


Abstract
The risk of idiosyncratic drug toxicity (IDT) is of great concern to the pharmaceutical industry. Current hypotheses based on retrospective studies suggest that the occurrence of IDT is related to covalent binding and daily dose. We determined the covalent binding of 42 radiolabeled drugs in three test systems (human liver microsomes and hepatocytes in vitro and rat liver in vivo) to assess the risk of IDT. On the basis of safety profiles given in official documentation, tested drugs were classified into the safety categories of safe, warning, black box warning, and withdrawn. The covalent binding in each of the three test systems did not distinguish the safety categories clearly. However, when the log-normalized covalent binding was plotted against the log-normalized daily dose, the distribution of the plot in the safety categories became clear. An ordinal logistic regression analysis indicated that both covalent binding and daily dose were significantly correlated with safety category and that covalent binding in hepatocytes was the best predictor among the three systems. When two separation lines were drawn on the correlation graph between covalent binding in human hepatocytes and daily dose by a regression analysis to create three zones, 30 of 37 tested drugs were located in zones corresponding to their respective classified safety categories. In conclusion, we established a zone classification system using covalent binding in human hepatocytes and daily dose for the risk assessment of IDTs.
Idiosyncratic drug toxicity (IDT) occurs rarely but is often very serious and appears as severe hepatotoxicity, agranulocytosis, neutropenia, Stevens-Johnson syndrome (SJS), or other illnesses. Because of its low frequency of occurrence (1/1000–1/100,000), IDT is often found late in drug development or in the postmarketing phase (Kaplowitz, 2005; Baillie, 2006; Uetrecht, 2007). In recent years, several drugs, including troglitazone, zomepirac, and tienilic acid, have been withdrawn from the market because of IDT, or the use of drugs has been limited by the addition of black box warnings to the label, as in the cases of flutamide, nevirapine, and valproic acid. For the pharmaceutical industry, it is important that drugs with the potential risk of IDT be screened out in the early phase of discovery and/or the development process. Unfortunately, conventional animal models of toxicity are poor predictors for clinical situations and the mechanisms of IDT are not fully understood despite many efforts to clarify them (Evans et al., 2004; Walgren et al., 2005; Masubuchi et al., 2007; Takakusa et al., 2008).
Current hypotheses based on retrospective studies suggest that the metabolic activation of a drug to a reactive metabolite and its covalent binding to cellular macromolecules are involved in the occurrence of IDT (Uetrecht, 2001; Zhou et al., 2007). Estimation of covalent binding to cellular macromolecules by using radiolabeled drugs is a direct and reliable method. There are several examples of reactive metabolites forming covalent bonds with IDT-causing drugs, such as tienilic acid, acetaminophen, and clozapine (Lecoeur et al., 1994; Hinson et al., 1995; Gardner et al., 1998). Evans et al. (2004) proposed a threshold level of 50 pmol/mg protein as a screening criterion of covalent binding to human liver microsomes (HLMs) in vitro and rat liver in vivo. A previous study by our group determined the covalent binding of a variety of drugs to HLMs in vitro and rat liver in vivo; these included drugs withdrawn from the market, drugs with black box warnings in the United States labeling, and some safe drugs. It was found that most of the problematic drugs exhibited higher HLM in vitro covalent binding than “safe” drugs (Takakusa et al., 2008).
Some reports suggest that the exposure to or daily dose of a drug may be related to the occurrence of IDT. Uetrecht (1999) reported that the occurrence of IDT is rare with drugs given at a daily dose of 10 mg or less. Walgren et al. (2005) also pointed out the contribution of high daily dose to IDT risk. For example, in the case of antidiabetic “glitazone” drugs, troglitazone caused a high incidence of IDT in patients and had to be withdrawn from the market, whereas rosiglitazone and pioglitazone do not show significant IDT risk even though they have similar chemical structures. The daily dose of troglitazone is 400 to 600 mg, whereas the daily doses of rosiglitazone and pioglitazone are 4 to 8 and 15 to 45 mg, respectively.
These retrospective studies suggest that the occurrence of IDT may be related to both covalent binding and exposure. To date, however, only limited numbers of systematic investigations have been reported with regard to the relationship between covalent binding, daily dose, and IDT (Obach et al., 2008). In this study, to assess the risk of IDT, we determined the covalent binding of 42 radiolabeled drugs in three test systems. These systems were HLMs, which are most commonly used for oxidative metabolism; human hepatocytes, which have a full component of cellular enzyme systems; and rat liver in vivo, which includes many biological processes such as absorption or tissue distribution and is realistic in the assessment of reactive metabolite formation in the body. From these data, we clarified the relationship between covalent binding, daily dose, and the safety profile. Finally, we established a zone classification system for the risk assessment of IDTs.
