


Abstract
An investigation was conducted to follow up on the apparent species-dependent toxicity reported for 6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinoline (SGX523), a mesenchymal-epithelial transition factor (c-MET) inhibitor that entered clinical development for the treatment of solid tumors. Patients treated with SGX523 exhibited compromised renal function presumably resulting from crystal deposits in renal tubules. Our independent metabo‘lite profiling of SGX523 indicates that a major NADPH-independent, late-eluting metabolite [6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinolin-2(1H)-one (M11)] was generated by monkey and human liver S-9, and to a lesser extent by rat S-9, whereas M11 was absent in dog S-9 incubations. We confirmed the identity of M11 as 2-quinolinone-SGX523. Experiments with various molybdenum hydroxylase inhibitors showed that aldehyde oxidase (AO), and not xanthine oxidase, metabolized SGX523 to M11 in monkey and human liver cytosol. In addition, the oxygen incorporated into M11 was derived from water rather than atmospheric oxygen, corroborating M11 formation via AO. After oral dosing in monkeys, metabolite profiling of plasma and urine showed that SGX523 was indeed metabolized to M11 and its N-demethyl analog (M8). In urine, M11 levels were ∼70-fold greater than that of SGX523, and the solubility of M11 in urine was only 3% of that of SGX523. In summary, SGX523 is metabolized by AO in a species-specific manner to a markedly less-soluble metabolite, M11. We propose that M11 is likely involved in the observed obstructive nephropathy reported in clinical studies. Moreover, this study illustrates the need to conduct thorough metabolic evaluations early in drug development to select the most relevant nonclinical species for toxicological evaluation.
There has been much effort during the past several years to develop targeted therapy for oncological indications to improve efficacy and tolerability. One compelling therapeutic target is mesenchymal-epithelial transition factor (c-MET), a receptor tyrosine kinase implicated in the control of multiple signal transduction pathways involved in tumor growth and metastasis (Liu et al., 2008). 6-(6-(1-Methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinoline (SGX523) is an orally bioavailable, potent, and selective small molecule inhibitor of c-MET, and this compound was one of the first selective c-MET inhibitors to be evaluated in patients (Buchanan et al., 2009). Because the microsomal metabolism profile of SGX523 was similar among rats, dogs, monkeys, and humans, investigational new drug-enabling studies were conducted in rats and dogs (Burley, 2009). The SGX523 Phase I study was started at a dose of 40 mg in patients. After escalating to doses >80 mg of SGX523 in patients, acute renal failure was observed as evidenced by increased serum creatinine. The analysis of samples from the discontinued clinical trial revealed a metabolism profile different from that of the preclinical species studied. Subsequent in vivo studies using monkeys showed obstructive nephropathy with intratubular crystal formation, consistent with human data, but the mechanism involved in the toxicity has not been reported (Burley, 2009). In an effort to better understand this apparent species difference in metabolism and the consequent development of drug-induced crystal nephropathy in a species-specific manner, we prepared SGX523 and studied its metabolite profile.
Materials and Methods
Chemicals and Reagents.
All the chemicals, including allopurinol, oxipurinol, methotrexate, menadione, raloxifene, proadifen hydrochloride (SKF525-A), and H218O (97 atom % 18O), were purchased from Sigma-Aldrich (St. Louis, MO). Male and female pooled liver S-9 fractions prepared from IGS Sprague-Dawley rats, beagle dogs, cynomolgus monkeys, or humans were purchased from XenoTech, LLC (Lenexa, KS). Human hepatic cytosol and cynomolgus monkey hepatic cytosol and microsomes were purchased from XenoTech, LLC and Celsis In Vitro, Inc. (Baltimore, MD).
SGX523 was synthesized by Incyte Corporation (Wilmington, DE) according to the patent (WO 2008/051808 A2) by Bounaud et al. (2008) and was isolated as an amorphous free base [dimethyl sulfoxide (DMSO)-d6, 500 MHz]: δ 8.88 (dd, J = 4.1, 1.7, 1H), 8.43 (d, J = 9.8, 1H), 8.42 (s, 1H), 8.33 (dd, J = 8.4, 1.5, 1H), 8.13 (d, J = 2.2, 1H), 7.99 (s, 1H), 7.97 (d, J = 8.8, 1H), 7.76 (d, J = 9.7, 1H), 7.73 (dd, J = 8.9, 2.2, 1H), 7.53 (dd, J = 8.4, 4.2, 1H), 3.88 (s, 3H); m/z 360 [M+H]+ [literature (DMSO-d6, 500 MHz): δ 8.86 (dd, 1H), 8.34 (dd, 1H), 8.26 (s, 1H), 8.24 (d, 1H), 8.17 (d, 1H), 8.01 (d, 1H), 7.99 (s, 1H), 7.82 (dd, 1H), 7.74 (d, 1H), 7.57 (dd, 1H), 3.95 (s, 3H)].
