


Abstract
The ability to predict in vivo clearance from in vitro intrinsic clearance for compounds metabolized by aldehyde oxidase has not been demonstrated. To date, there is no established scaling method for predicting aldehyde oxidase-mediated clearance using in vitro or animal data. This challenge is exacerbated by the fact that rats and dogs, two of the laboratory animal species commonly used to develop in vitro-in vivo correlations of clearance, differ from humans with regard to expression of aldehyde oxidase. The objective of this investigation was to develop an in vitro-in vivo correlation of intrinsic clearance for aldehyde oxidase, using 11 drugs known to be metabolized by this enzyme. The set consisted of methotrexate, XK-469, (±)-4-(4-cyanoanilino)-5,6-dihydro-7-hydroxy-7H-cyclopenta[d]pyrimidine (RS-8359), zaleplon, 6-deoxypenciclovir, zoniporide, O6-benzylguanine, N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide (DACA), carbazeran, PF-4217903, and PF-945863. These compounds were assayed using two in vitro systems (pooled human liver cytosol and liver S-9 fractions) to calculate scaled unbound intrinsic clearance, and they were then compared with calculated in vivo unbound intrinsic clearance. The investigation provided a relative scale that can be used for in vitro-in vivo correlation of aldehyde oxidase clearance and suggests limits as to when a potential new drug candidate that is metabolized by this enzyme will possess acceptable human clearance, or when structural modification is required to reduce aldehyde oxidase catalyzed metabolism.
Aldehyde oxidase is a molybdenum cofactor-containing soluble enzyme present in the liver and other tissues of several mammalian species (Beedham et al., 2003). It is involved in the oxidation of nitrogen-containing heterocyclic compounds to lactams, as well as oxidation of aldehydes (as the name implies) and reduction reactions of N-O and N-S bonds. In the metabolism of drugs, aldehyde oxidase has been shown to catalyze the metabolic activation of the antiviral agent famciclovir to penciclovir (Rashidi et al., 1997), and it plays a role in the metabolic clearance of the hypnotic agent zaleplon (Lake et al., 2002). It has also been shown to be involved in the oxidation of intermediary metabolites of drugs such as the conversion of cyclic iminium ions arising from the cytochrome P450 (P450)-catalyzed oxidation of pyrrolidines and piperidines to lactams as well as the oxidation of aldehydes that have arisen from 1° alcohol containing drugs to carboxylic acids. In humans, aldehyde oxidase is represented by a single gene product, although three inactive pseudogenes for this enzyme are also present in the human genome (Garattini et al., 2008). Some inhibitors of aldehyde oxidase have been identified in vitro, including raloxifene (Obach, 2004), hydralazine (Johnson et al., 1985), phenothiazine drugs, estradiol, and menadione (Johns, 1967); however, clinical drug interactions via inhibition of aldehyde oxidase has not been established. In early drug research efforts, it is commonplace for newly synthesized compounds to be tested for proclivity to high clearance by incubating with human liver microsomes and cofactors to support cytochrome P450 activity (Obach, 2001). Compounds demonstrating high lability are either discarded or structurally modified to impart improved metabolic stability. This type of activity has now been occurring for several years, and our ability to find and design new drugs with low cytochrome P450-catalyzed lability is well established. An unintended consequence of such an effort has been that the number of new drug candidates that are cleared via other pathways, such as drug transporters and non-P450 enzymes, has increased. One of the enzymes that has become increasingly important is aldehyde oxidase. Aldehyde oxidase is a cytosolic enzyme, and thus its potential contribution to the metabolic clearance of new compounds is not addressed in standard metabolic lability screens using liver microsomes.