Materials and Methods
Materials. A total of 42 radiolabeled drugs were used. Amodiaquine, benzbromarone, carbamazepine, clozapine, clopidogrel, donepezil, flutamide, furosemide, imipramine, nevirapine, olanzapine, pioglitazone, rosiglitazone, sulfamethoxazole, tienilic acid, tacrine, valsartan, zafirlukast, and zomepirac (all 14C-labeled) were obtained from BlyChem Ltd. (Billingham, UK). Levofloxacin, olmesartan, pravastatin, and ticlopidine (all 14C-labeled) were obtained from Sekisui Medical Co., Ltd. (Ibaraki, Japan). 14C-Labeled atorvastatin was obtained from MDS Pharma Services (Montreal, QC, Canada). Celecoxib and warfarin (both 14C-labeled) and propranolol and tamoxifen (both 3H-labeled) were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Acetaminophen, aminopyrine, caffeine, diclofenac, erythromycin, procainamide, and valproic acid (all 14C-labeled) and ethinylestradiol and fluoxetine (both 3H-labeled) were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Indomethacin and phenytoin (both 14C-labeled) and 3H-labeled verapamil were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). 14C-Labeled amlodipine and 3H-labeled ritonavir were purchased from Morvek Biochemicals (Brea, CA). The specific radio activities of the 14C-radiolabeled compounds were 13 to 58 mCi/mmol, and 3H-radiolabeled compounds were diluted with cold compounds at the final activity of 200 mCi/mmol. Unlabeled acetaminophen, amodiaquine, benzbromarone, carbamazepine, clozapine, diclofenac, erythromycin, ethinylestradiol, fluoxetine, flutamide, furosemide, imipramine, indomethacin, phenytoin, propranolol, sulfamethoxazole, tacrine, tamoxifen, ticlopidine, verapamil, warfarin, and zomepirac were obtained from Sigma-Aldrich (St. Louis, MO). Unlabeled aminopyrine, amlodipine, and valproic acid were purchased from Wako Pure Chemicals (Osaka, Japan). Unlabeled nevirapine and olanzapine were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Unlabeled caffeine and zafirlukast were obtained from Fluka (Buchs, Switzerland) and Cayman Chemical (Ann Arbor, MI), respectively. Unlabeled atorvastatin, celecoxib, clopidogrel, donepezil, levofloxacin, olmesartan, pioglitazone, pravastatin, procainamide, rosiglitazone, tienilic acid, ritonavir, and valsartan were synthesized by Daiichi Sankyo Co., Ltd. (Tokyo, Japan) Pooled human microsomes (n = 50, mixed gender) were purchased from XenoTech, LLC (Lenexa, KS). NADP and glucose-6-phosphate dehydrogenase were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan) and glucose 6-phophate (G6P) was obtained from Sigma-Aldrich. Cryopreserved human hepatocytes (lots HH-286, HH-281, and HH-288) were purchased from BD Biosciences (San Jose, CA), and lot POP (pooled from five individuals) and lot SKI (pooled from 20 individuals) were purchased from In Vitro Technologies (Baltimore, MD). Williams' E medium was purchased from Sigma-Aldrich. All other reagents and solvents were of the highest grade commercially available.
Animals. Male Crj:CD(SD)IGS rats (4–8 weeks) were obtained from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). The rats were acclimatized for 1 week with a 12-h light/dark cycle in a humidity- and temperature-controlled environment and allowed free access to food and tap water until experimental use, whereupon food was withdrawn for 16 to 18 h before administration of the radiolabeled drugs. The rats were cared for and treated in accordance with the National Institute of Health Guidelines for Laboratory Animal Welfare (Institute of Laboratory Animal Resources, 1996). The Institutional Animal Care and Use Committee approved the protocols.
