


Abstract
The selective estrogen receptor modulator, raloxifene, has been demonstrated as a potent uncompetitive inhibitor of human liver aldehyde oxidase-catalyzed oxidation of phthalazine, vanillin, and nicotine-Δ1′(5′)-iminium ion, with Ki values of 0.87 to 1.4 nM. Inhibition was not time-dependent. Raloxifene has also been shown to be a noncompetitive inhibitor of an aldehyde oxidase-catalyzed reduction reaction of a hydroxamic acid-containing compound, with a Ki of 51 nM. However, raloxifene had only small effects on xanthine oxidase, an enzyme related to aldehyde oxidase. In addition, several other compounds of the same therapeutic class as raloxifene were examined for their potential to inhibit aldehyde oxidase. However, none were as potent as raloxifene, since IC50 values were orders of magnitude higher and ranged from 0.29 to 57 μ M. In an examination of analogs of raloxifene, it was shown that the bisphenol structure with a hydrophobic group on the 3-position of the benzthiophene ring system was the most important element that imparts inhibitory potency. The relevance of these data to the mechanistic understanding of aldehyde oxidase catalysis, as well as to the potential for raloxifene to cause drug interactions with agents for which aldehyde oxidase-mediated metabolism is important, such as zaleplon or famciclovir, is discussed.
Aldehyde oxidase is an enzyme involved in the metabolism of drugs and other xenobiotics that possess aldehyde and azaheterocyclic substituents, among others (Beedham, 2002). It is a member of a family of enzymes referred to as molybdenum cofactor-containing enzymes that also includes xanthine oxidase, due to the presence of the unique molybdopterin prosthetic group. It is present in the liver, as well as some other tissues of humans and other mammalian species such as the rat, guinea pig, monkey, and rabbit. Aldehyde oxidase converts aldehydes to carboxylic acids and azaheterocyclic compounds to lactams using molecular oxygen as an electron acceptor. It can also catalyze reduction reactions of some drugs, with the drug that is reduced replacing oxygen as the electron acceptor.
Drugs for which aldehyde oxidase plays the predominant role in metabolic clearance are few. A notable example is the recently introduced azaheterocyclic hypnotic agent, zaleplon (Kawashima et al., 1999; Lake et al., 2002), in which aldehyde oxidase generates the major metabolite 5-oxozaleplon as well as the corresponding 5-oxo analog of the N-dealkylated metabolite of zaleplon. Also, the bioactivation of famciclovir to the active antiviral agent penciclovir requires aldehyde oxidase-mediated metabolism (Clarke et al., 1995; Rashidi et al., 1997). Famciclovir is converted to the 6-oxo metabolite, which undergoes deesterification to penciclovir; also, the deesterified analog of famciclovir, 6-deoxypenciclovir, is oxidized by aldehyde oxidase to the active compound. Although it is not frequently involved in initial pathways of metabolism of drugs, aldehyde oxidase may play a greater role in the intermediary metabolism of compounds, since aldehydes are frequent metabolic intermediates in the oxidative metabolism of alcohols to carboxylic acids (Ruenitz and Bai, 1995; Rochat et al., 1998). However, compared with the cytochrome P450 family, the number of drugs for which aldehyde oxidase is the major enzyme responsible for clearance is few. Thus, as a target of drug-drug interactions, the importance of aldehyde oxidase is substantially less than that of cytochrome P450 enzymes. One drug interaction attributed to inhibition of aldehyde oxidase is the increase in zaleplon exposure, with a concomitant decrease in the 5-oxozaleplon metabolite, brought about by coadministration of cimetidine (Renwick et al., 2002). However, the importance of this enzyme in drug metabolism may increase in the coming years. As efforts in drug design strive to reduce or remove cytochrome P450 metabolic lability, new drugs will be cleared by other mechanisms, such as aldehyde oxidase-mediated metabolism.
In a previous report, over 200 frequently prescribed drugs and other compounds were tested as inhibitors of human liver cytosolic aldehyde oxidase activity (O. Obach, et al., 2004) The most notable inhibitors included estrogens, phenothiazines, and tricyclic antidepressants, among others. The most potent inhibitor observed was the selective estrogen receptor modulator (SERM1), raloxifene, which demonstrated an extremely low IC50 value of 2.9 nM for phthalazine oxidase in human liver cytosol. The objectives of the present study are to further characterize the inhibition of human aldehyde oxidase by raloxifene. Specifically, objectives include 1) determination of the mechanism of raloxifene inhibition of aldehyde oxidase, 2) determination of whether raloxifene will inhibit the metabolism of different classes of aldehyde oxidase substrates (i.e., aldehydes, aromatic azaheterocyclics, quaternary iminium ions; Fig. 1), 3) determination of whether raloxifene will inhibit an aldehyde oxidase-catalyzed reduction reaction (Fig. 1), 4) an assessment of the capabilities of other drugs of the same therapeutic class, i.e., estrogens, to inhibit aldehyde oxidase, and 5) determination of the structural elements of raloxifene that impart the high potency for inhibition of aldehyde oxidase. A discussion of the data in the context of the mechanism of aldehyde oxidase, as well as relevance for drug-drug interactions that could occur by inhibition of aldehyde oxidase, is presented.