Although an ability to predict in vivo clearance from in vitro intrinsic clearance (CLint) data measured in human liver microsomes is well established for P450-cleared compounds, an ability to do the same for aldehyde oxidase-cleared compounds has not been demonstrated. The inability to predict in vivo clearance from in vitro intrinsic clearance for aldehyde oxidase-cleared compounds poses a challenge, because the prediction of human pharmacokinetics is an important need in drug research; however, if a new compound is metabolized by aldehyde oxidase, there is no established scaling method by which a prediction of clearance can be made. This challenge is exacerbated by the fact that two of the laboratory animal species commonly used to develop in vitro-in vivo correlations of clearance have highly variable (rat) or no (dog) expression of human aldehyde oxidase. Our objective was to develop an in vitro-in vivo correlation of intrinsic clearance for aldehyde oxidase, using drugs metabolized by this enzyme. In this study, the set of drugs was limited to 1) the number of data establishing them as aldehyde oxidase substrates and 2) those that also have human pharmacokinetic data (Fig. 1). Two in vitro systems, pooled human liver cytosol and liver S-9 fractions, were tested for their ability to calculate clearance.

Structures of the 11 aldehyde oxidase substrates used in this analysis.
Materials and Methods
Materials.
Acetonitrile was obtained from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). O6-Benzylguanine, methotrexate, XK-469, zaleplon, buspirone, 1 M potassium phosphate dibasic solution, 1 M potassium phosphate monobasic solution, and dimethyl sulfoxide were purchased from Sigma-Aldrich (St. Louis, MO). Carbazeran, N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide (DACA), PF-945863, PF-4217903, 4-(4-cyanoanilino)-5,6-dihydro-7-hydroxy-7H-cyclopenta[d]pyrimidine (RS-8359), fluconazole, and zoniporide were obtained from Pfizer's internal compound library (Milwaukee, WI). 6-Deoxypenciclovir was purchased from US Biological (Swampscott, MA). Matching human liver cytosol and S-9 fractions, pooled from 10 donors, were previously prepared and frozen (Celsis, IVT, Chicago, IL). Plasma for the plasma protein-binding assay was pooled from six donors (three female and three male) and were obtained from Bioreclamation Inc. (Westbury, NY). Human plasma specifically for the blood/plasma ratio was obtained from Richmond Pharmacology (London, UK), and fresh blood was collected from human volunteers (in-house) on the day of the experiment. All other materials were of the highest quality attainable.
In Vitro Incubations in Liver Cytosol and S-9 Fractions.
The in vitro incubations to determine clearance were performed by using an automated procedure on a Beckman Biomek FX system (Beckman Coulter, Inc., Fullerton, CA). Incubations from previously frozen, matching pooled human liver S-9 and cytosol from 10 donors contained 1 mg/ml cytosol or 2.5 mg/ml S-9 fraction in 100 mM potassium phosphate buffer (pH 7.4) and initiated with substrate [(1 μM) final organic vehicle concentration was 0.01% dimethyl sulfoxide and 0.6% acetonitrile], with a final reaction volume of 200 μl. Samples (25-μl aliquots) were removed from the S-9 assay at 0, 10, 20, 30, 45, and 60 min and from the cytosol assay at 0, 10, 30, 60, 120, 240 min and quenched with 75 μl of chilled acetonitrile containing 0.1 μM buspirone as an internal standard. Samples were then centrifuged at 3000 rpm (GH3.8A rotor; Beckman Coulter) for 15 min at room temperature. A 50-μl aliquot of supernatant was transferred to a clean, shallow 96-well plate and combined with 50 μl of Milli-Q water (Millipore Corporation, Billerica, MA), mixed, and analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS). All incubations were performed at least in duplicate.
Plasma Protein Binding and Blood/Plasma Ratios.
The protein binding assay was performed using an automated procedure on a Beckman Biomek FX system. In brief, plasma with the addition of 1 μM compound was placed in one side of a divided 96-well dialysis membrane apparatus, and 100 mM potassium phosphate buffer was aliquoted into the other side. Plasma samples were dialyzed against buffer for 6 h at 37°C on an oscillating platform in a 5% CO2 atmosphere. Incubation samples (20 μl of plasma study sample and 80 μl of blank buffer, or 80 μl of buffer study sample and 20 μl of blank matrix) were quenched with 300 μl of 50:50 methanol/acetonitrile containing internal standard (0.1 μM buspirone). Quenched samples were centrifuged for 15 min at 3200 rpm to pellet the precipitated proteins. The supernatant was collected and analyzed by LC-MS/MS. All incubations were performed in triplicate.