In Vitro HLM Covalent Binding Study. The experimental procedure was based on that used in a study reported previously (Masubuchi et al., 2007). The incubation mixture consisted of the following: 10 μM radiolabeled test drug (substrate), 2 mg/ml HLMs, 100 mM potassium phosphate buffer (pH 7.4), 25 mM glucose 6-phosphate, 2 units/ml glucose-6-phosphate dehydrogenase, and 10 mM MgCl2. The mixture was preincubated for 3 min at 37°C. A reaction was initiated by the addition of β-NADP+ to reach a final concentration of 2.5 mM, and the final incubation volume was 0.5 ml. Because the substrates were dissolved in acetonitrile, the final incubation mixture contained 1% (v/v) acetonitrile. Radiolabeled drugs of at least 95% purity were used. After incubation of the mixture for 1 h, the reaction was terminated by the addition of 0.5 ml of ice-cold acetonitrile. After vortexing, sonication was performed in an ultrasonic bath, and the mixture was centrifuged. The precipitated protein was serially washed twice with the following solvents: 80% (v/v) aqueous methanol containing 10% (w/v) trichloroacetic acid, diethyl ether-methanol (1:1, v/v), and 80% methanol. The resulting precipitated protein was dissolved in 0.5 ml 1.0 M NaOH, and aliquots were taken for a protein assay with a DC Protein Assay Kit (Bio-Rad, Hercules, CA) and also for the determination of radioactivity using a liquid scintillation counter after mixing of the aliquot with Hionic-Fluor scintillation cocktail (PerkinElmer Life and Analytical Sciences). The amount of the test drug-related material, as radioactivity covalently bound to the microsomal protein, was determined as the covalent binding (picomoles per milligram of protein). All of the experiments were performed in triplicate.
In Vitro Human Hepatocyte Covalent Binding Study. Cryopreserved human hepatocytes were carefully thawed in a water bath set at 37°C and were suspended in Williams' E medium at a final cell concentration of 1.0 × 106 cells/ml. The total cell count and the number of viable cells were determined by the trypan blue exclusion method, and hepatocytes with more than 70% viability were used. The final incubation volume was 1.5 ml on a six-well plate. The hepatocytes were preincubated for 5 min in a humidified 37°C incubator (5% CO2). Because the radiolabeled drugs were dissolved in methanol, the final incubation mixture contained 1% v/v methanol. Radiolabeled drugs of more than 95% purity were used. Reactions were initiated by adding the radiolabeled drugs at the final concentration of 10 μM. After 2 h of incubation in a humidified 37°C incubator, 0.45 ml of the suspension was sampled into 1 ml of 1 mM unlabeled solution in ice-cold methanol. After vortexing, sonication was applied in an ultrasonic bath, and then the mixture was centrifuged. The precipitated protein was immediately washed with 1 mM unlabeled solution in ice-cold methanol. After centrifugation, the precipitated protein was serially washed three times with each of the following solvents: dimethyl sulfoxide-methanol (1:4, v/v), methanol containing 25% (w/v) trichloroacetic acid, 100% methanol, and 80% methanol. The resulting precipitated protein was dissolved in 0.5 N NaOH and neutralized by adding 5 N HCl. Aliquots were taken, and the protein amount and radioactivity were determined as described above. All of the experiments were performed in triplicate.
Rat Liver in Vivo Covalent Binding Study. The experimental procedure was based on a study reported previously (Masubuchi et al., 2007). Radiolabeled drugs of more than 95% purity were used. Radiolabeled and unlabeled drugs were dissolved or suspended in 0.5% methylcellulose (400 centipoise) to prepare a solution at a concentration of 2 mg/ml as a free base or acid form for oral administration to fasted rats. After a single oral administration of each test drug at a dose of 20 mg/kg, the rats were exsanguinated at 2, 6, or 24 h (n = 3 animals for each time point), and liver samples were collected and stored frozen until analysis. The liver samples were weighed and then homogenized with aqueous 1.15% (v/v) KCl. In the same way as in the in vitro covalent binding study using HLMs, the liver homogenate was washed with organic solvents, followed by protein assay and determination of radioactivity covalently bound to the protein, as described above. The highest value of three time points was used for the further analysis of IDT risk assessment.