6-(6-(1-Methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinolin-2(1H)-one (2-quinolinone-SGX523, M11) was synthesized by Incyte Corporation from 6-(1-methyl-1H-pyrazol-4-yl)[1,2,4]triazolo[4,3-b]pyridazine-3-thiol and 6-bromoquinolin-2(1H)-one (Zaragoza et al., 2005) by methods analogous to those described in patent WO 2008/051808 A2 applied for by SGX Pharmaceuticals. The material was isolated as amorphous free base (DMSO-d6, 500 MHz): δ 8.49 (s, 1H), 8.37 (d, J = 9.6, 1H), 8.08 (d, J = 0.6, 1H), 7.97 (d, J = 2.1, 1H), 7.87 (d, J = 9.6, 1H), 7.73 (d, J = 9.5, 1H), 7.67 (dd, J = 8.6, 2.1, 1H), 7.26 (d, J = 8.7, 1H), 6.49 (d, J = 9.5, 1H), 3.92 (s, 3H); m/z 376 [M+H]+.
In Vitro Enzyme Assays.
SGX523 was incubated in triplicate at 37°C in a shaking water bath with liver S-9 fractions prepared from rats (n = 400, mixed gender), beagle dogs (n = 10, mixed gender), cynomolgus monkeys (n = 10, mixed gender), or humans (n = 50, mixed gender). Incubations included liver S-9 fraction (1 mg of protein/ml), potassium phosphate buffer (50 mM, pH 7.4) with or without NADPH (1.0 mM), and SGX523 (5 μM), which was used to initiate the reaction. In a second experiment, SGX523 was incubated in triplicate at 37°C in a shaking water bath with hepatic microsomes, S-9, or cytosol (1 mg of protein/ml) prepared from cynomolgus monkeys (n = 10, mixed gender for each preparation) and potassium phosphate buffer (50 mM, pH 7.4) with or without NADPH (1.0 mM) at the final concentrations indicated. For these incubations, SGX523 (5 μM) was used to initiate the reaction. In a third experiment, the effects of various molybdenum hydroxylase inhibitors on the formation of M11 in monkey and human liver cytosol were examined. These chemical inhibitor studies were carried out at 37°C in a shaking water bath and included monkey or human liver cytosol (1 mg/ml), potassium phosphate buffer (50 mM, pH 7.4), and SGX523 (5 μM) with or without the chemical inhibitor. The chemical inhibitors allopurinol (Massey et al., 1970), oxipurinol (Massey et al., 1970), and methotrexate (Lewis et al., 1984) were used to selectively inhibit cytosolic xanthine oxidase (XO), whereas menadione (Johns, 1967), raloxifene (Obach, 2004), and SKF525-A (Robertson and Bland, 1993) were used to selectively inhibit cytosolic aldehyde oxidase (AO). These chemical inhibitors were added in DMSO at final concentrations of 100 μM, except for raloxifene, which was used at a final concentration of 20 μM. For all the experiments, the final concentration of DMSO was less than 0.15% (v/v). All the in vitro metabolism incubations were terminated by the addition of 2 volumes of cold methanol/acetonitrile (50:50, v/v). After mixing, samples were centrifuged at ∼1500g for 30 min, and the resulting supernatants were analyzed by liquid chromatography/mass spectrometry (LC/MS).
H218O Incorporation into Metabolites.
Incorporation of oxygen from the medium into phase I metabolites was assessed by incubation of SGX523 (10 μM) at 37°C in a shaking water bath with cynomolgus monkey liver S-9 fraction (n = 10, mixed gender), potassium phosphate buffer (62 mM, pH 7.4), with or without NADPH (1.0 mM), and H218O (33% v/v). Reactions were terminated at 0 and 30 min by the addition of 1 volume of cold acetonitrile/methanol/DMSO (1:1:1, v/v). After mixing, samples were centrifuged, and the resulting supernatants were analyzed by LC/MS using the method detailed below for metabolite profiling. Fractional H218O content of oxygenated metabolites was calculated using the metabolite isotopic profile, determined using information-dependent acquisition-triggered enhanced resolution scans and correcting for natural isotopic abundance, as determined from the corresponding metabolite in H216O incubations.
Pharmacokinetics of SGX523 in Monkeys.
Male cynomolgus monkeys (5 kg, n = 2 for each route of administration) received a single intravenous or oral dose of SGX523 after an overnight fast (approximately 10 h) in a parallel study design. The intravenous and oral doses were 1.6 and 6.2 mg/kg, respectively, and the monkeys were fed 4 h after dosing. SGX523 was formulated in 5% dimethyl acetamide, 10% propylene glycol in saline for intravenous dosing and using 5% dimethyl acetamide in 0.5% methylcellulose for oral dosing. SGX523 and M11 concentrations in plasma and urine samples collected from 0 to 24 h postdose were determined by a quantitative LC/MS/MS analysis method. In addition, these samples were used for metabolite profiling by LC/MS/MS.
Equilibrium Solubility Determinations.
The solubilities of SGX523 and M11 were determined in 0.1 N HCl, pH 1.0; water, pH 6.8; potassium phosphate buffer, 0.1 M, pH 7.4; and in blank control monkey urine, pH 8.4. Compound was accurately weighed, and 1 ml of the appropriate matrix was added. Vials were capped and incubated at 37°C overnight. Samples were then centrifuged and filtered using a 0.45-μm filter, and the concentrations of SGX523 or M11 in the filtrate were determined by the quantitative LC/MS/MS analysis method described below. For both SGX523 and M11, experiments were conducted to determine the nonspecific binding to the filter. Experiments showed that SGX523 was not retained on the filter. For M11, 20 to 40% of the compound was retained on the filter in various matrices; therefore, filtrate concentrations were corrected accordingly.
