


Abstract
CYP2D6 and CYP3A4 represent two particularly important members of the cytochrome P450 enzyme family due to their involvement in the metabolism of many commercially available drugs. Avoiding potent inhibitory interactions with both of these enzymes is highly desirable in early drug discovery, long before entering clinical trials. Computational prediction of this liability as early as possible is desired. Using a commercially available data set of over 1750 molecules to train computer models that were generated with commercially available software enabled predictions of inhibition for CYP2D6 and CYP3A4, which were compared with empirical data. The results suggest that using a recursive partitioning (tree) technique with augmented atom descriptors enables a statistically significant rank ordering of test-set molecules (Spearman's ρ of 0.61 and 0.48 for CYP2D6 and CYP3A4, respectively), which represents an increased rate of identifying the best compounds when compared with the random rate. This approach represents a valuable computational filter in early drug discovery to identify compounds that may have P450 inhibition liabilities prior to molecule synthesis. Such computational filters offer a new approach in which lead optimization in silico can occur with virtual molecules simultaneously tested against multiple enzymes implicated in drug-drug interactions, with a resultant cost savings from a decreased level of molecule synthesis and in vitro screening.
The likelihood of unfavorable drug-drug interactions (DDIs1), although small in some clinical populations, remains an important factor in lead optimization and product differentiation for pharmaceutical companies. DDI discovered in the late stages of the drug discovery process could have severe consequences and, in many cases, cause the abandonment of late-stage clinical candidates. Many pharmaceutical companies, in an attempt to minimize this dilemma, are screening for DDIs in vitro in the early stages of drug discovery (Wynalda and Wienkers, 1997; Gao et al., 2002). Although many of these in vitro tests can be performed in a high-throughput fashion, they require the molecule to be synthesized or isolated, yet drug discovery today is becoming reliant on in silico testing of large virtual compound collections (Leach and Hann, 2000). Predictive computational models that identify liabilities such as DDIs will have an enormous impact on minimizing the advancement of undesirable compounds and ultimately will help to shorten drug discovery timelines when used with other filters for metabolism and toxicology (Ekins and Rose, 2002). Therefore, identifying an efficient method of computational filtering for potential interaction with drug-metabolizing enzymes is of paramount importance.
One particular area of focus for DDIs is the interactions with the cytochromes P450 (P450s). The key P450s that have been identified and modeled extensively include CYP2D6 (Ekins et al., 1999a) and CYP3A4 (Ekins et al., 1999b). These enzymes metabolize a vast array of structurally diverse commercially available drugs and represent major routes of drug clearance. Therefore, it has become a widely accepted practice that potent inhibition of these enzymes should be avoided where possible. Many approaches to predicting inhibition have been taken. A comprehensive analysis of existing physicochemical data, protein homology, nuclear magnetic resonance, site-directed mutagenesis, and quantitative structure-activity relationship (QSAR) approaches for prediction of P450 inhibition have all been modestly effective (Smith et al., 1997a,b). Recently, a neural network model was applied to differentiate between substrates and inhibitors of CYP3A4 using the data collated in the human P450 metabolism data base (Molnar and Kesuru, 2002). This model was then used to predict previously published data on structurally diverse CYP3A4 inhibitors (Ekins et al., 1999b). The model was not able to correctly rank the inhibition constants and misclassified a potent inhibitor as a noninhibitor. A major reason for the failures of these approaches could be due to the generally small sets of compounds used for model building for which empirical data exist. Even with this considerable shortcoming, recent reviews have described and compared many pharmacophores and QSARs for key P450s (Ekins et al., 2001; de Groot and Ekins, 2002), including those for CYP2D6 (Ekins et al., 1999a) and CYP3A4 (Ekins et al., 1999b; Riley et al., 2001). The pharmacophore models have provided structural insight into key features for inhibition. Despite these relative successes, it is obvious that there is still a need for a rapid computational approach that provides a realistic rank ordering of inhibitor potency that is suitable for early drug discovery. The achievement of such a goal may sacrifice some of the interpretability of a pharmacophore and homology model but will more than make up for this in terms of its utility for rapidly scoring large virtual data bases.