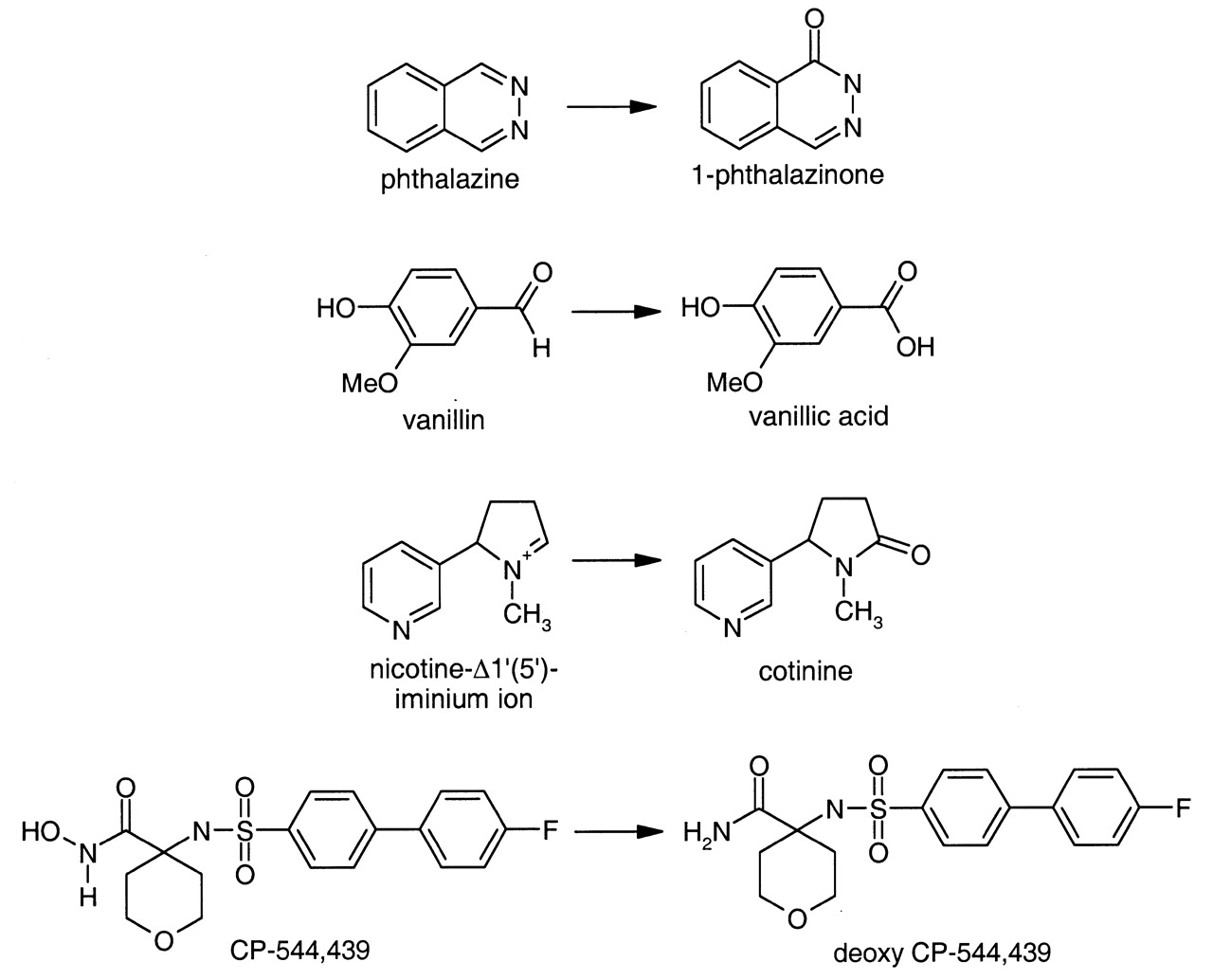
Reactions catalyzed by aldehyde oxidase.
Materials and Methods
Materials. Phthalazine, 1-phthalazinone, 4-methyl-1-phthalazinone, vanillin, vanillic acid, and 3-chloro-4-hydroxybenzoic acid were obtained from Aldrich Chemical Co. (Milwaukee, WI). Cotinine, pterin, isoxanthopterin, N-methylnicotinamide, and amantadine were purchased from Sigma-Aldrich (St Louis, MO). Nicotine-Δ1′(5′)-iminium bisperchlorate was prepared as described (Peterson et al., 1987). Compounds 1 to 5 were obtained from the Pfizer sample bank (Pfizer, Groton, CT). CP-544,439 was obtained from the Pfizer sample bank, and the deoxy product was prepared by reduction of CP-544,439 with TiCl3, followed by purification on silica. The resulting amide was pure by thin-layer chromatography and contained no detectable starting material as assessed by HPLC-MS. Human liver cytosol prepared from 10 individual donors was purchased from BD Gentest (Woburn, MA).
Phthalazine Oxidase Assay. Phthalazine (0.6–10 μM for Ki determinations; 2.0 μM for IC50 determinations) was incubated with human liver cytosol (0.05 mg/ml) in a total volume of 0.2 ml of 25 mM KH2PO4, pH 7.4, containing 0.1 mM EDTA, in the presence and absence of raloxifene and other inhibitors. Raloxifene and other inhibitors were added in dimethyl sulfoxide such that the final concentration of solvent was 1% (v/v). Incubations were conducted open to the air for 2.5 min in a water bath set at 37°C. Initial time course experiments established the linearity of reaction velocity versus protein concentration and incubation time (Obach et al., 2004). In preincubation experiments, raloxifene was incubated with cytosol for 30 min at 37°C before addition of phthalazine. The incubations were terminated by the addition of 0.05 ml of formic acid (1 M) containing 4-methyl-1-phthalazinone (25 ng) as an internal standard. Terminated incubation mixtures were subjected to filtration through a Millipore Multiscreen-HA 0.45-μm mixed cellulose ester (Millipore Corporation, Bedford, MA) 96-well membrane vacuum filtration module into 96-well plates. The resulting filtrate was analyzed by an HPLC-MS system comprising a PE-Sciex API100 mass spectrometer with Turbo Ionspray source (PerkinElmerSciex Instruments, Boston, MA), an Agilent quaternary HPLC pump with membrane degasser (Agilent Technologies, Palo Alto, CA), and Leap autosampler (CTC Analytics, LEAP Technologies Inc., Carrboro, NC). Samples (0.03 ml) were injected onto an Aqua reverse-phase C18 column (2.5 × 50 mm; 5 μm; MetaChem Technologies Inc., Division of ANSYS Diagnostics Inc., Lake Forest, CA) preequilibrated in 20 mM acetic acid (adjusted to pH 4.0 with NH4OH) containing 14% CH3CN at a flow rate of 0.5 ml/min. Elution was effected with a solvent program of holding at 14% CH3CN over the first 3 min, followed by a linear gradient to 77% CH3CN over 0.5 min, and reequilibration to initial conditions over the next 6.5 min. The state file settings on the mass spectrometer were set at following values: ionspray voltage, 4500; source temperature, 450°C; orifice voltage, 35; ring potential, 100; Q1 potential, –5, with other settings set to optimize system response. 1-Phthalazinone and 4-methyl-1-phthalazinone were detected by monitoring ion current at m/z 147 and 161, respectively. Retention times for analyte and internal standard were 1.1 and 2.0 min, respectively. Quantitation was done using a 1-phthalazinone standard curve ranging from 1.0 to 100 ng/ml. Assay standards were within ±15% of nominal values.