Analytical Methods for Quantitative Analysis.
The concentration of M11 in samples generated from in vitro studies was determined using electrospray ionization LC/MS with a Thermo Finnigan LCQ Deca-XP Plus ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) operated in positive ionization mode. Separation of SGX523 and its metabolites was achieved using a Jones Chromatography Genesis AQ high-performance liquid chromatography column (2.0 × 150 mm, 4 μm; Grace, Deerfield, IL) with a gradient elution scheme consisting of mobile phase A, 5 mM ammonium formate in water, pH 3.2; and mobile phase B, acetonitrile. Mobile phase B was increased linearly from 25% to 95% over 8 min using a flow rate of 600 μl/min. The M11 metabolite (m/z 376.0) peak was integrated from extracted ion chromatograms, and the amounts were determined from a standard curve based on peak area. Calibration curves generally were linear from 0.020 to 1.0 μM.
The concentration of SGX523 and M11 in plasma samples and solubility studies were determined by another LC/MS/MS method. The monkey plasma samples were subjected to protein precipitation with 4 volumes of 90% acetonitrile in methanol, centrifuged, and the resulting supernatants were dried down and reconstituted with a 25% acetonitrile in water and then subjected to LC/MS/MS analysis. Plasma supernatants and filtrates from solubility studies were injected onto a Phenomenex (Torrance, CA) Synergi 4-μm polar-RP 50 × 2-mm 80A coupled to Sciex API 3000 triple quadruple mass spectrometer (AB Sciex, Foster City, CA), and separation was achieved using a gradient elution consisting of mobile phase A, 0.1% formic acid in water, and mobile phase B, 0.1% formic acid in acetonitrile. Mobile phase B was increased linearly from 5% to 65% over 5 min using a flow rate of 300 μl/min. The ionization was optimized using electrospray ionization, and detection was via multiple reaction monitoring (MRM) of the characteristic ion dissociation transition m/z 360.0 → 160.0 for SGX523 and m/z 376.0 → 176.0 for metabolite M11. This method was linear over the range 0.002 to 2.5 μM.
The monkey urine samples were precipitated with 1 volume of acetonitrile/methanol/DMSO (1:1:1, v/v), sonicated, vortex-mixed, and centrifuged. The supernatants were then diluted 10-fold with 1:1 control urine/organic solvents (acetonitrile/methanol/DMSO, 1:1:1, v/v) and analyzed using electrospray ionization LC/MS/MS with an Applied Biosystems (Foster City, CA) 4000 Q TRAP hybrid triple quadrupole/linear ion trap mass spectrometer operated in positive ionization mode. SGX523 and its metabolites were separated on an ACE C18 column (4.6 × 150 mm, 3 μm; Advanced Chromatography Technologies, Aberdeen, Scotland, UK) using a gradient composed of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) that increased linearly from 10 to 35% over 50 min with a flow rate of 600 μl/min. Quantitation was accomplished via MRM of the characteristic ion dissociation transition m/z 360.0 → 160.0 for SGX523 and m/z 376.0 → 176.0 for M11. The standard curve ranged from 0.06 to 4.9 μM and 0.37 to 30 μM for SGX523 and M11, respectively.
Analytical Methods for Metabolite Profiling.
For metabolite profiling, plasma samples from individual monkeys were pooled according to the Hamilton pooling method (Hamilton et al., 1981), whereas urine samples were analyzed individually. Aliquots of plasma and urine samples were precipitated with 1 or 2 volumes of acetonitrile/methanol/DMSO (1:1:1, v/v), placed in a sonicating water bath for 10 min, vortex-mixed, and then centrifuged. The resulting supernatants were then subjected to direct LC/MS/MS analysis. SGX523 metabolism samples from both the in vitro S-9 incubations and the in vivo studies were analyzed using electrospray ionization LC/MS/MS with an Applied Biosystems 4000 Q TRAP hybrid triple quadrupole/linear ion trap mass spectrometer operated in positive ionization mode. SGX523 and its metabolites were separated on an ACE C18 column (4.6 × 150 mm, 3 μm; Advanced Chromatography Technologies) using a gradient composed of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B) that increased linearly from 10% to 35% over 50 min with a flow rate of 600 μl/min. Samples were analyzed using a linear ion trap-enabled enhanced Q1 MS or MRM survey scans in conjunction with information-dependent acquisition–triggered enhanced resolution and/or enhanced product ion scans. In the case of MRM analysis, MS/MS transitions for metabolites were selected based on the fragmentation pattern of the parent molecule.
Results
Marked Species Difference in the Metabolism of SGX523 by Rat, Dog, Monkey, and Human Liver S-9 Fractions.