Data Analysis. Ordinal logistic regression analysis was performed to assess the relationship between covalent binding, daily dose, and safety category by the following equation using JMP 5.0.1 statistical software (SAS Institute, Cary, NC), 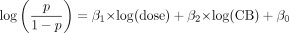 where p is the probability of each category, and the left side of the equation is the logit value between two categories. Dose is the daily dose of the tested drug, CB is the covalent binding in each test system, and β0, β1, and β2 are coefficient values of the equation. When the odds were unity between the categories, lines separating the zone were drawn where the logit values were zero and the equation was rearranged to yield the following:
where p is the probability of each category, and the left side of the equation is the logit value between two categories. Dose is the daily dose of the tested drug, CB is the covalent binding in each test system, and β0, β1, and β2 are coefficient values of the equation. When the odds were unity between the categories, lines separating the zone were drawn where the logit values were zero and the equation was rearranged to yield the following: 
Results
Classification of Tested Drugs. The tested drugs were classified into four categories on the basis of their safety profiles in the Physician's Desk Reference (1995, 2000, 2004, 2008) and Japanese drug labeling (Table 1). The first safety category, WDN, included drugs withdrawn from the market because of IDT in forms such as severe hepatotoxicity, agranulocytosis, neutropenia, and SJS. This category included four drugs: aminopyrine, amodiaquine, tienilic acid, and zomepirac. The second safety category, BBW, included drugs that had a black box warning for IDT in the Physicians' Desk Reference (2000, 2004, 2008). This category included eight drugs: benzbromarone, carbamazepine, clozapine, flutamide, nevirapine, ticlopidine, ritonavir, and valproic acid. Ritonavir was placed in the BBW category because of its black box warning about serious drug-drug interactions based on mechanism-based inhibition related to covalent binding; however, its labeling does not carry any alert regarding IDT (Koudriakova et al., 1998; Zhou et al., 2007). The third safety category, WNG, included drugs that did not have a black box warning but had a warning for IDT in the Physicians' Desk Reference (1995, 2004, 2008) or in Japanese labeling. This category included 18 drugs: acetaminophen, atorvastatin, celecoxib, clopidogrel, diclofenac, erythromycin, fluoxetine, furosemide, imipramine, indomethacin, phenytoin, procainamide, propranolol, sulfamethoxazole, tacrine, tamoxifen, verapamil, and zafirlukast. The last safety category, SAFE, included drugs with no warnings in the Physicians' Desk Reference (2004, 2008) or Japanese labeling. This category included 12 drugs: amlodipine, caffeine, donepezil, ethinylestradiol, levofloxacin, olanzapine, olmesartan, pioglitazone, pravastatin, rosiglitazone, valsartan, and warfarin. For the analysis below, the WDN and BBW categories were combined as BBW/WDN, because the difference between BBW and WDN was considered to depend on the clinical risk-benefit balances or the safety profile of the other drugs in the same class.
Information on tested drugs, typical daily doses, and safety profiles in relation to IDT

Daily doses of the test drugs categorized by safety profile. Numbers associated with symbols correspond to drug names as follows: 1, aminopyrine; 2, amodiaquine; 3, tienilic acid; 4, zomepirac; 5, benzbromarone; 6, carbamazepine; 7, clozapine; 8, flutamide; 9, nevirapine; 10, ritonavir; 11, ticlopidine; 12, valproic acid; 13, acetaminophen; 14, atorvastatin; 15, celecoxib; 16, clopidogrel; 17, diclofenac; 18, erythromycin; 19, fluoxetine; 20, furosemide; 21, imipramine; 22, indomethacin; 23, phenytoin; 24, procainamide; 25, propranolol; 26, sulfamethoxazole; 27, tacrine; 28, tamoxifen; 29, verapamil; 30, zafirlukast; 31, amlodipine; 32, caffeine; 33, donepezil; 34, ethinylestradiol; 35, levofloxacin; 36, olanzapine; 37, olmesartan; 38, pioglitazone; 39, pravastatin; 40, rosiglitazone; 41, valsartan; 42, warfarin.
Daily Dose of Tested Drugs. The daily doses of the tested drugs are shown in Table 1. Almost all of the daily dose data were obtained from the Physicians' Desk Reference (1995, 2000, 2004, 2008), except in the cases of amodiaquine and benzbromarone, because these two drugs have not been sold on the market in the United States. The data of daily dose for amodiaquine were obtained from a publication by Van den Broek et al. (2003) and for benzbromarone from Japanese drug labeling. For the analysis, the maximum dose in clinical use was used as the daily dose. Figure 1 shows the daily dose of the tested drugs in each safety category: SAFE, WNG, and BBW/WDN. Although the daily doses of WNG and BBW/WDN drugs tended to be higher than those of SAFE drugs, daily dose could not be used to distinguish the safety categories clearly.