Vanillin Oxidase Assay. Vanillin (0.6–10 μM for Ki determinations) was incubated with human liver cytosol (0.05 mg/ml) in a total volume of 0.5 ml of 25 mM KH2PO4, pH 7.4, containing 0.1 mM EDTA, in the presence and absence of raloxifene, as described above. Incubations were conducted open to the air for 5 min in a water bath set at 37°C. Preliminary experiments established the linearity of reaction velocity under these conditions. The incubations were terminated by the addition of 0.05 ml of HCl (1 M). To each sample was added 3-chloro-4-hydroxybenzoic acid as an internal standard (0.02 ml of a 0.5 μg/ml solution in CH3CN). Samples were extracted with ethyl acetate (3 ml), the solvent was transferred and evaporated under N2 at 35°C, and the residue was reconstituted in 0.04 ml of 20 mM acetic acid, (adjusted to pH 4.0 with NH4OH). The resulting material was analyzed by HPLC-MS using the system described above. Samples (0.025 ml) were injected onto an Aqua reverse-phase C18 column (2.5 × 50 mm; 5 μm) preequilibrated in 20 mM acetic acid (adjusted to pH 4.0 with NH4OH) containing 11% CH3CN at a flow rate of 0.5 ml/min. Elution was achieved with a solvent program of holding at 11% CH3CN over the first 0.8 min, followed by a linear gradient to 77% CH3CN over 2.5 min, holding at this composition for 0.5 min, followed by reequilibration to initial conditions over the next 6 min. The state file settings on the mass spectrometer were set at the following values: ionspray voltage, –3500; source temperature, 450°C; orifice voltage, –25; ring potential, –80; Q1 potential, 5, with other settings set to optimize system response. Vanillic acid and 3-chloro-4-hydroxybenzoic acid were detected by monitoring ion current at m/z 167 and 171, respectively. Retention times for analyte and internal standard were 1.1 and 3.0 min, respectively. Quantitation was done using a standard curve ranging from 10 to 1000 ng/ml. Assay standards were within ±12% of nominal values.
Nicotine-Δ1′(5′)-Iminium Ion Oxidase Assay. Nicotine-Δ1′(5′)-iminium bisperchlorate (0.3–5.0 μM for Ki determinations) was incubated in the presence of human liver cytosol (0.03 mg/ml) in a total volume of 0.2 ml of 25 mM KH2PO4, pH 7.4, containing 0.1 mM EDTA, in the presence and absence of raloxifene. Incubations were conducted for 2 min at 37°C and terminated by the addition of 0.05 ml of HCl (1 M) containing amantadine as an internal standard (250 ng/ml). Terminated incubation mixtures were subject to filtration through a Millipore Multiscreen-HA 0.45-μm mixed cellulose ester 96-well membrane vacuum filtration module into 96-well plates. The resulting filtrate was analyzed by HPLC-MS using the equipment described above. Samples (0.02 ml) were injected onto a Phenomenex Luna C18 column (2.5 × 50 mm; 5 μm; Phenomenex, Torrance, CA) preequilibrated in 20 mM acetic acid (adjusted to pH 4.0 with NH4OH) containing 5% CH3CN at a flow rate of 0.5 ml/min. Elution was effected with a solvent program of holding at 5% CH3CN over the first 0.4 min, followed by a linear gradient to 95% CH3CN over 0.6 min, holding at this composition for 1 min, and reequilibration to initial conditions over the next 6 min. The state file settings on the mass spectrometer were set at the following values: ionspray voltage, 2000; source temperature, 450°C; orifice voltage, 30; ring potential, 90; Q1 potential, –5, with other settings set to optimize system response. Cotinine and amantadine were detected by monitoring ion current at m/z 177 and 151, respectively, and retention times were 1.6 and 2.3 min, respectively. Quantitation was done using a cotinine standard curve ranging from 3.0 to 300 ng/ml. Assay standards were within ±9.9% of nominal values.
CP-544,439 Reductase Assay. CP-544,439 (10–100 μM for Ki determinations), N-methylnicotinamide (0.5 mM), and pooled human liver cytosol (0.5 mg/ml) were incubated in 0.2-ml incubations containing 0.1 mM EDTA and 25 mM KH2PO4, pH 7.4, in the presence and absence of raloxifene. Incubations were commenced with the addition of cytosol and were conducted at 37°C for 10 min. Reactions were terminated with the addition of 3 ml of methyl tert-butyl ether (extraction solvent) and internal standard (10 ng of CP-472,434, an analog of the product possessing a geminal dimethyl group in place of the tetrahydropyran ring, in 0.05 ml of methanol). The organic fraction was evaporated under N2 and the residue was reconstituted in 0.04 ml of CH3CN/H2O (1:2). Samples (0.02 ml) were injected onto an HPLC-MS system described above. The column was a Phenomenex C18 Luna (2.0 × 50 mm; 5-μm particle size) equilibrated in 20 mM CH3COOH, pH 4 (with NH4OH) containing 37% CH3CN at a flow rate of 0.5 ml/min. Detection was accomplished in the negative ion mode, monitoring m/z 393.3 and 351.3 for deoxy-CP-544,439 and internal standard, respectively. The retention time for deoxy-CP-544,439 was 1.3 min and for internal standard was 1.4 min. Quantitation was accomplished with a standard curve of deoxy-CP-544,439 ranging from 0.2 to 20 ng/ml. Assay standards were within 12% or less of nominal values.
Xanthine Oxidase Assay. Pterin (10–1000 μM for KM determination; 35 μM for inhibition experiments) was incubated with pooled human liver cytosol (0.5 mg/ml) in 0.1 ml of 25 mM KH2PO4, pH 7.4, containing 0.3 mM EDTA. Incubations were commenced with the addition of cytosol and were conducted in a shaking water bath maintained at 37°C open to air. (In initial experiments, reaction velocity linearity was demonstrated out through 2.5 h.) After 60 min, the incubations were terminated by addition of 0.03 ml of HCl (1 M). The samples were analyzed by HPLC-MS using the instrumentation described above. A Waters (Milford, MA) Polarity column (2.0 × 50 mm; 5 μm) was used with a mobile phase of 20 mM acetic acid at 0.5 ml/min. Detection of isoxanthopterin was accomplished in the negative ion mode monitoring m/z 178. The retention time was 2.8 min. Quantitation was accomplished with a standard curve of isoxanthopterin ranging from 0.1 to 10 μM. Mean assay inaccuracy was 6%.
Data Analysis. Enzyme kinetic and inhibition data were analyzed using Sigma Plot (v8.0) with the Enzyme Kinetics Module (v1.1) (SPSS Inc., Chicago, IL). Data were fit to various models of inhibition (i.e., competitive, noncompetitive, uncompetitive, mixed) and goodness of fit was rank-ordered using the Aikake Information Criteria. For CP-544,439 reduction activity, there appeared to be some reaction in human cytosol that was not aldehyde oxidase-catalyzed such that inhibition of the reaction by menadione reached a maximum of about 70%. Thus, data measuring the effect of raloxifene on CP-544,439 reduction activity was corrected for this factor.