SGX523 was metabolized via both NADPH-dependent and non–NADPH-dependent enzymes. Figure 1 shows that after incubation of SGX523 in rat, dog, monkey, and human liver S-9 fractions, the NADPH-dependent metabolites are relatively polar compared with parent and similar among species. Metabolites M5 (N-desmethyl, m/z 346 [M+H]+), M6 (monooxygenation, m/z 376 [M+H]+), and M9 (monooxygenation, m/z 376 [M+H]) were held in common across the species. In addition, trace amounts of M3 (N-desmethyl monooxygenation, m/z 362 [M+H]+) were detected in all the species examined. M4 (dioxygenation, m/z 392 [M+H]+) was detected at trace levels in all but the dog liver S-9 incubations. The abundance of these metabolites was decreased by the pan-cytochrome P450 (P450) inhibitor, aminobenzotriazole (data not shown). In contrast, a major late-eluting and therefore presumed to be less polar metabolite (M11, m/z 376 [M+H]+) was detected in monkey and human liver S-9 incubations. This metabolite was formed in the absence of NADPH. The identity of this oxidative metabolite was established as 2-quinolinone-SGX523 (m/z 376 [M+H]+) by cochromatography and comparison of mass spectral data with a synthetic standard prepared by Incyte Corporation. Whereas metabolite M11 was not detected in dog liver S-9, it was present in trace amounts in rat liver S-9 incubations. In addition, M8 (desmethyl 2-quinolinone SGX523, m/z 362 [M+H]+), a minor metabolite, was detected in monkey and human liver S-9 incubations but not in rat or dog liver S-9 incubations.

The in vitro metabolism of SGX523 by hepatic S-9 fractions from rats, dogs, monkeys, and humans in the presence of NADPH. SGX523 (5 μM) was incubated at 37°C with S-9 fractions from various species (1 mg of protein/ml) and potassium phosphate buffer (50 mM, pH 7.4) with NADPH (1.0 mM). Incubations were terminated with organic, centrifuged, and the resulting supernatants analyzed by LC/MS.
Metabolite M11 Is Formed by Aldehyde Oxidase.
As shown in Fig. 2, M11 was formed by monkey liver cytosol and monkey liver S-9 independent of NADPH. Molybdenum hydroxylases were the likely enzymes producing M11, given that this reaction was cytosolic and NADPH-independent. We were surprised to find that small amounts of M11 were produced by monkey liver microsomes in the absence of NADPH, which is likely a result of the contamination of the microsomes with cytosolic enzymes. It is likely that less M11 was produced by S-9 in the presence of NADPH (versus in the absence of NADPH) because of metabolism via P450 pathways. To further identify the specific molybdenum hydroxylase enzyme responsible for the formation of M11, SGX523 was incubated with either monkey or human liver cytosol along with chemical inhibitors of either XO or AO (Table 1). Allopurinol, oxipurinol, and methotrexate, all selective inhibitors of XO, did not inhibit the formation of M11. However, menadione, a selective inhibitor of AO, significantly inhibited the formation of M11 to 9 and 11% of the control (no inhibitor) in monkey and human cytosol, respectively. Raloxifene inhibited the formation of this metabolite to 43 and 16% of control in monkey and human cytosol, respectively. SKF525-A, a reported inhibitor of AO, did not inhibit the formation of this metabolite in monkey cytosol (95% of control), whereas M11 formation was inhibited by SKF525-A in human cytosol (62% of control). It should be noted that inhibition of molybdenum hydroxylases is less understood than is the case with P450s, and the same AO inhibitor can exhibit different inhibitory potentials among species and for different marker substrates (Sahi et al., 2008). Together, these data suggested that AO and not XO is the enzyme responsible for the formation of the M11 metabolite in the monkey and human.