The blood partitioning assay was performed using an automated procedure on a Hamilton Star robotic system (Bonaduz GR, Switzerland). In brief, blood and plasma were aliquoted into appropriate wells of the incubation block. Test compounds (5 μl) were then spiked into these solutions (final concentration of 1 μM) and incubated for 60 min at 37°C. After incubation, samples were centrifuged at 3000 rpm for 15 min at 20°C. Aliquots from both blood and plasma wells (20 μl) were then taken and added to acetonitrile plus internal standard (0.05 μM fluconazole) (1:4 v/v). Samples were then analyzed by LC-MS/MS. All incubations were performed in duplicate.
Bioanalytical Methods.
The compounds of interest were introduced to a Synergi Polar-RP (2 × 30 mm, 4 μ; Phenomenex, Torrance, CA) high-performance liquid chromatography (HPLC) column with a CTC PAL autosampler (Leap Technologies, Carrboro, NC) and an integrated HPLC pumping system (Shimadzu Scientific Instruments, Columbia, MD). These compounds were then eluted and detected by an API 4000-triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA) fitted with a TurboIonSpray interface. The mobile phase consisted of 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B), and the flow rate was 0.8 ml/min. The starting condition for the HPLC gradient was 99:1 (A/B), and this was held for 0.3 min. From 0.3 to 1.2 min, the mobile phase composition changed linearly to 1:99 (A/B). This condition was held until 1.9 min. The gradient was returned in a linear fashion to 99:1 (A/B) from 1.9 to 1.95 min and re-equilibrated until 2 min. The injection volume was 20 μl. Multiple reaction monitoring was used to monitor the compounds. Table 1 lists the ionization mode, m/z transitions, and retention times for all the compounds used in this analysis. The peak area ratio of the analyte to the internal standard (buspirone or fluconazole) was determined for each injection and used to measure substrate depletion in the clearance, the plasma protein binding, or the blood/plasma ratio assays.
Mass/charge ratios, declustering potential, collision energies, and retention times for aldehyde oxidase substrates used in clearance, plasma protein binding, and blood/plasma ratio experiments
In Vivo Data.
Human pharmacokinetic data were gathered from the scientific literature. We used data from studies in which drugs were dosed orally or intravenously. For orally administered drugs, the maximal free in vivo intrinsic clearance due to aldehyde oxidase (CL′int,AO) was calculated as follows:
 where CLpo is the apparent oral clearance (dose/area under the curve), CLrenal is the renal clearance of unchanged drug, and fu is the fraction of the drug unbound in blood (calculated from Table 2). This assumes that the fraction absorbed equals unity, to yield an upper limit estimate, and that the liver is the primary organ of metabolic clearance. In some cases, other data were available on the amount of drug-related material excreted as unchanged drug plus drug converted to other nonaldehyde oxidase-generated metabolites. In these instances, CL′int,AO was calculated as follows:
where CLpo is the apparent oral clearance (dose/area under the curve), CLrenal is the renal clearance of unchanged drug, and fu is the fraction of the drug unbound in blood (calculated from Table 2). This assumes that the fraction absorbed equals unity, to yield an upper limit estimate, and that the liver is the primary organ of metabolic clearance. In some cases, other data were available on the amount of drug-related material excreted as unchanged drug plus drug converted to other nonaldehyde oxidase-generated metabolites. In these instances, CL′int,AO was calculated as follows:
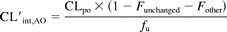 where Funchanged is the fraction of the drug excreted as unchanged parent compound in urine or feces, and Fother refers to the fraction of the dose excreted as metabolites other than those generated by aldehyde oxidase.
where Funchanged is the fraction of the drug excreted as unchanged parent compound in urine or feces, and Fother refers to the fraction of the dose excreted as metabolites other than those generated by aldehyde oxidase.