Results
The enzyme kinetic parameters for phthalazine oxidase, vanillin oxidase, nicotine-Δ1′(5′)-iminium ion oxidase, and CP-544,439 reductase are listed in Table 1. The conversion of nicotine-Δ1′(5′)-iminium ion to cotinine occurred with the greatest intrinsic clearance, followed by vanillin oxidation, phthalazine oxidation, and CP-544,439 reduction. Vanillin and nicotine-Δ1′(5′)-iminium ion oxidations followed simple Michaelis-Menten kinetics, whereas the other two reactions demonstrated substrate inhibition at high substrate concentrations (Fig. 2).
Enzyme kinetic parameters for aldehyde oxidase- and xanthine oxidase-catalyzed reactions in pooled human liver cytosol
Values represent the mean (±standard error) of triplicate determinations.

Substrate saturation plots for vanillin oxidase (A), nicotine-Δ1′(5′)-iminium ion oxidase (B), and CP-544,439 reductase (C).
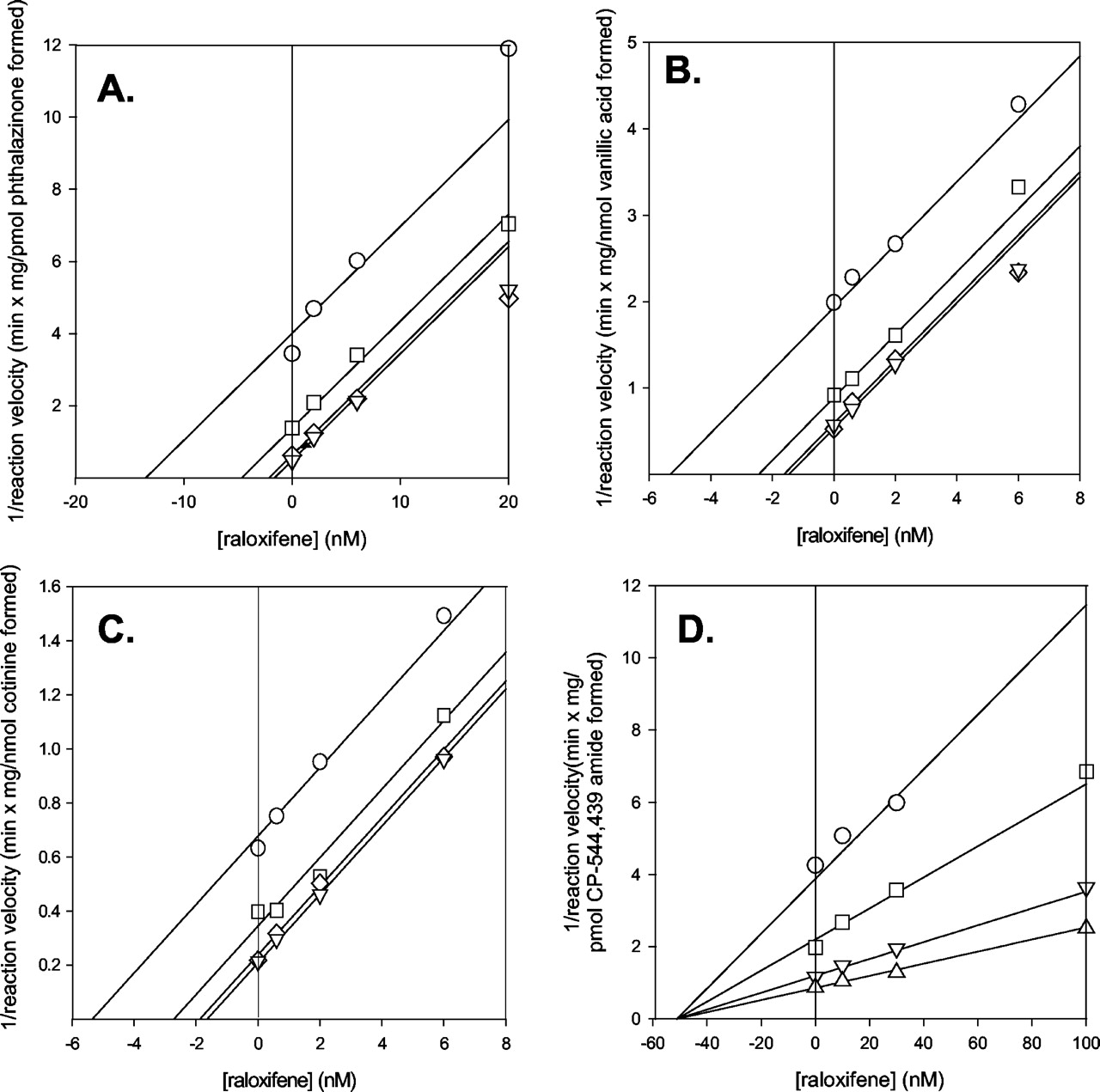
Raloxifene was a potent inhibitor for the three oxidation reactions. Ki values ranged between 0.87 and 1.4 nM (Table 2), and in all three cases the mechanism of inhibition was uncompetitive. This is illustrated by the parallel line pattern in the Dixon plots for these inhibition data (Fig. 3). However, for the reduction reaction, the Ki value of raloxifene was substantially greater (51 nM) and the mechanism of inhibition was noncompetitive (Fig. 3). Preincubation of raloxifene with human cytosol did not increase the capability of raloxifene to inhibit aldehyde oxidase (Fig. 4). At a concentration of 50 μM (approximately 1000 and 50,000 times that of the Ki values for aldehyde oxidase reduction and oxidation reactions), raloxifene caused 58% inhibition of pterin oxidation, a marker of activity for human xanthine oxidase. Allopurinol, an established inhibitor of xanthine oxidase, yielded 86% inhibition at 100 μM, whereas menadione, an aldehyde oxidase-specific inhibitor, actually caused stimulation of pterin oxidation (Table 3).
Inhibition of aldehyde oxidase-catalyzed reactions by raloxifene
Values represent the mean (±standard error) of triplicate determinations.