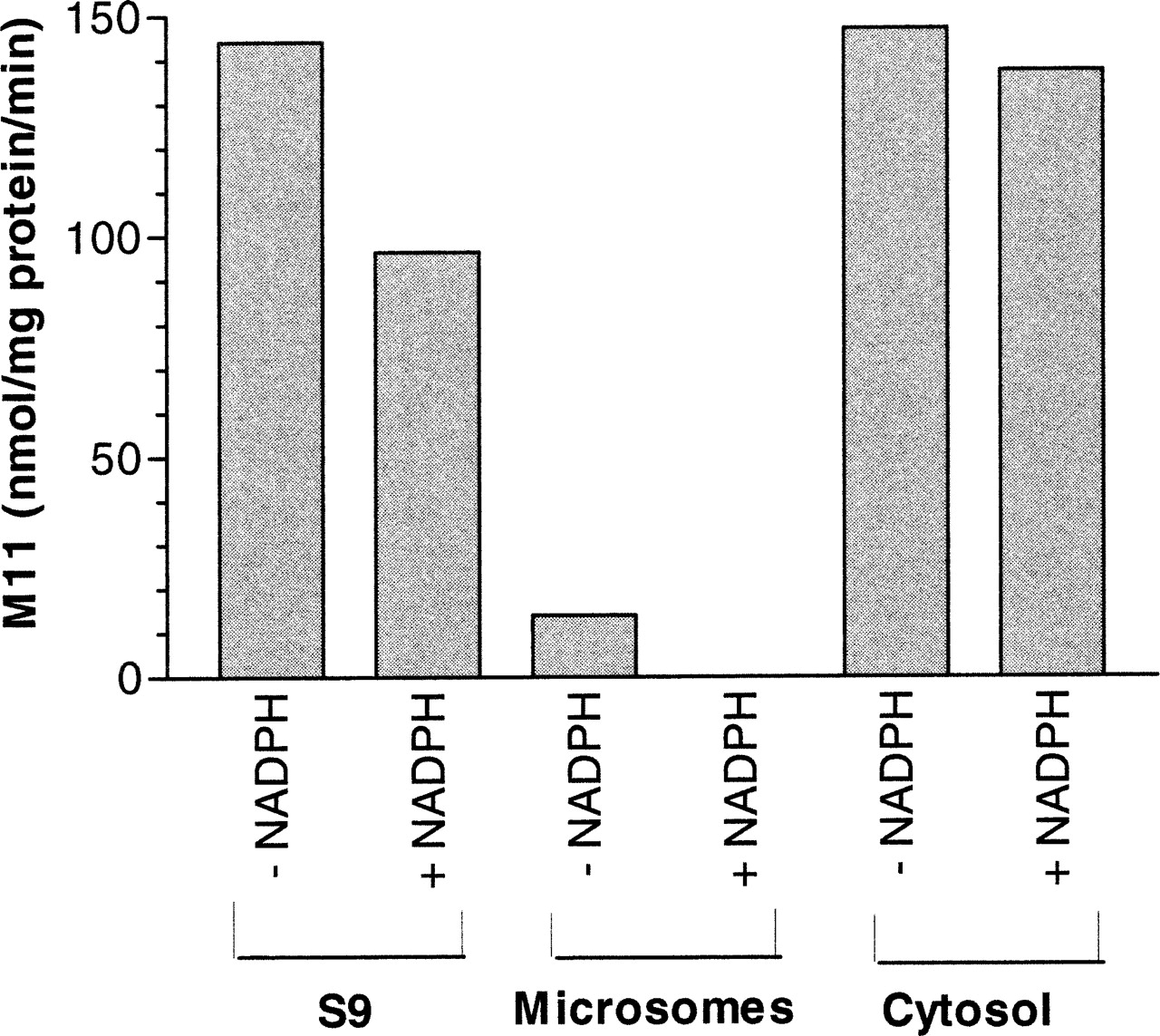
The effect of NADPH on the formation of M11 by monkey liver subcellular fractions. SGX523 (5 μM) was incubated at 37°C with microsomes, S-9, or cytosol (1 mg of protein/ml) prepared from cynomolgus monkeys, as well as potassium phosphate buffer (50 mM, pH 7.4) with or without NADPH (1.0 mM). Incubations were terminated after 30 min with the addition of methanol/acetonitrile, centrifuged, and the resulting supernatants analyzed by LC/MS.
Effect of various molybdenum hydroxylase inhibitors on the formation of metabolite M11 in monkey and human liver cytosol
These chemical inhibitor studies were carried out at 37°C in triplicate and included liver cytosol (1 mg/ml), potassium phosphate buffer (50 mM, pH 7.4), and SGX523 (5 μM) with or without the chemical inhibitors of AO and XO at the final concentrations shown. Incubations were terminated with methanol/acetonitrile after 30-min incubation. After centrifugation, the resulting supernatants were analyzed by LC/MS.
Water Is the Source of Oxygen Atom in M11.
LC/MS analysis of SGX523 incubations with cynomolgus monkey liver S-9 fraction in the presence of H218O indicates that 100% of the oxygen incorporated into M11 was sourced from the medium (Fig. 3). MS/MS analysis of the molecular ions of 16O- and 18O-incorporated M11 ([M+H]+ = 376 and 378, respectively) clearly shows the 2-atomic mass unit shift in the parent ion and in the mercaptoquinolinone fragment. Other oxygenated metabolites were NADPH-dependent and clearly did not contain any oxygen from water (data not shown). Because AO is an oxidase and not an oxygenase, the oxygen incorporated into an AO substrate is derived from water rather than atmospheric oxygen; therefore, these results corroborate M11 formation via AO metabolism of SGX523.

MS/MS spectra of M11 formed after incubation of SGX523 with monkey liver S-9 fraction, potassium phosphate buffer, pH 7.4, and either H216O (A) or H218O (B).
M11 Is a Major Metabolite Detected in Monkey Plasma and Urine.
LC/MS/MS analysis of plasma and urine from cynomolgus monkeys dosed orally with SGX523 shows that M11 is a major metabolite in plasma and urine for this species (Fig. 4). In addition to M11, M5 (N-desmethyl, m/z 346 [M+H]+), M6 (monooxygenation, m/z 376 [M+H]+), and M8 (desmethyl 2-quinolinone SGX523, m/z 362 [M+H]+) were the most abundant metabolites detected in plasma. Other phase I metabolites were observed at trace to minor levels: M1 (N-desmethyl dioxygenation, m/z 378 [M+H]+), M2 (dioxygenation, m/z 392 [M+H]+), M3 (N-desmethyl monooxygenation, m/z 362 [M+H]+), M4 (dioxygenation, m/z 392 [M+H]+), M7 (N-desmethyl monooxygenation, m/z 362 [M+H]+), M9 (monooxygenation, m/z 376 [M+H]+), M10 (monooxygenation, m/z 376 [M+H]+), and M12 (dioxygenation, m/z 392 [M+H]+). In urine, only small amounts of intact SGX523 were detected (peak area = 3% of total chromatographed), whereas the M11 metabolite and its desmethyl analog (M8) were the most abundant metabolites observed, representing 23 and 18% of the total area chromatographed, respectively. In addition, M1, M2, M3, M4, M5, M6, M7, M9, M10, M12, M13 (N-desmethyl monooxygenation, m/z 362 [M+H]+), and M14 (N-desmethyl dioxygenation, m/z 378 [M+H]+) were detected, albeit to a lesser extent. Estimation of the quantities of metabolites is based solely on full-scan mass spectral response, which is highly analyte-specific and considered semiquantitative in the absence of authentic standards.
