–1 increases and k2 decreases. This result suggests that CYP2D6 destabilizes the E-S complex, causing its dissociation to the individual components (E and S). It should be noted that Km ≅ KS when k2 is very small compared with k–1/k1; however, these values (Km ≅ KS) do not approach equality but rather KS becomes larger than Km. The reasons for these results are unclear but may be related to the slightly different conditions in which the two experiments (Km versus KS determinations) were performed such as slight differences in lipid concentrations.
KS is the ratio of k–1/k1, where k–1 is the dissociation of the enzyme (E)-substrate (S) complex, k1 is the formation of the E-S complex, and k2 is the rate of product formation. Because Km is a sum of KS and the fraction k2/k1, for Km to remain the same, while KS increases 20-fold would require that k1 remains unchanged while k–1 increases and k2 decreases. This result suggests that CYP2D6 destabilizes the E-S complex, causing its dissociation to the individual components (E and S). It should be noted that Km ≅ KS when k2 is very small compared with k–1/k1; however, these values (Km ≅ KS) do not approach equality but rather KS becomes larger than Km. The reasons for these results are unclear but may be related to the slightly different conditions in which the two experiments (Km versus KS determinations) were performed such as slight differences in lipid concentrations.

Spectral binding studies of (S)-flurbiprofen with CYP2C9 in the presence of CYP2D6. The inset shows the absorbance (Abs) difference as a function of (S)-flurbiprofen concentration used to calculate KS.

Spectral difference binding studies of (S)-flurbiprofen with CYP2D6. No change in binding states was observed.
The order of combination of the enzymes also has not been explored in a systematic manner previously. Kaminsky and Guengerich (1985) briefly evaluated the order of addition of the competing P450 after the addition of CPR, lipids, and substrate. This order of addition was found to have no influence on the amount of inhibition observed. In the current work presented here, Km was unaffected by protein-protein interactions; however, the order of combination of enzymes influenced the effect CPR had on the inhibition. The degree of inhibition was similar at subsaturating levels of CPR, regardless of the mixing scheme (Table 2). However, differences in the degree of inhibition were noted at saturating levels of CPR. In the Allmix1 and Allmix2 cases (enzymes + CPR mixed concurrently, sit for 10 min, and then add lipids), less inhibition was noted when additional CPR was added. These findings suggest that some portion of the reduction in CYP2C9 activity is due to the two enzymes competing for the CPR, because all constituents are allowed to equilibrate simultaneously. However, with sequential mixing (two of the components added first, allowed to equilibrate, and then the third component added) in all cases (SeqAdd1 and SeqAdd2) but one (SeqAdd3), additional CPR had no effect on the degree of inhibition.
Although the order of addition of enzymes does not have any in vivo significance, it can provide information on whether irreversible or tightly bound quasi-irreversible heteromers are formed. Gorsky and Coon (1986) demonstrated that when only P450 and CPR are combined, the order of mixing the enzymes and lipids was insignificant. However, when P450, CPR, and b5 were combined, the order of addition of b5 had a significant effect on the extent of metabolism. In the present study, three enzyme proteins were also being combined (CYP2C9, CYP2D6, and CPR), and the order of mixing of these enzymes also seemed to affect the rate of metabolism. In theory, reversible interactions between proteins should not be influenced by the order of addition, whereas the formation of tightly bound heteromers (quasi-irreversible interactions) would be affected by the order of mixing. In the current study this was also able to be probed by altering mixing order. Because mixing order affected the rate of metabolism in the current study, these results suggest that quasi-irreversible interactions are occurring when CYP2C9, CYP2D6, and CPR are combined.
In an attempt to explain the effects of order of addition observed in our system, a simplistic kinetic model is proposed below. The basis for the proposed model lies in the following assumptions: 1) P450s mixed together form heteromers with each other and CPR (Alston et al., 1991; Backes et al., 1998; Backes and Kelley, 2003; Hazai et al., 2005); 2) heteromer complexes retain catalytic activity (Backes et al., 1998); 3) only one P450 binding site exists on a single CPR molecule (Backes et al., 1998); and 4) additional combinations of proteins (e.g., CPR-CYP2C9-CYP2D6-CPR) can be formed but are found in lower amounts than the primary species (CYP2D6-CYP2C9-CPR) (Backes and Kelley, 2003).
The following mixing schemes represent the order of addition (only primary species presumed to be formed are shown).
1. SeqAdd1: 2C9_2D6_5mins_CPR 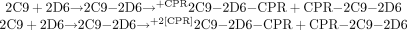 In this mixing scheme, doubling the amount of CPR does not produce a new species, but simply more of the same two species.
In this mixing scheme, doubling the amount of CPR does not produce a new species, but simply more of the same two species.
2. SeqAdd2: 2C9_CPR_5mins_2D6 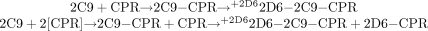 Doubling the amount of CPR results in a new species being formed, namely 2D6-CPR. However, no CYP2D6 substrate is present.
Doubling the amount of CPR results in a new species being formed, namely 2D6-CPR. However, no CYP2D6 substrate is present.