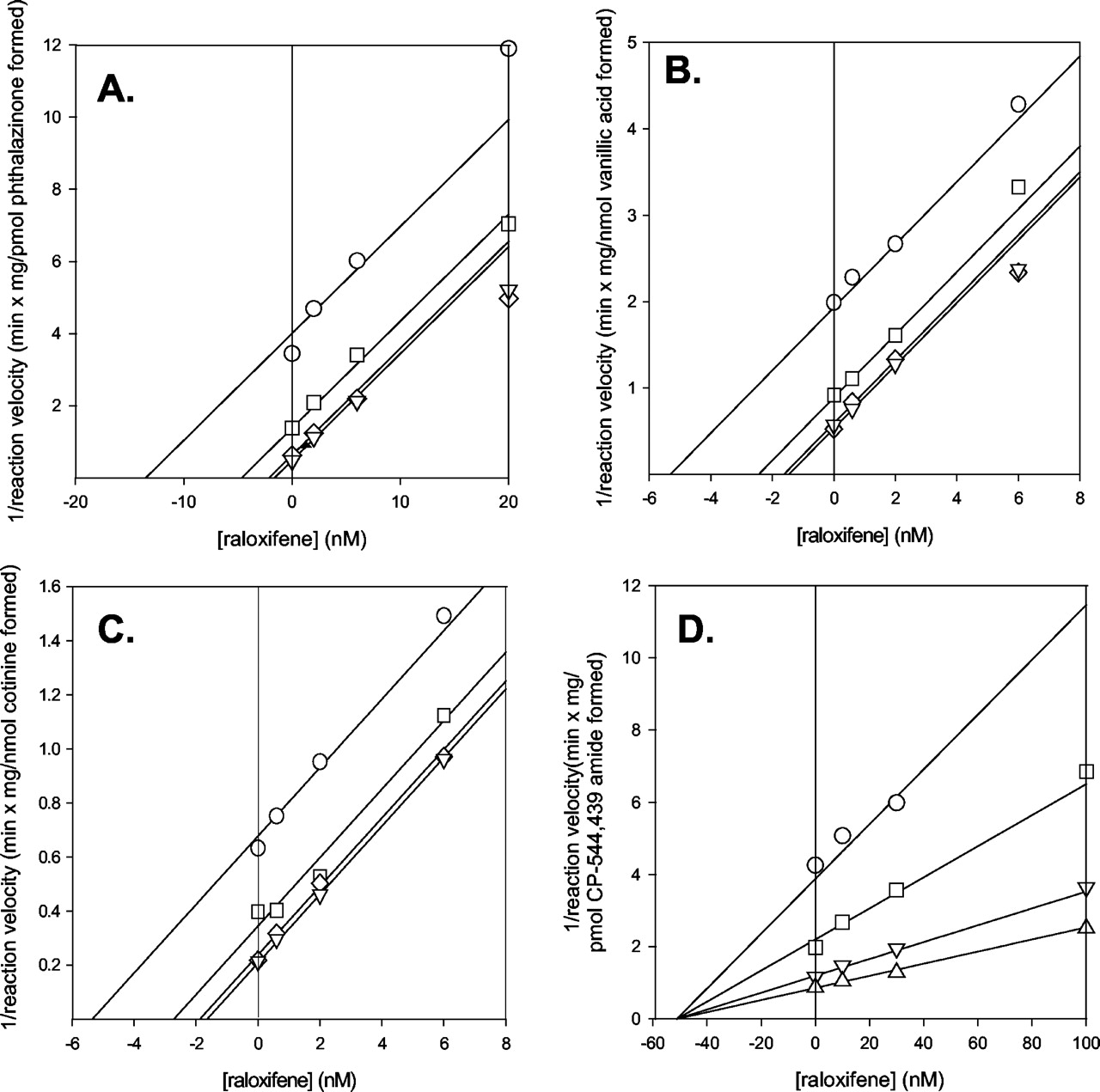
Dixon plots for the inhibition of aldehyde oxidase-catalyzed reactions by raloxifene.
A, phthalazine oxidase; B, vanillin oxidase; C, nicotine-Δ1′(5′)-iminium ion oxidase; D, CP-544,439 reductase.

Effect of preincubation on inhibition of phthalazine oxidase activity by raloxifene.
Solid bars represent no preincubation; open bars represent reaction velocities measured after a 30-min preincubation.
Effect of raloxifene, allopurinol, and menadione on human liver cytosolic xanthine oxidase-catalyzed pterin oxidation
Values represent the mean ± S.D. of three determinations.
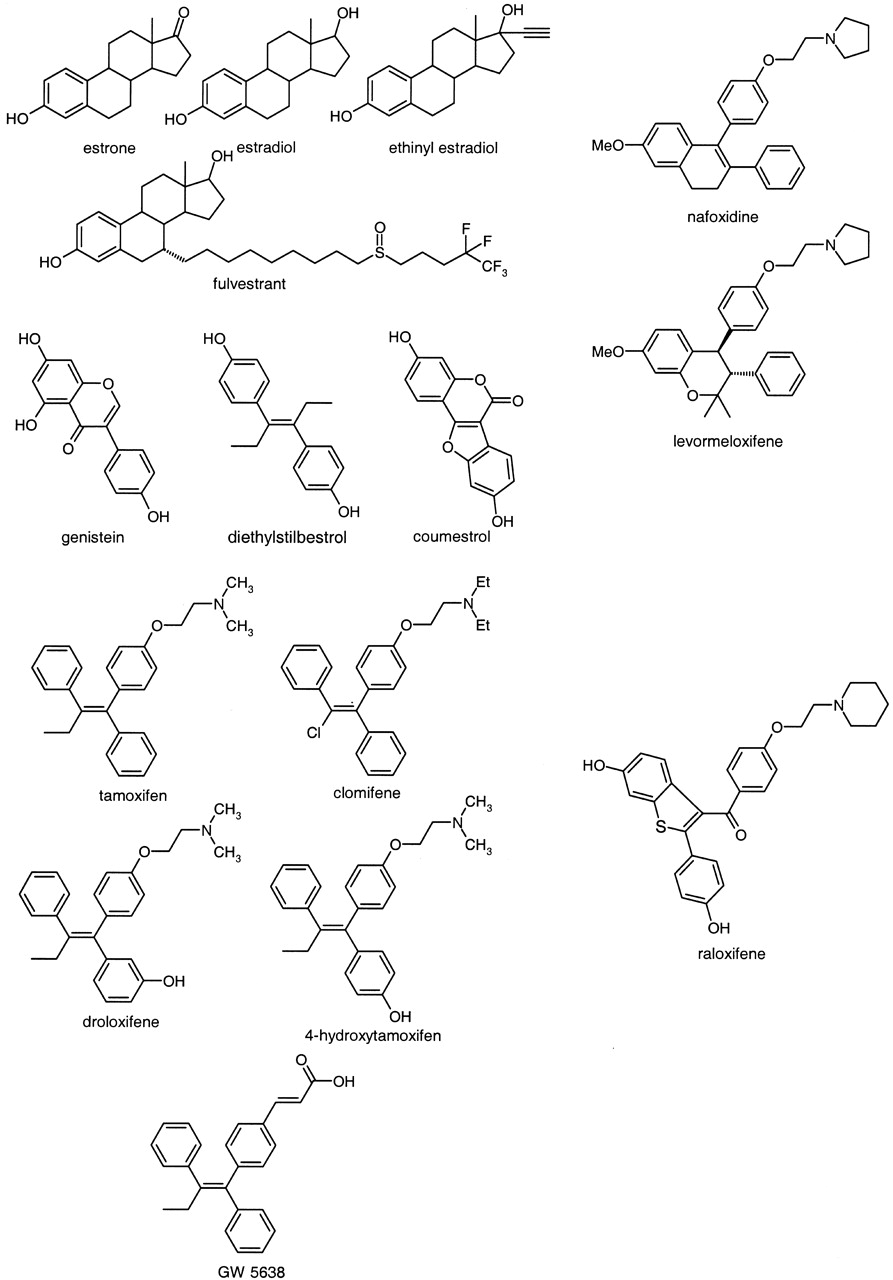
In previous experiments, other estrogens (estradiol, ethinyl estradiol, and tamoxifen) were also demonstrated to be potent inhibitors of aldehyde oxidase, albeit substantially less potent than raloxifene (Obach et al., 2004) To better understand the potential for estrogens and SERMs to inhibit aldehyde oxidase, 11 additional compounds were tested for their ability to inhibit phthalazine oxidase activity, and the resulting data were combined with data from the previous report (Obach et al., 2004). Data are listed in Table 5, and the chemical structures are shown in Fig. 5. Most had IC50 values in a narrow range between 0.29 and 2.5 μM. Raloxifene was clearly more potent than any other compound of this therapeutic class. GW5638, an analog of tamoxifen that possesses an acidic group in place of the weakly basic pyrrolidine substituent, was substantially less potent as an inhibitor of aldehyde oxidase. Bisphenolic compounds such as diethylstilbestrol, coumestrol, and genistein appeared to be potent inhibitors, and addition of a phenolic group to tamoxifen (i.e., 4-hydroxytamoxifen, droloxifene) caused a slight increase in potency from tamoxifen.
Inhibition of human cytosolic aldehyde oxidase-catalyzed phthalazine oxidation activity by estrogen antagonists and selective estrogen receptor modulators
IC50 values are means ± S.E.

Structures of raloxifene and other estrogens.
Elements of the structure of raloxifene that are essential to the inhibitory potency were tested by determination of the inhibitory potency of structural analogs (Table 4). Methylation of the two phenols resulted in a >1000× decrease in potency (compound 1; IC50 = 8.0 μM). Replacement of one phenol with –CF3 (compound 5) resulted in a much smaller decrease in potency (16×), and moving the phenol on the benzothiophene by one position (compound 4) resulted in a 9× decrease in potency. Compound 3, in which only the 2-phenylbenzothiophene portion containing both phenolic groups remained, was 40× less potent than raloxifene and had potency comparable to that of the bisphenolic compounds genistein and coumestrol (Table 5). Interestingly, removing the basicity of the piperidine nitrogen of raloxifene by conversion to an amide (compound 2) had little effect on the inhibitory potency.
Inhibition of human cytosolic aldehyde oxidase-catalyzed phthalazine oxidation activity by raloxifene and structural analogs
Values for IC50 are means ± S.E.
Discussion