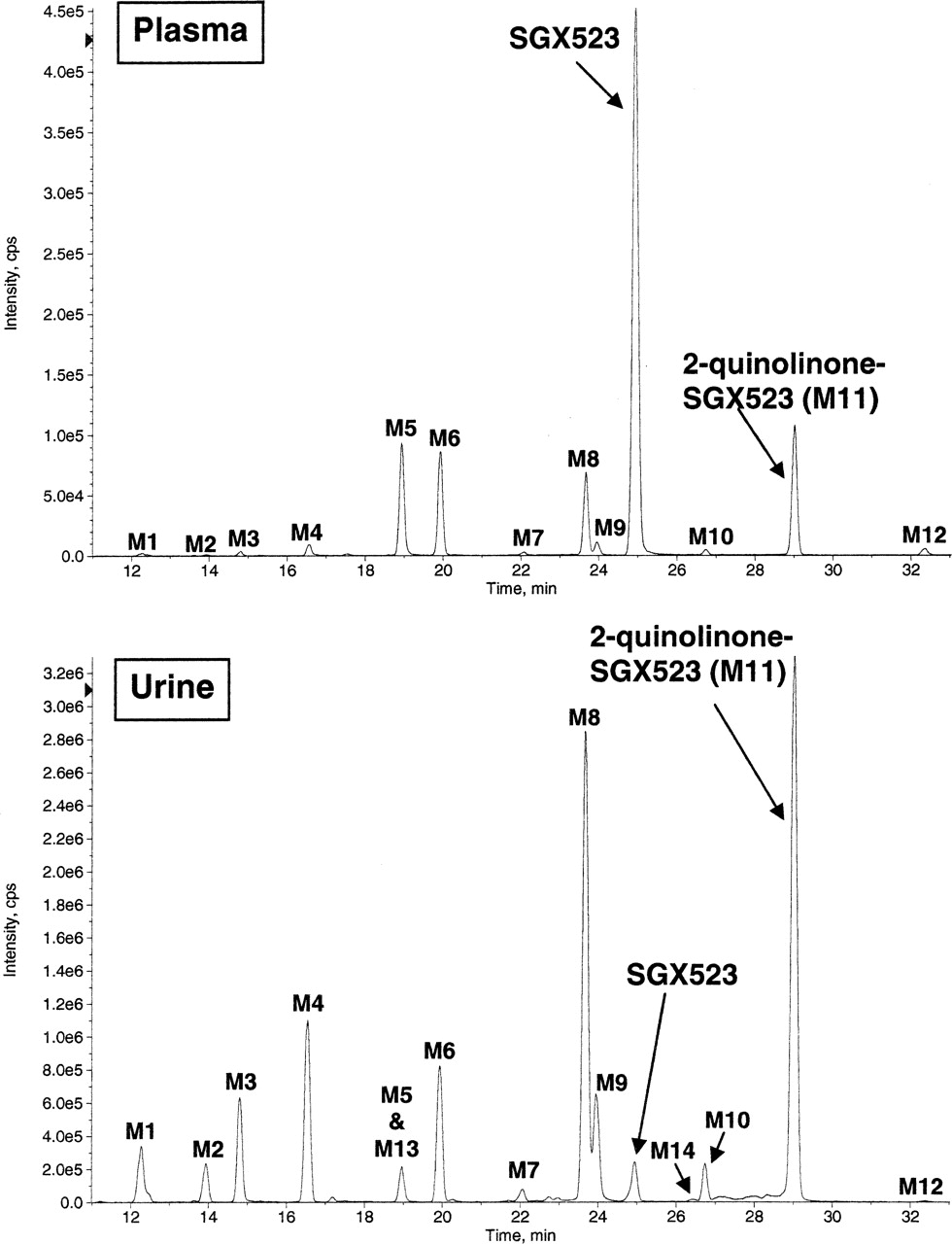
The metabolite profile (LC/MS ion chromatograms) of SGX523 in monkey plasma and urine. Pooled plasma and urine samples from monkeys given a single oral dose of SGX523 were precipitated with acetonitrile/methanol/DMSO and analyzed by LC/MS/MS.
Pharmacokinetics and Urinary Excretion.
Table 2 shows that after intravenous dosing in monkeys, SGX523 exhibited low clearance (0.22 l/h/kg) and a moderate volume of distribution (0.65 l/kg). The half-life value for SGX523 was 4.1 h. Approximately 0.2% of the administered intravenous dose was excreted in monkey urine as intact SGX523. After oral dosing, the average plasma Cmax value was 4.8 μM, and the oral bioavailability was moderate (39%). In urine samples solubilized with a mixture of organic solvents and DMSO, the concentration of M11 (29.2 μM) was ∼70-fold greater than that of parent compound (0.4 μM) after a single oral dose of SGX523.
Pharmacokinetics of SGX523 in cynomolgus monkeys and the urinary excretion of SGX523 and metabolite M11
Male cynomolgus monkeys (n = 2 for each route of administration) received a single intravenous or oral dose of SGX523 after an overnight fast using a parallel study design. SGX523 and M11 concentrations in plasma and urine samples were determined by LC/MS/MS analysis.
M11 Is Less Soluble than SGX523.