Plasma protein binding and blood/plasma ratio data used in scaling
For some drugs in this analysis, the human in vivo pharmacokinetic data were available after intravenous administration. In these cases, free intrinsic clearance was back-calculated from the well stirred model of hepatic clearance, assuming that the liver was the primary metabolic clearing organ:
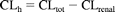
 where CLh is hepatic blood clearance and Qh is hepatic blood flow [21 ml/(min · kg)]. If other data were available on the total metabolic profile in humans, the unbound in vivo intrinsic clearance by aldehyde oxidase was calculated as follows:
where CLh is hepatic blood clearance and Qh is hepatic blood flow [21 ml/(min · kg)]. If other data were available on the total metabolic profile in humans, the unbound in vivo intrinsic clearance by aldehyde oxidase was calculated as follows:

For this study, sufficient data were available in the scientific literature to carry out this analysis for nine drugs: methotrexate, XK-469, RS-8359, zaleplon, 6-deoxypenciclovir, zoniporide, O6-benzylguanine, DACA, and carbazeran. An additional two drugs were added for which unpublished human pharmacokinetic data were available in-house (PF-4217903 and PF-945863). The in vivo CL′int data calculated from in vivo pharmacokinetic data are listed in Table 3.
Human pharmacokinetic data for drugs metabolized by aldehyde oxidase
In vitro intrinsic clearance data were scaled to in vivo data using the following equations:
 and
and
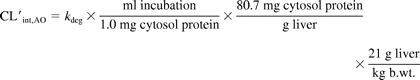 for S-9 and cytosol data, respectively. The S-9 and cytosolic recovery terms in the above equations were taken from the work of Houston and Galetin (2008).
for S-9 and cytosol data, respectively. The S-9 and cytosolic recovery terms in the above equations were taken from the work of Houston and Galetin (2008).
Results
In vitro t1/2 values measured in pooled human liver cytosol and S-9 fractions are listed in Table 4, along with intrinsic clearance values calculated using the scaling factors described under Materials and Methods. Carbazeran was the most rapidly consumed drug, with a t1/2 value of under 4 min. For two drugs, methotrexate and XK-469, consumption was not detected in S-9 or cytosol, thus upper limit t1/2 estimates of 120 and 480 min were used in scaling to give minimal estimates of in vivo intrinsic clearance of 5.9 and 2.5 ml/min/kg, respectively. These three examples represent the bounds of high and low intrinsic clearance among the 11 drugs tested. The scaled intrinsic clearance values generated from S-9 and cytosol fractions correlated with each other (r2 = 0.918) (Fig. 2).
In vitro intrinsic clearance data for drugs in pooled human liver cytosol and S-9 fractions
The cytosol reaction values are an average of n = 4; therefore, an average %CV of 4% was observed.

Correlation between scaled in vitro CL′int measured in pooled human liver cytosol and S-9 fraction.
A comparison of the scaled in vivo CL′int values to those back-calculated from in vivo data showed that the in vitro data underestimated the in vivo values in almost every case. The magnitude of the underestimate was greatest for the high CL′int compounds. For example, the in vivo CL′int values for carbazeran and DACA were underestimated by at least 20-fold. On average, CL′int was underestimated by 11-fold.
A bar graph of scaled CL′int values plotted in the order of back-calculated in vivo CL′int values is shown in Fig. 3. Whereas absolute values of in vivo CL′int were not calculable from in vitro data, Fig. 3 shows that the rank order is generally aligned and permits categorization of aldehyde oxidase-cleared compounds—essentially a calibration scale. Compounds that possessed metabolic lability in cytosol or S-9 that matched or exceeded that of 6-deoxypencyclovir will be high CL′int compounds. Those with lability that matched or are lower than PF-4217903 will have low CL′int. Zaleplon resides are in the middle. Zaleplon is a high clearance drug in humans [16 ml/(min · kg); ∼80% of hepatic blood flow], but approximately two thirds of its clearance is via aldehyde oxidase. RS-8359 was an outlier, with in vitro CL′int relatively more rapid than other compounds with higher back-calculated in vivo CL′int (zaleplon and PF-4217903), irrespective of the system used.