In an earlier study in which over 200 drugs were examined for their potential to inhibit human liver cytosolic aldehyde oxidase activity, as assessed through measurement of phthalazine oxidation, raloxifene was identified as the most potent inhibitor, with an IC50 value of 2.9 nM. The objective of this study was to characterize raloxifene inhibition with regard to mechanism, determine potency versus various classes of aldehyde oxidase-catalyzed reactions, determine specificity for aldehyde oxidase versus the related enzyme xanthine oxidase, and test whether the inhibition was time-dependent. Furthermore, other estrogen compounds, tamoxifen, estradiol, and ethinyl estradiol, were also demonstrated to be inhibitors of aldehyde oxidase, albeit weaker than raloxifene (Obach et al., 2004). Thus, in the present experiments, other estrogens and SERMs were tested for their capability to inhibit this enzyme.
Aldehyde oxidase can catalyze both oxidation and reduction reactions on drugs and xenobiotics. For oxidation reactions, substrates have been classified into three structural types: aldehydes, which undergo oxidation to carboxylic acids; aromatic azaheterocyclic compounds, which undergo oxidation to lactams; and quaternary azaheterocyclic compounds, which also undergo oxidation to lactams (Beedham, 2002). Three aldehyde oxidase substrates examined in this study, vanillin, phthalazine, and nicotine-Δ1′(5′)-iminium ion, exemplify these three classes of oxidized substrates, respectively. Ordinarily, aldehyde oxidase utilizes oxygen as the terminal electron acceptor yielding hydrogen peroxide as the by-product of aldehyde oxidase-catalyzed drug oxidation reactions. However, in some cases, aldehyde oxidase can utilize other compounds in place of oxygen as terminal acceptors of reducing equivalents, resulting in reduction reactions. Examples of reduction reactions catalyzed by aldehyde oxidase include N–O bond reductions in nitro groups (Wolpert et al., 1973), hydroxamic acids (Sugihara and Tatsumi, 1986), and benzisoxazoles (Sugihara et al., 1996), as well as N–S bond reductions in benzisothiazoles (Kamel et al., 2002). In many cases, these reactions are observed when incubations are conducted under anaerobic conditions, since the reducing substrates would be competing with oxygen. In this study, the reduction of CP-544,439, a hydroxamic acid, to the corresponding amide was used as a probe reduction reaction for aldehyde oxidase. This reaction was observed under aerobic conditions.
For all three oxidation reactions, raloxifene was determined to be a potent, uncompetitive inhibitor. The inhibition constants for the three oxidation reactions were consistent and ranged from 0.87 to 1.4 nM. Uncompetitive inhibition is unusual for drug-metabolizing enzymes. It implies that the inhibitor can only bind to the enzyme in the substrate-bound state. Raloxifene did not exhibit any time-dependent inhibition (Fig. 4). Additionally, raloxifene was tested as a potential inhibitor of the related molybdenum cofactor enzyme, xanthine oxidase. At a concentration of 50 μM, raloxifene only caused 58% inhibition of pterin oxidation to isoxanthopterin. This indicates a high selectivity of raloxifene for aldehyde oxidase among these two molybdenum cofactor enzymes.