SGX523 exhibits marked pH-dependent solubility, with a solubility value that ranged from 2734 μg/ml in acid at pH 1.0 to only 4 μg/ml at pH 7.4 (Table 3). In addition, SGX523 was soluble in monkey urine at a concentration limit of 13 μg/ml. The intrinsic solubility of M11 was remarkably lower than that of the parent compound, with solubility values in water (pH 6.8), buffer (pH 7.4), acid (pH 1.0), and monkey urine (pH 8.4) of only 0.13, 0.10, 0.25, and 0.37 μg/ml, respectively. Therefore, in urine, the solubility value for M11 (0.37 μg/ml) was only 3% of that of SGX523 (13 μg/ml). Moreover, this metabolite did not exhibit the striking pH-dependent solubility noted for intact SGX523 because the solubility range was only ∼3-fold between acidic and neutral conditions for M11.
Solubility (μg/ml) of SGX523 versus metabolite M11
Compound was accurately weighted, and 1 ml of the appropriate matrix was added. Vials were capped and incubated at 37°C overnight. Samples were then centrifuged and filtered using a 0.45-μm filter, and the concentrations of SGX523 and M11 in the supernatant were determined by LC/MS/MS analysis.
Discussion
During drug discovery, the metabolism of new drug candidates is typically evaluated using hepatic microsomes because the vast majority of drugs are cleared via P450s. This P450-focused evaluation can lead researchers to underestimate the contribution from non-P450 enzymes such as cytosolic AO. Understanding the metabolic pathways and enzymes responsible for metabolite formation across species can improve predictions for pharmacokinetic, pharmacological, and toxicological extrapolations to humans. Ultimately, metabolite profiling data should be incorporated into the decision to select the most clinically relevant species for nonclinical safety assessment, thereby potentially avoiding those situations in which metabolites require further independent toxicological evaluations as outlined in the Food and Drug Administration metabolite safety guidance (“MIST”). This is especially important to consider for AO because activity in humans is relatively high compared with the commonly used preclinical safety species of rats and dogs (Smith and Obach, 2009).
The current investigation provides evidence that SGX523 is metabolized by AO, in a species-specific manner, to a markedly less-soluble metabolite, 2-quinolinone-SGX523 (M11). We propose that M11 may have precipitated in the renal tubule leading to crystal nephropathy and obstructive renal failure in patients. Acute renal failure attributed to precipitation of drug has been reported for a number of pharmaceuticals, including acyclovir, sulfonamide, methotrexate, and indinavir (Perazella, 1999). In such cases, nephrotoxicity occurs when drug-related material exceeds its solubility limit and precipitates in the tubular lumen of the nephron. Such a clinical finding was not anticipated for SGX523 based on initial preclinical studies conducted in rats and dogs, whereas subsequent studies using monkeys showed obstructive nephropathy with intratubular crystal formation (Burley, 2009).
Our in vitro studies using liver S-9 fractions from rats, monkeys, dogs, and humans showed that SGX523 underwent species-dependent metabolism. A late-eluting metabolite (M11, 2-quinolinone-SGX523) was generated by monkey and human liver S-9, and to a lesser extent by rat liver S-9, whereas this metabolite was completely absent in dog liver S-9 incubations. Although additional NADPH-dependent polar metabolites have been detected, these metabolites were not characterized because a marked species difference was not evident. It is important to note that this species-specific pattern of metabolism, obvious when using S-9 incubations, would not be apparent if microsomes were used. Because M11 was produced by NADPH-independent cytosolic enzymes and water supplied the oxygen atom incorporated into the substrate, the subsequent in vitro experiments focused on molybdenum hydroxylases. There are two major molybdenum hydroxylases that participate in the metabolism of drugs: AO and XO. In vitro experiments using monkey and human liver cytosol with selective chemical inhibitors of these enzymes showed that AO and not XO metabolized SGX523 to M11. This enzyme is implicated by the fact that the formation of M11 was inhibited significantly by two (menadione and raloxifene) of the three known selective inhibitors of AO studied in monkey cytosol and all three AO inhibitors in human cytosol (menadione, raloxifene, and SKF525A). In contrast, none of the selective inhibitors of XO inhibited the formation of M11 in monkey or human cytosol.
More important, the species differences noted for the levels of this AO-mediated metabolite of SGX523 generated during in vitro S-9 incubations are consistent with the reported species-dependent differences described for hepatic AO expression. For most N-heterocycles that are substrates for AO, the monkey and human exhibit the highest activity of hepatic AO, with lower activities reported for rat, and little to no AO activity in beagle dogs (Beedham et al., 1987; Kawashima et al., 1999; Sugihara et al., 2000; Austin et al., 2001). The extent of the species differences reported for AO activity is substrate-dependent because the size of the active and binding sites of this enzyme varies among species (Beedham et al., 1990, 1995). An example of another quinoline-containing small molecule that was metabolized in a species-specific manner by AO is trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011) (Austin et al., 2001). The high clearance of SB-277011 in monkeys, and not in rats and dogs, was attributed to a major metabolic pathway via oxidation on the quinoline ring by AO. These authors also noted that not all N-heterocyclic–containing compounds from the same series as SB-277011 were substrates for AO, suggesting it is not always possible to predict metabolism via AO within a chemical series. Despite the remarkable differences noted, often AO is not rigorously examined or considered because there is no known case of a drug exclusively metabolized by AO, and few drugs are predominantly metabolized by AO.