Covalent Binding Study with Three Test Systems. We determined the covalent binding of as many as 42 radiolabeled drugs in HLMs, human hepatocytes in vitro, and rat liver in vivo (Table 2). Covalent binding of the 42 radiolabeled drugs in HLMs was determined by incubation for 1 h with the HLMs. The covalent binding of SAFE drugs ranged from 0.1 (levofloxacin) to 937.5 (ethinylestradiol) pmol/mg protein, that of WNG drugs ranged from 3.2 (sulfamethoxazole) to 417.4 (clopidogrel) pmol/mg protein, and that of BBW/WDN drugs ranged from 3.7 (carbamazepine) to 858.0 (ticlopidine) pmol/mg protein. We compared the covalent binding in HLMs of the drugs in all of the safety categories (Fig. 2A). This comparison was unable to distinguish the safety categories.
Covalent binding of tested drugs in HLMs and human hepatocytes in vitro and rat liver in vivo
Data are the mean ± S.D.
The covalent binding of 37 radiolabeled drugs in human hepatocytes was determined by incubation for 2 h with human hepatocytes. Tienilic acid, carbamazepine, erythromycin, furosemide, and indomethacin were not tested in the hepatocyte system because of insufficient purity of the radiolabeled drugs. The covalent binding of SAFE drugs ranged from 0.0 (levofloxacin) to 80.6 (ethinylestradiol) pmol/mg protein, that of WNG drugs ranged from 0.8 (sulfamethoxazole) to 209.2 (atorvastatin) pmol/mg protein, and that of BBW/WDN drugs ranged from 1.0 (aminopyrine) to 91.3 (amodiaquine) pmol/mg protein. We compared the covalent bindings in human hepatocytes of the drugs in all of the safety categories (Fig. 2B). As was the case in HLMs, comparison of covalent binding in human hepatocytes was not useful for distinguishing the safety categories.
Covalent binding of 42 drugs in rat liver in vivo was determined after a single administration of a 20-mg/kg dose of radiolabeled drug. A relatively high dose was chosen to highlight the potential of metabolic bioactivation and to balance maximizing analytical sensitivity with standardizing protocol. The covalent binding of SAFE drugs ranged from 0.0 (levofloxacin and olmesartan) to 210.2 (ethinylestradiol) pmol/mg protein, that for WNG drugs ranged from 1.4 (celecoxib) to 326.8 (imipramine) pmol/mg protein, and that for BBW/WDN drugs ranged from 13.6 (benzbromarone) to 555.7 (aminopyrine) pmol/mg protein. We compared the covalent bindings in rat liver in vivo of the drugs in all of the safety categories (Fig. 2C). Covalent binding in rat liver in vivo was not useful for distinguishing the safety categories.
Correlations among Covalent Bindings in the Three Test Systems. To clarify the correlations among the three test systems, the covalent bindings from the different systems were plotted in pairs. Figure 3 shows representative results for HLMs and rat liver in vivo. Application of a log-linear regression analysis revealed that between HLM and human hepatocytes the correlation coefficient (r) was 0.25, between HLMs and rat liver in vivo r = 0.56, and between human hepatocytes and rat liver in vivo r = 0.20. Weak correlations were therefore observed.