3. SeqAdd3: 2D6_CPR_5mins_2C9 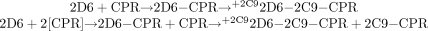 Analogous to the mixing scheme 2 above, doubling the amount of CPR results in a new species, namely 2C9-CPR. This is an active species because substrate is present, and, thus, this 2C9-CPR species also contributes to the formation of the 4-hydroxyflurbiprofen metabolite. Hence, in this mixing scheme, increasing the amount of CPR reduces the amount of inhibition because a “noninhibited species” i.e., 2C9-CPR is present, whereas no change in the degree of inhibition would be observed with mixing schemes 1 and 2 under the conditions of added CPR.
Analogous to the mixing scheme 2 above, doubling the amount of CPR results in a new species, namely 2C9-CPR. This is an active species because substrate is present, and, thus, this 2C9-CPR species also contributes to the formation of the 4-hydroxyflurbiprofen metabolite. Hence, in this mixing scheme, increasing the amount of CPR reduces the amount of inhibition because a “noninhibited species” i.e., 2C9-CPR is present, whereas no change in the degree of inhibition would be observed with mixing schemes 1 and 2 under the conditions of added CPR.
Inconsistent or differential incorporation of enzymes and CPR into the lipid matrix (DLPC) when two enzymes are coincubated in reconstituted systems could lead to changes in activity. Furthermore, differences in mobility of aggregated heteromers, compared with monomers, into the lipid matrix could also affect activity. Differences observed among the various mixing schemes could, in part, be due to lipid incorporation differences. However, triple-expression (Tan et al., 1997; Li et al., 1999) systems and microsomal systems (Kaminsky and Guengerich, 1985; Cawley et al., 2001) have also exhibited inhibition and activation, suggesting that differences in incorporation into lipids provides an incomplete explanation for the observed changes. It has been suggested that in vivo the ratio of total P450/CPR is 20:1, whereas in reconstituted systems the ratio used is 1:2 (Estabrook et al., 1971). What role the ratio (in vivo versus in vitro) of P450 to its redox partner CPR plays in this interaction is unclear. However, two studies (Kaminsky and Guengerich, 1985; Cawley et al., 2001) did observe a protein-protein interaction in microsomes from rats treated in vivo with enzyme-specific enzyme inducers, suggesting that the observations are not an artifact of the in vitro system. The other P450 redox partner, cytochrome b5, also can alter the catalytic activity and kinetic profile of P450 metabolism through mechanisms independent of provision of electrons (Jushchyshyn et al., 2005; Locuson et al., 2007), purportedly via conformational changes in the P450 induced by cytochrome b5. For simplicity, cytochrome b5 was omitted from the experiments described herein, but it should be recognized that the situation may be even more complicated than currently presented.
Several explanations have been put forth to explain, at least in part, P450 protein-protein interactions, including the role of ionic bonds between P450 proteins. Kelley et al. (2005) used high concentrations of buffer salts to eliminate any ionic interactions between CYP1A2-CYP2B4 and were able to regain most of the activity that had been diminished by the purported interactions. However, circular dichroism studies have demonstrated conformational changes in proteins at high salt concentrations (Yun et al., 1996), rendering the aforementioned studies somewhat ambiguous. It is possible that ionic interactions play a role in P450-P450 interactions but additional work in this area is needed.
Studies of P450 protein-protein interactions using either denatured or apoproteins also demonstrated inhibition of catalytic activity (Kaminsky and Guengerich, 1985), lending additional credence to the hypothesis that conformational changes occur as a result of the protein interactions. However, it must be recognized that these inactive proteins may also bind CPR, albeit to a nonproductive endpoint. Locuson et al. (2007), demonstrated that apo-b5 could stimulate CYP2C9-mediated metabolism despite its inability to provide electrons (Locuson et al., 2007). In addition, these results demonstrated that the apoprotein was not competing with the CPR for binding to the enzyme, because activity was stimulated rather than inhibited.
Cytochrome P450 2C9 has a relative large and flexible active site architecture that has been proposed to accommodate more than one substrate molecule simultaneously (Hummel et al., 2003), whereas it has been reported that CYP2D6 is characterized by a smaller active site volume and accepts primarily basic nitrogen and planar aromatic ring substrates (Rowland et al., 2006). This larger active site volume and flexibility of CYP2C9 compared with CYP2D6 may explain in part the differential inhibition of CYP2C9 if conformational changes induced by P450-P450 interactions are critical to the observed effects. The change in CYP2C9 affinity (KS) for substrate observed in the current studies is consistent with a conformational change in the active site architecture.
The current studies demonstrate in vitro interactions between two important human P450 enzymes (CYP2C9 and CYP2D6), resulting in a differential reduction in catalytic activity. The order of combining these enzymes yielded unexpectedly different results that could be explained by a simple kinetic model consistent with previous modeling results and, thus, providing indirect evidence for the formation of CYP2C9-CYP2D6 heteromers that further complex with CPR to form a reactive entity that results in altered metabolism rates and the potential to confound in vitro-in vivo correlations.
Footnotes
-
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM063215].
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.026500.
-
ABBREVIATIONS: P450, cytochrome P450; CPR, NADPH-cytochrome P450 reductase; b5, cytochrome b5; DLPC, dilauroylphosphatidylcholine; HPLC, high-performance liquid chromatography; KS, spectrally determined binding affinity.
- Accepted May 15, 2009.
- Received January 3, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}