Additional information supporting the generation of M11 by AO comes from the chemical structure of SGX523 and what is known in terms of affinity of AO for substrates. AO is a molybdozyme that can oxidize a number of nitrogen-containing heterocycles, including quinolines (Kitamura et al., 2006). A main characteristic of this cytosolic enzyme is that it catalyzes nucleophilic oxidation at an electron-deficient carbon atom usually adjacent to the nitrogen atom of the heterocycle. This is unlike P450, which prefers to oxidize carbon atoms with high electron density. Therefore, it can be inferred that AO is the more likely enzyme to produce the 2-quinolinone metabolite of SGX523. The M11 metabolite exhibits markedly lower solubility than parent compound under acid, neutral, and basic conditions, with solubility values well below 1 μg/ml. In particular, the solubility of M11 (0.37 μg/ml) in urine was only 3% of that of parent SGX523 (13 μg/ml). Moreover, this metabolite did not exhibit the striking pH-dependent solubility noted for intact SGX523 because the solubility value between acidic and neutral conditions for the metabolite only differed ∼3- versus 684-fold for parent compound. The dramatic difference in aqueous solubility and pH-dependent solubility between SGX523 and M11 is because the basic nitrogen of the quinoline functional group is converted to the neutral lactam species in M11. Thus, the resulting change in pKa would adversely affect ionization and solubility of the 2-quinolinone metabolite at physiological conditions. AO-generated lactam functional groups are inherently less water-soluble than their precursors. For example, methotrexate is metabolized via AO to the less soluble 7-hydroxymethotrexate metabolite because the aqueous solubility of this metabolite (1.55 mg/ml) at pH 7 is ∼20% of that of parent (8.90 mg/ml) solubility (Jacobs et al., 1976). However, the effects of the lactam on aqueous solubility of the metabolite as a whole may be mitigated by other polar or ionizable functionalities. Therefore, poor water solubility is not always a hallmark characteristic of lactam metabolites.
SGX523 exhibited low clearance (∼10% hepatic blood flow) in cynomolgus monkeys after intravenous dosing, as well as moderate oral bioavailability (∼39%). Although metabolite profiling of plasma showed that intact parent compound was the major moiety detected, this compound was significantly metabolized via AO-mediated pathways as evidenced by the presence of both the M11 metabolite and its N-demethyl analog (M8). SGX523 was presumably cleared via the hepatic route because less than 1% of the administered intravenous dose was excreted in urine as intact SGX523. In urine solubilized with a mixture of organic solvents and DMSO, M11 was the major moiety detected, and this metabolite was detected at levels ∼70-fold greater than that of parent compound. The concentration of the M11 metabolite measured in monkey urine after an oral dose was 29.2 μM, ∼29-fold above the in vitro solubility limit in drug-free monkey urine at body temperature (37 μg/ml = 1.0 μM). It is sometimes difficult to predict compound precipitation by simply comparing compound solubility in drug-free urine with the concentration measured in urine samples from subjects after oral dosing, presumably because of supersaturation in the kidney (Merschman et al., 2005). In addition, the kidney may have contributed to the AO-mediated metabolism of SGX523. Although the tissue distribution of AO in the cynomolgus monkeys has not been reported, the proximal, distal, and collecting tubules of the human kidney, but not the glomerulus, have been shown to express AO (Moriwaki et al., 2001).
In summary, a predominant metabolic pathway of SGX523 in monkeys has been identified both in vitro and in vivo as oxidation of the quinoline ring (Fig. 5). The major metabolite has been confirmed as 2-quinolinone SGX523 (M11), and it was formed in vitro in a species-specific manner. This metabolite showed markedly lower solubility than that of parent compound and in particular did not show the enhanced solubility at acidic conditions that the parent compound did. In addition, the extent of the urinary excretion of M11 was significantly greater than that of SGX523. Taken together, these data suggest that this 2-quinolinone metabolite is likely involved in the observed obstructive nephropathy in humans and also provides an explanation for the species difference in the renal toxicity of SGX523. This case of AO-mediated toxicity serves to underline the importance of conducting thorough metabolism evaluations of new chemical entities using multiple in vitro systems. In particular, compounds with an aromatic aza-heterocycle should be evaluated for metabolism via cytosolic molybdenum oxidases across multiple species to select the most appropriate species for toxicological evaluation.

Summary of SGX523 metabolism in the monkey.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032375.
-
ABBREVIATIONS:
- c-MET
- mesenchymal-epithelial transition factor
- SGX523
- (6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinoline
- SKF525-A
- proadifen hydrochloride
- DMSO
- dimethyl sulfoxide
- M11/2-quinolinone-SGX523
- 6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinolin-2(1H)-one
- XO
- xanthine oxidase
- AO
- aldehyde oxidase
- LC/(MS)/MS
- liquid chromatography/(tandem) mass spectrometry
- MRM
- multiple reaction monitoring
- P450
- cytochrome P450
- SB-277011
- trans-N-[4-[2-(6-cyano-1,2,3,4-tetrahydroisoquinolin-2-yl) ethyl]cyclohexyl]-4-quinolinecarboxamide.
- Received January 25, 2010.
- Accepted April 23, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}