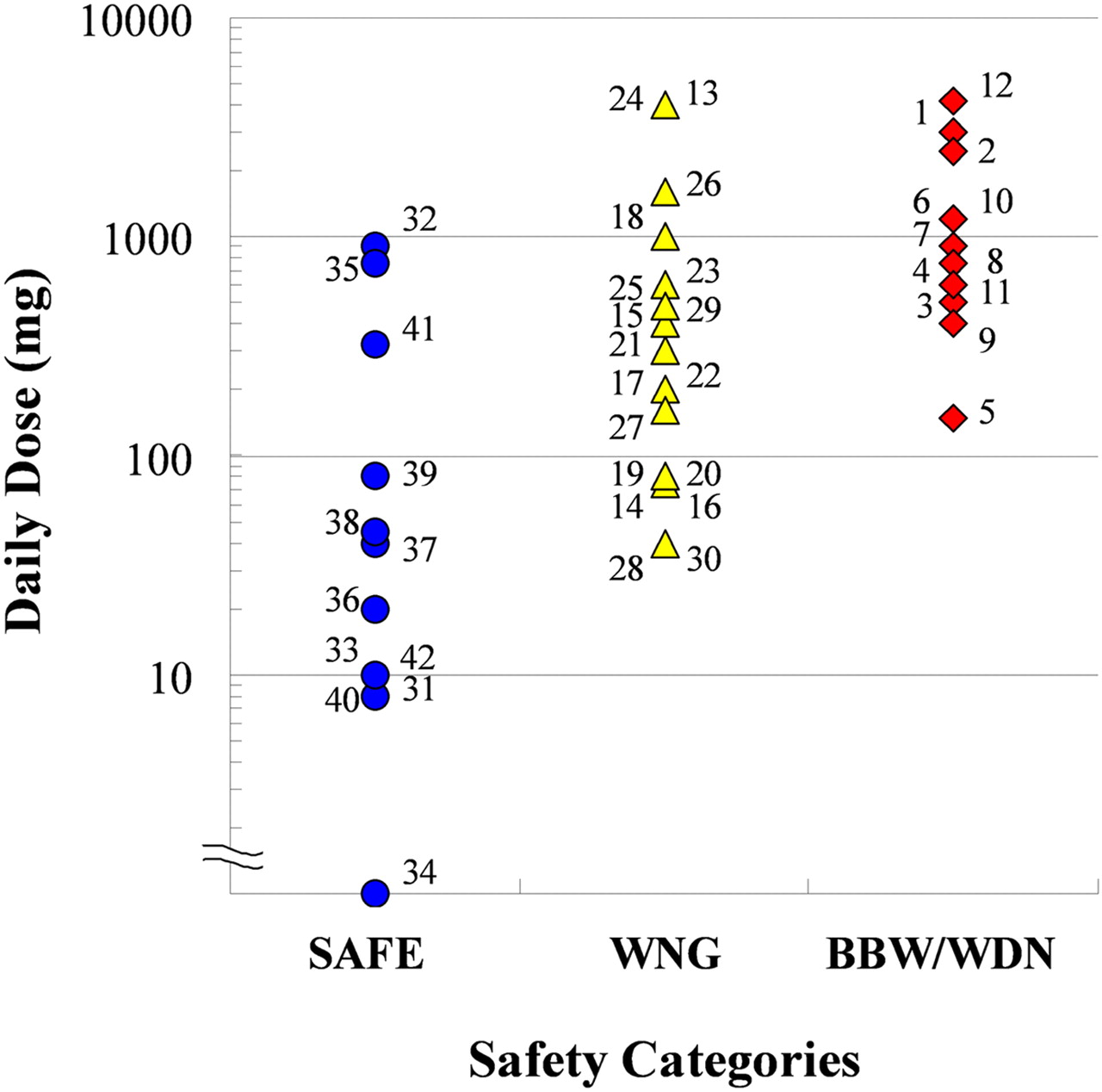
Relationships among Covalent Binding, Daily Dose, and Safety Category. The log-normalized covalent bindings in HLM, human hepatocytes, and rat liver in vivo were plotted against the log-normalized daily dose. Figure 4 is a representative result for human hepatocytes. Drugs with lower daily doses and lower covalent binding were safer, whereas drugs with higher doses and higher covalent binding were relatively problematic. To investigate the correlations between covalent binding, daily dose, and safety categories statistically, an ordinal logistic regression analysis was performed (Table 3). The results indicated that both covalent binding and daily dose were statistically significant in each of the three test systems and that daily dose was the more important factor, because the value of |β1| (the daily dose coefficient) was higher than that of |β2| (the covalent binding coefficient) in HLMs or human hepatocytes. Among the three test systems, classification using human hepatocytes showed the largest logit r2, with a value of 0.49, from the results of a whole-model test. From the results of the ordinal logistic regression analysis, two separation lines were drawn in each correlation figure for which the odds were unity between SAFE and WNG and between WNG and BBW/WDN (Fig. 4). We assigned these zones separated by lines as “acceptable,” “problematic,” and “unacceptable,” corresponding, respectively, to the safety categories SAFE, WNG, and BBW/WDN. Twelve of 14 SAFE drugs, 12 of 14 WNG drugs, and 5 of 8 BBW/WDN drugs were located in the acceptable, problematic, and unacceptable sections, respectively (Fig. 4).
Ordinal logistic regression analysis of daily dose and covalent binding in HLMs and human hepatocytes in vitro and rat liver in vivo
Interlot Differences in Covalent Binding in Human Hepatocytes. To investigate interlot differences in covalent binding, we used eight drugs to evaluate four lots of hepatocytes, including two lots from a single donor (HH-281 and HH-288) and two lots pooled from 5 or 20 individual donors (POP and SKI) to add to the data shown in Fig. 2B from lot HH-286. The eight drugs were zomepirac, clozapine, acetaminophen, atorvastatin, diclofenac, ethinylestradiol, olanzapine, and warfarin. Although there were 3-fold interlot differences in the covalent bindings of both acetaminophen and olanzapine, the covalent bindings of drugs that showed high-level binding, such as atorvastatin, clozapine, and ethinylestradiol, were not variable among lots (Fig. 5).