For the reduction reaction studied, raloxifene was also a potent inhibitor, albeit the Ki value was 50-fold greater and the mechanism of the inhibition was noncompetitive. In aldehyde oxidase-catalyzed reduction reactions, the system is supplemented with a saturating concentration of a reducing substrate as a source of electrons, in this case N-methylnicotinamide, which complicates the mechanistic interpretation of the kinetic data. Additionally, oxygen may also be competing with the substrate to be reduced (i.e., CP-544,439 in this case). Noncompetitive inhibition suggests that the inhibitor can bind both substrate-bound and substrate-free forms of the enzyme. Thus, raloxifene could be competing with the binding of CP-544,439 to the site of the enzyme at which substrates that are reduced by the enzyme bind, as well as binding a substrate-bound intermediate form of the enzyme. With saturating N-methylnicotinamide, it could be suggested that raloxifene is simply binding to the same reduced enzyme form as when it exhibits effects on oxidized substrates. However, if this were the case, it would be expected that the Ki value for CP-544,439 reduction would also be in the range of 1 nM. Proposed schemes for the inhibition of aldehyde oxidase by raloxifene are presented in Fig. 6 and the accompanying legend. It is clear that other types of physical data need to be gathered to better understand the mechanism of inhibition of aldehyde oxidase by raloxifene. This will be the effort of further experimentation.
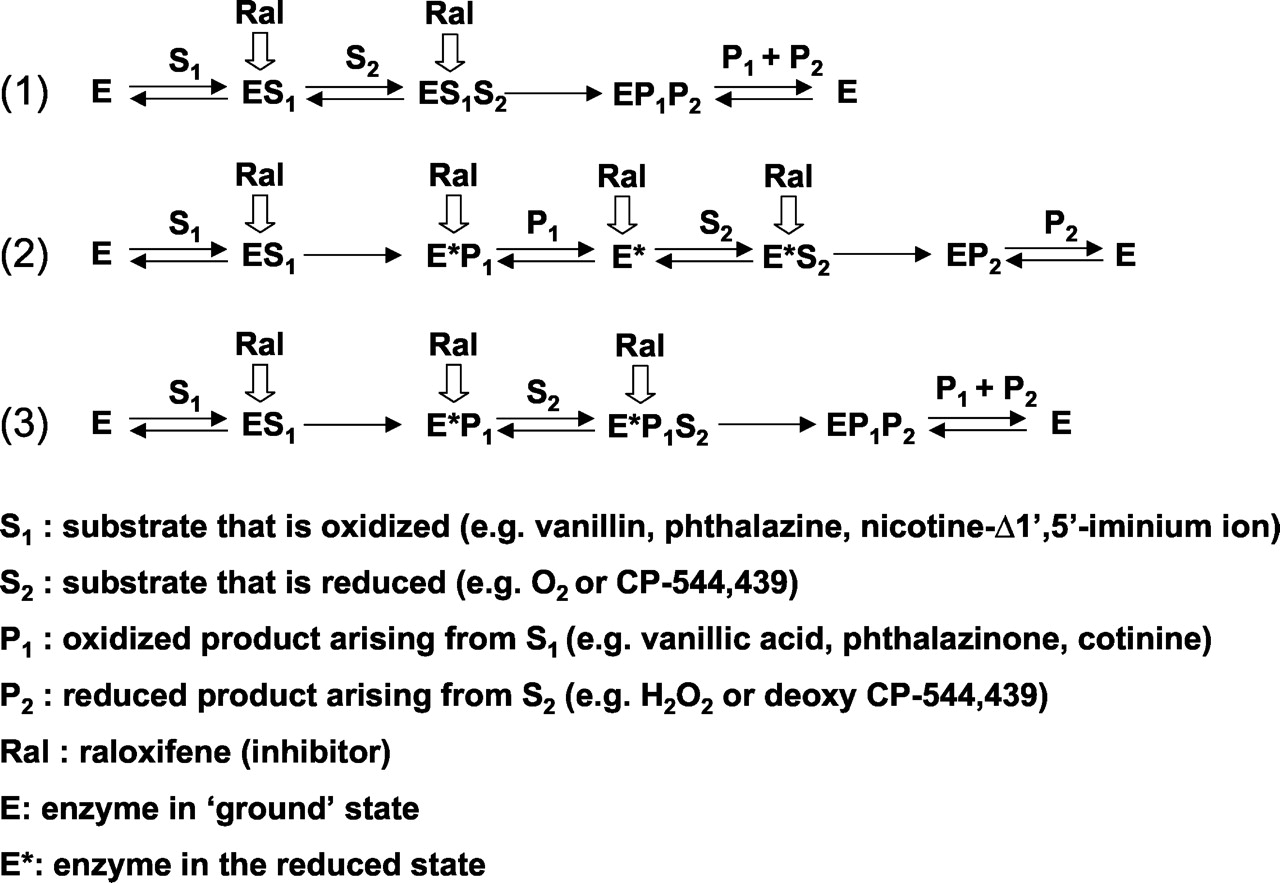
Three possibilities illustrating mechanisms by which raloxifene can inhibit aldehyde oxidase.
Each illustration accounts for the uncompetitive mechanism of inhibition for the substrate that is oxidized and the noncompetitive mechanism for the substrate that is reduced. The bold arrows indicate intermediate species that could be bound by raloxifene. In (1), a sequential mechanism is presented in which both substrates bind before the redox reaction occurs, and raloxifene can bind to substrate-bound forms of the enzyme. In (2) and (3), the substrate that is oxidized (S1) transfers reducing equivalents to the enzyme before binding of the substrate that is reduced. In (2), the oxidized product dissociates before binding of the substrate (S2) that is reduced. In (3), the substrate that is reduced binds before release of the oxidized product. The data suggest that raloxifene cannot bind to aldehyde oxidase unless S1 has already bound the enzyme (i.e., uncompetitive for phthalazine, etc.). Raloxifene is noncompetitive for the substrate that is to be reduced (S2), so that it either binds to a form of the enzyme that lacks S2 [such as E* in (2)] or already possesses it [e.g., E*P1 in (2) or (3)].