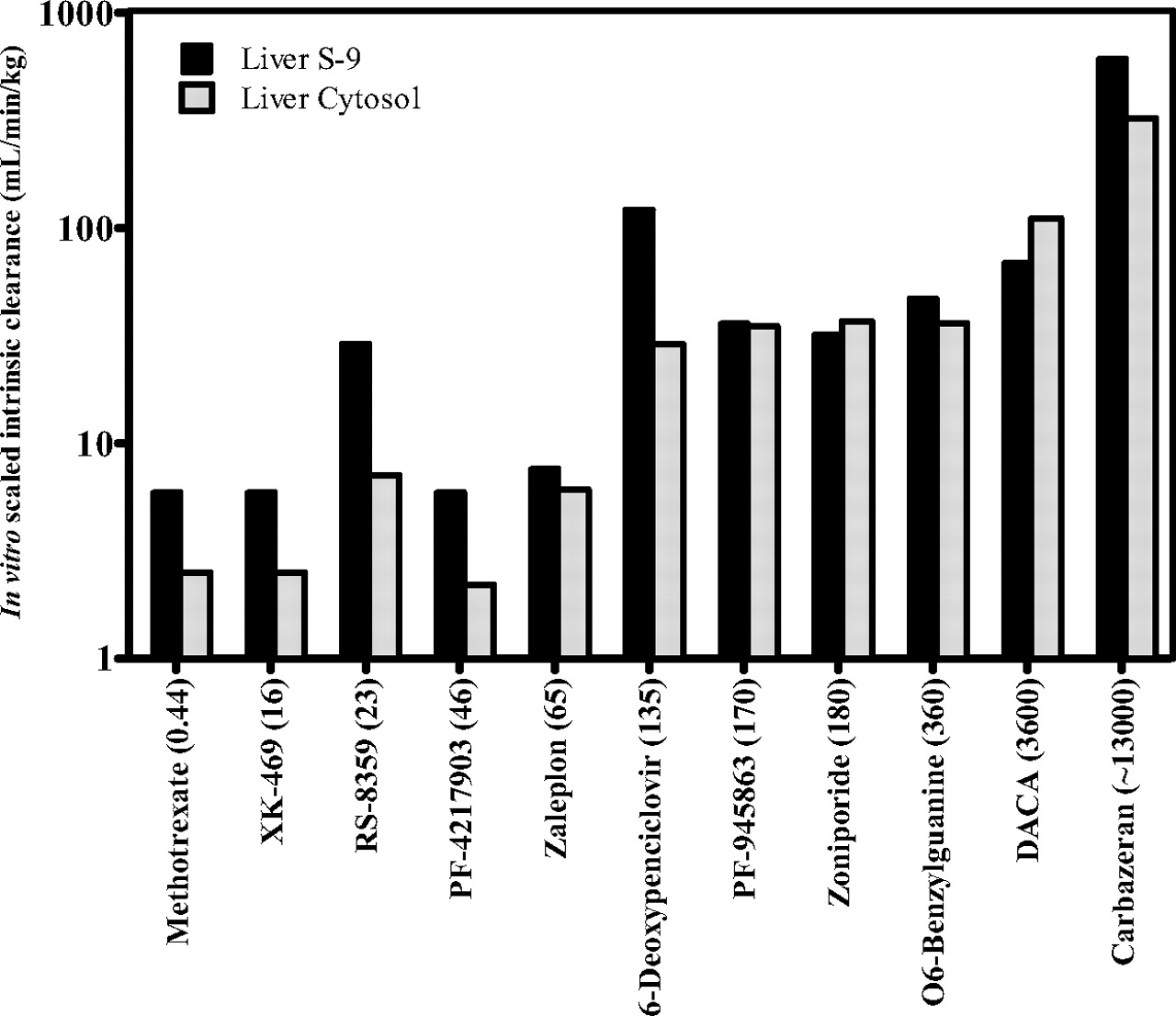
Bar graph calibrating in vitro-scaled intrinsic clearance with in vivo free intrinsic clearance for 11 compounds metabolized by aldehyde oxidase in humans. Numbers in parentheses refer to estimated in vivo free intrinsic clearance. Bars are placed in the order of in vivo free intrinsic clearance.
Discussion
The prediction of human clearance from in vitro data are a well established and important process in the search for new drugs. Drugs that possess optimal pharmacokinetics are sought, and clearance is an important pharmacokinetic attribute that helps define half-life and oral bioavailability (Obach, 2001). Clearance that is mediated by cytochrome P450 enzymes is well understood such that screening for P450-mediated lability has become routine in drug research (Zhang et al., 2007). Furthermore, recent advances have also been made in predicting clearance for compounds metabolized by glucuronyl transferases (Kilford et al., 2009). In our laboratories, we found that the number of compounds for which aldehyde oxidase plays a greater role has increased, especially if the compounds possess a substituent amenable to metabolism by this enzyme (e.g., aromatic azaheterocyclic rings). This increased occurrence of aldehyde oxidase metabolism poses a challenge in developing a method whereby clearance can be predicted for such compounds using in vitro metabolism data.