Discussion
To assess the risk of IDT caused by reactive metabolites, we established a zone classification system using daily dose and covalent binding in human hepatocytes, which was indicated by an ordinal logistic regression analysis as the best predictor. The zones, each separated by a border line along which the logit value was zero, were defined acceptable, problematic, and unacceptable. The safety categories were well separated by these zones. Although 7 of 37 tested drugs were located falsely, 4 of these 7 compounds were plotted near a border line. This good correlation means that this zone system can be used for IDT risk assessment. For example, a drug with a covalent binding of 50 pmol/mg protein may be acceptable at a dose less than 25 mg, problematic at a dose between 25 and 250 mg, and unacceptable at a dose greater than 250 mg (Fig. 4). Although this zone classification system is not an absolute criterion, because other factors such as therapeutic area, unmet medical needs, and the dosing period of the drug should also be taken into account, this assessment system should help in the screening of compounds at the drug discovery stage or in making a decision for further drug development using the covalent binding data and the range of the daily dose predicted from preclinical or clinical study data. The advantage of using this risk assessment system is the ability to estimate the risk at an earlier stage.
This study was based on the hypothesis that metabolic activation of a drug to a reactive metabolite and its covalent binding to cellular macromolecules are involved in the occurrence of IDT. Evans et al. (2004) proposed a “threshold” level of 50 pmol/mg protein for the screening criteria of covalent binding to HLMs in vitro and rat liver in vivo. This is the only previous study to have shown a targeted threshold for covalent binding. In our study, we determined the covalent bindings of as many as 42 radiolabeled drugs in three test systems: HLMs, human hepatocytes in vitro, and rat liver in vivo. Almost all of the tested BBW/WDN drugs (the exception being zomepirac) exceeded the 50 pmol/mg protein threshold in HLM and rat liver in vivo studies (Fig. 3). However, many of the WNG drugs did not exceed the threshold in each test system, and 4 of the 12 SAFE drugs were falsely classified as problematic. These results indicate that risk assessment by using only a threshold of 50 pmol/mg protein can lead to false judgments. On the other hand, when covalent binding was plotted against daily dose, the separation of the safety categories was clearer in terms of each drug's covalent binding. This finding means that covalent binding was an important risk factor in the occurrence of IDT.

Covalent bindings in HLMs (A) and human hepatocytes (B) in vitro and rat liver in vivo (C), categorized by safety profile. Numbers associated with symbols correspond to the same drug names as in the legend to Fig. 1. Each point is the mean of triplicate analyses.
Daily dose was also an important factor in the occurrence of IDT. Recent research suggests that IDT seems to accompany higher daily dosing drug regimens. Lammert et al. (2008), using two pharmaceutical databases and serious drug-induced injury case reports, reported a statistically significant relationship between daily dose and idiosyncratic drug-induced liver injury. As stated in the Introduction, Uetrecht (1999) reported that the occurrence of IDT is rare with drugs given at daily doses of 10 mg or less. On the basis of the results we obtained here with human hepatocytes, if the daily dose is 10 mg or less, then it will fall in the acceptable zone (up to a covalent binding of more than 250 pmol/mg protein) (Fig. 4)—higher than any of the covalent bindings observed, including those of clozapine and ticlopidine. Our results may, therefore, support Uetrecht's theory.

Correlation of covalent binding in HLMs and rat liver in vivo. Safety profiles of tested drugs are as follows: BBW/WDN, red; WNG, yellow; and SAFE, blue. Numbers associated with the symbols correspond to the same drug names as in the legend to Fig. 1.

Zone classification of IDT risk assessment in accordance with daily dose and covalent binding in human hepatocytes. Safety profiles of the tested drugs are as follows: BBW/WDN, red; WNG, yellow; and SAFE, blue. Numbers associated with the symbols correspond to the same drug names as in the legend to Fig. 1. The daily dose of ethinylestradiol was 0.035 mg, but to make the figure easy to see this drug has been plotted by using a larger value.