Structural analogs of raloxifene were examined to gain some understanding of the structural elements that impart inhibitory potency. The two phenolic groups appear to be important, since compounds 1 (no phenols) and 5 (one phenol) were less potent than raloxifene (raloxifene > compound 5 > compound 1). The precise position of the phenol on the benzothiophene ring also had an effect on inhibition (raloxifene > compound 4). Finally, the presence of the 3-aroyl-p-ethoxypiperidinyl substituent is important for the potency of raloxifene, since its removal (compound 3) resulted in a marked loss of potency. Interestingly, the basicity of the piperidine ring nitrogen does not appear to be required inasmuch as the analogous amide (compound 2) possessed inhibitory potency similar to that of raloxifene.
Raloxifene, a member of the SERM class of compounds, is a frequently prescribed agent for the long-term treatment of osteoporosis. Since raloxifene demonstrated such a high degree of inhibitory potency for human liver aldehyde oxidase, an investigation of the effects of other compounds of this class, as well as estrogen analogs, was undertaken to address the potential for drug-drug interactions that could be mediated by these compounds. The other compounds examined also demonstrated inhibition of aldehyde oxidase-catalyzed phthalazine oxidation, although none approached the potency of raloxifene (IC50 of 5.7 nM versus 0.29–57 μM). Clearly, raloxifene is unique among these compounds in its capability to inhibit aldehyde oxidase, and it is also unique in its chemical structure (Fig. 5). Raloxifene combines a bisphenolic structure with an alkylamino chain. The other inhibitors, except the steroids, possess either one of these entities or the other, but not both. Genistein, diethylstilbestrol, and coumestrol possess the bisphenol structure; tamoxifen, 4-hydroxytamoxifen, clomifene, droloxifene, nafoxidine, and levormeloxifene possess the alkylamino side chain. Also, comparison of tamoxifen with 4-hydroxytamoxifen and droloxifene, which are monophenol analogs of tamoxifen, shows that the introduction of one phenol group increases the inhibitory potency to a nominal degree. The analog in which the alkylamino group was replaced with a carboxylic acid, GW5638, demonstrated markedly less inhibition, suggesting that the side chain cannot possess an anionic character for inhibition of aldehyde oxidase by this class of drugs.
Among the sterols, all four demonstrated comparable inhibitory potency. In previous work, the importance of the phenol group for aldehyde oxidase inhibition by steroids was demonstrated (Obach et al., 2004). Comparable inhibition by these four sterols is consistent with this notion. A greater number of analogs need to be tested to develop any structure-activity relationships for sterols.
Drug-drug interactions that occur via a mechanism of aldehyde oxidase inhibition have not been demonstrated, with the exception of one: cimetidine/zaleplon (Renwick et al., 2002). This is due to the fact that not many drugs have been shown to be primarily cleared by aldehyde oxidase-mediated metabolism, nor are there many drugs that are potent inhibitors. Zaleplon oxidation has been recently established as an aldehyde oxidase-mediated major clearance mechanism for this drug (Lake et al., 2002; Renwick et al., 2002). The activation of famciclovir to the active antiviral agent penciclovir requires aldehyde oxidase-mediated oxidation (Clarke et al., 1995; Rashidi et al., 1997). Furthermore, there are intermediary metabolic reactions attributed to aldehyde oxidase, including such agents as nicotine (Gorrod and Hibberd, 1982), tamoxifen (Ruenitz and Bai, 1995), and citalopram (Rochat et al., 1998), and it is likely that there are others yet to be revealed. Nevertheless, compared with cytochrome P450, there is a paucity of information on aldehyde oxidase and its potential to be responsible for drug clearance and a target for drug-drug interactions. The in vitro data reported herein for raloxifene, in addition to being of interest in furthering our understanding of this enzyme (see above), also suggest that investigation of raloxifene as a potential precipitant of drug-drug interactions is warranted. The inhibitory potency for raloxifene on aldehyde oxidase (1 nM for oxidation reactions; 50 nM for reduction reaction) is comparable to or even greater than in vitro potencies reported for such well known precipitants of cytochrome P450-based drug interactions such as quinidine and ketoconazole.
The projection of the magnitude of in vivo drug interactions from in vitro inhibition data is not easily done (Lin and Pearson, 2002; Yao and Levy, 2002). There appear to be many variables that are needed, some of which are impossible to reliably estimate. Attempts have been made based on [I]/Ki calculations, in which values for [I] refer to relevant in vivo concentrations of inhibitor (e.g., unbound systemic concentrations, estimates of liver concentrations, etc.). Empirically, values of [I]/Ki greater than unity suggest a high risk for drug-drug interactions, those below 0.1 a low risk, and those between 0.1 and 1 an area of uncertainty (Tucker et al., 2001). Raloxifene concentrations in the liver during first-pass, after a standard oral dose of 60 mg, are likely to exceed its Ki, 1 nM, which would suggest that raloxifene would have a high potential to cause drug interactions with compounds for which aldehyde oxidase is important, e.g., zaleplon and famciclovir. However, the [I]/Ki approach makes the assumption that the mechanism of inhibition is competitive or noncompetitive, but not uncompetitive, like the mechanism of raloxifene inhibition of aldehyde oxidase. In uncompetitive inhibition, both Vmax and KM are affected, but the ratio, Vmax/KM, remains the same. The extent of drug interactions exhibited by an uncompetitive inhibitor would be more magnified as the concentration of the substrate relative to its KM value is greater, the opposite of that which occurs for competitive inhibitors. Also, at lower substrate concentrations, relative to KM, that would be more likely to be attained at low doses, the magnitude of a drug interaction for an uncompetitive inhibitor will be lower. Because of this unusual mechanism of inhibition, any prediction of a raloxifene drug interaction on an aldehyde oxidase-cleared compound from in vitro data is particularly tenuous. Verification with an in vivo interaction study is needed.
Acknowledgments
Helpful discussions of these data with Prof. Christine Beedham, University of Bradford, Bradford, UK, and suggestion of the use of CP-544,439 as an aldehyde oxidase reduction substrate by Dr. Deepak Dalvie, Pfizer, are gratefully acknowledged.
Footnotes
-
↵1 Abbreviations used are: SERM, selective estrogen receptor modulator; HPLC-MS, high pressure liquid chromatography-mass spectrometry; CP-544,439, 4-[4-(fluorophenoxy)-benzenesulfonylamino]-tetrahydro-pyran-4-carbvlic acid hydroxyamide; CP-472,434, 2-[4-(fluorophenoxy)-benzenesulfonylamino]-isobutyramide; GW5638, (2E)-3-[4-(1Z)-1,2-diphenyl-1-butenyl]phenyl]-2-propenoic acid.
- Received August 26, 2003.
- Accepted September 25, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}