This study represents the first report to correlate in vitro metabolism data with in vivo pharmacokinetic data for compounds metabolized by aldehyde oxidase. This process is challenging because, unlike cytochrome P450 enzymes, there are only a few drugs for which this enzyme is known to play a role and for which enough information exists (i.e., pharmacokinetics in humans and knowledge of metabolic pathways) to permit estimates to be made for in vivo CL′int mediated by aldehyde oxidase. Eleven compounds, which had been reported in the literature to be aldehyde oxidase substrates, had adequate information in the scientific literature or from in-house investigations permitting crude estimates of aldehyde oxidase-mediated intrinsic clearance to be made. Given the limited information available, various assumptions were made in estimating aldehyde oxidase-mediated in vivo CL′int. In drugs for which only oral pharmacokinetic data were available, fraction absorbed equals unity. Moreover, aldehyde oxidase metabolism was assumed to occur exclusively in the liver, despite the knowledge that the enzyme is expressed in some extrahepatic tissues (Beedham, 2001).
Despite these aforementioned shortcomings and assumptions, a cross-compound in vitro-in vivo correlation was attainable (Fig. 3). This correlation offers a means by which new compounds that are metabolized by aldehyde oxidase can be used to make a prediction of whether it will have a high, medium, or low CL′int. The in vitro system, either pooled human liver cytosol or S-9 fraction, can be calibrated using high, medium, and low CL′int selected from the 11 compounds described here, and the CL′int for the new compound can be measured side by side with a few strategically chosen compounds from this test set. Compounds with aldehyde oxidase-mediated CL′int less than that of zaleplon would be predicted to be low CL′int in vivo. Compounds with CL′int equal to or greater than 6-deoxypenciclovir, zoniporide, or O6-benzylguanine would be predicted to be high CL′int in vivo. Of course, total clearance will be a function of both CL′int and protein binding such that a high CL′int compound could still have an acceptable half-life or hepatic extraction due to high protein binding. For example, zoniporide has a lower CL′int than DACA, but it has a higher in vivo clearance due to a marked difference in free fraction. Thus, it is always important to consider multiple essential input parameters when attempting to predict clearance.
No one explanation can be pinpointed to understand the underestimation of the in vitro-scaled intrinsic clearance compared to the in vivo values. One can hypothesize that because aldehyde oxidase is expressed extrahepatically (Beedham, 2001), the abundance found outside the liver may influence total clearance of the compound. In addition, because the lability of the enzyme has been observed in our laboratories and others to occur during the homogenization and storage process, the true comparable in vivo activity may not be realized in vitro (Duley et al., 1985). Therefore, additional research may be needed to establish a stable and reproducible source of aldehyde oxidase.
In conclusion, an in vitro-in vivo correlation for intrinsic clearance has been demonstrated for human aldehyde oxidase-metabolized compounds using pooled human liver cytosol and S-9 fractions. These findings should be of value to investigators who search for new drugs when aldehyde oxidase is encountered as a potentially important enzyme in clearance.
Acknowledgments.
We thank Andrea Clouser-Roche, Julian Haynes, Jennifer Tran, and Paul Wainwright for their technical expertise and contributions to this work. We also thank Drs. Deepak Dalvie, Caroline Lee, and Ellen Wu for useful conversations and review of this work.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.033555.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- CLint
- intrinsic clearance
- DACA
- N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide
- RS-8359
- (±)-4-(4-cyanoanilino)-5,6-dihydro-7-hydroxy-7H-cyclopenta[d]pyrimidine
- LC-MS/MS
- liquid chromatography/tandem mass spectrometry
- HPLC
- high-performance liquid chromatography
- CL′int,AO
- clearance due to aldehyde oxidase
- CLpo
- oral clearance
- CLrenal
- renal clearance
- CLh
- hepatic clearance.
- Received March 26, 2010.
- Accepted May 5, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}