Categorization of the safety levels of drugs associated with IDT was important in our study. IDT usually refers to adverse drug reactions that occur rarely, typically appearing as, for example, hepatotoxicity, agranulocytosis, or SJS. However, the definition of IDT differs from report to report. We defined severe hepatotoxicity, agranulocytosis, neutropenia, and SJS as IDT and then classified the tested drugs into three safety categories based on their safety profiles in the Physicians' Desk Reference (1995, 2000, 2004, 2008) and in Japanese drug labeling (Table 1). For example, atorvastatin is widely used and is thought to be safe. However, here we categorized atorvastatin as WNG, for the following reasons: the occurrence of SJS was reported in the Physicians' Desk Reference (2008) after the drug was marketed, and severe hepatotoxicity events have recently been reported (Andrade et al., 2006; Clarke and Mills, 2006). In addition, the Physicians' Desk Reference (2008) lists liver dysfunction as an adverse event. A recent report showed that HLM in vitro covalent binding of nine hepatotoxic drugs and nine nonhepatotoxic drugs was not predictive of hepatotoxicity, with or without consideration of daily dose (Obach et al., 2008). However, unlike in our study, these authors analyzed only hepatotoxicity as the target toxicity. This difference in categorization may have been the cause of the different conclusion.

Interlot differences in covalent binding in human hepatocytes. Symbols correspond to hepatocyte lots, as follows: circles, HH-286; squares, HH-266; triangles, HH-281; bar, POP; and diamonds, SKI. Safety categories of the tested drugs are as follows: BBW/WDN, red; WNG, yellow; and SAFE, blue. The daily dose of ethinylestradiol was 0.035 mg, but to make the figure easy to see this drug has been plotted by using a larger value.
In this study, we used three test systems of covalent binding: HLMs, human hepatocytes, and rat liver in vivo. The correlation between covalent binding from two in vitro systems and rat liver in vivo was unclear (Fig. 3). This low correlation may be explained by a species difference in metabolism or involvement of a bioactivation pathway. Species differences do not seem to significantly influence the covalent binding for drugs that are bioactivated by cytochromes P450, because we indicated that there was a good correlation of the in vitro covalent binding between HLMs and rat liver microsomes in the previous study (Takakusa et al., 2008). Differences in the bioactivation pathway may have an effect on covalent binding when the site of generation of active metabolites is considered. In terms of risk assessment, it is possible to obtain false-negative results using only in vitro data because all of the reactive metabolites cannot be detected with absolute certainty using hepatocytes or HLMs. Hence, several types of studies, including rat liver in vivo, will be needed for the risk assessment of IDTs. In particular, higher daily dose drugs should be tested using several test systems because the higher dose is the crucial risk factor for IDTs.
Covalent binding in human hepatocytes was the best predictor of IDT risk, but interlot differences in covalent binding was of concern in risk assessment using human hepatocytes. We evaluated four lots of hepatocytes in adding to the data from lot HH-286. The interlot differences in the covalent bindings of acetaminophen and olanzapine were large, but the overall variability of the covalent bindings in five lots of human hepatocytes seemed to be within an acceptable range. Seven of eight drugs tested were located within the same classification zone, regardless of the lot of hepatocytes used. Therefore, IDT risk assessment based on the zone classification in Fig. 4 could be reliable using different lots of hepatocytes.
In conclusion, we established a zone classification system for risk assessment of drugs by using data on daily dose and covalent binding to human hepatocytes. This classification system will help in the selection of drug candidates and in decision-making during the drug discovery and/or development processes.
Acknowledgments
We thank Katsuhiro Igeta, Masaru Iwasaki, Yoshihiro Miyaji, Dr. Tsuneo Deguchi, Akiko Watanabe, Dr. Nobuaki Watanabe, Dr. Rie Nakagomi, Yukiko Metsugi, Norio Suzuki, Ryoko Sawamura, Tsuyoshi Karibe, Yuichiro Imamura, Daigo Asano, Takeshi Shiiki, Kanae Suzuki, Katsunobu Hagihara, Dr. Shinichi Inoue, Ken-ichi Itokawa, and Dr. Yasushi Yoshigae for assistance with the assays.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.027797.
-
ABBREVIATIONS: IDT, idiosyncratic drug toxicity; SJS, Stevens-Johnson syndrome; HLM, human liver microsome; WDN, drugs withdrawn from the market; BBW, drugs with a black box warning for IDT in the PDR; WNG, drugs without a black box warning but with a warning for IDT (severe hepatotoxicity, neutropenia, agranulocytosis, or SJS) in either the PDR or Japanese labeling; SAFE, drugs without any warning in either the PDR or Japanese labeling.
- Accepted May 27, 2009.
- Received March 31, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}