The commercial availability of CYP2D6 and CYP3A4 in recombinant form along with substrate probes (Crespi et al., 1997, 1998) has allowed the development of high-throughput analysis of a large group of diverse molecules. A set of over 1750 compounds with inhibition data for CYP3A4 and CYP2D6 (Cerep, Redmond, WA) was used to develop computational models using recursive-partitioning approaches as implemented in commercially available software. Multiple tree models were generated in Chemtree (Golden Helix Inc., Bozeman, MT) using the continuous data set (percentage of inhibition). Chemtree used over 2500 of the augmented atom descriptors in which the focus atom and the attached atoms represent a feature, and the through bond count between these focus atoms is the distance (Young et al., 2002). Computational models were created and tested using additional data generated in our laboratories for a diverse set of 98 molecules purchased from external vendors. This approach represents, to our knowledge, the largest set of computational models for P450 inhibition that has been described and validated to date. The results of this study show that computational models can be generated to predict percentage of inhibition for both CYP2D6 and CYP3A4, and ultimately these techniques may be applied to other P450s.
Materials and Methods
Chemicals. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Molecules used for the validation set were purchased from Maybridge Chemicals (Trevillet, UK). 3-[2-N,N-Diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin (AMMC), 3-[2-N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin (AHMC), 7-benzyloxy-4-trifluoromethylcoumarin (BFC), 7-hydroxy-4-trifluoromethylcoumarin, and human recombinant CYP2D6 and CYP3A4 were purchased from PanVera Corporation (Madison, WI).
Enzyme Incubations. Recombinant CYP2D6 (20 nM) was incubated with 5 μM AMMC for 75 min at 37°C in the presence of the test compound (10 μM) in 1.25% final concentration of both acetonitrile and DMSO, with the NADPH-regenerating system (30 μM β-nicotinamide adenine dinucleotide phosphate, 3.3 mM d-glucose 6-phosphate, and 0.4 U/ml glucose-6-phosphate dehydrogenase). Recombinant CYP3A4 was incubated at 5 nM with 10 μM BFC for 20 min at 37°C in the presence of the test compound (10 μM) in a 1.5% final concentration of both acetonitrile and DMSO with the NADPH-regenerating system (100 μM β-nicotinamide adenine dinucleotide phosphate, 3.3 mM d-glucose 6-phosphate, and 0.4 U/ml glucose-6-phosphate dehydrogenase). Positive controls contained the complete reaction components and identical incubation conditions with the exception of compound added (1% DMSO added).
Analysis. The detection of the metabolites for AMMC and BFC, namely AHMC and HFC, respectively, were assessed by spectrofluorimetric measurement at an excitation wavelength of 405 nm for both AHMC and HFC and emission wavelengths of 460 nm for AHMC and 490 nm for HFC. All measurements were performed with a PerkinElmer Wallac Victor 5 multiplate reader (PerkinElmer Wallac, Gaithersburg, MD). The fluorescent intensity (fu) measured at (t = 0) was subtracted from that measured after the appropriate incubation time (t = 75 min for CYP2D6 and t = 20 min for CYP3A4). The ratio of signal-to-noise was calculated by comparing the fluorescence in incubations containing the test compound with the control samples containing the same solvent vehicle. The percentage of control activity was then calculated. Subsequently, the percentage of inhibition is calculated by subtracting the percentage of control activity from 100.
Computational Methods. The Chemtree recursive-partitioning software used was run on a Pentium 3 processor. Two data sets of percent-inhibition data were generated under the same conditions as described above and purchased from Cerep for CYP2D6 (1759 molecules) and CYP3A4 (1756 molecules). The molecules and experimental data for each enzyme were separately imported as an .sdf file in Chemtree to generate over 2500 augmented atom descriptors (Young et al., 2002), which were used along with the actual percent-inhibition value to generate 20 random tree models (with the following options: p value threshold for splits, 0.99; maximal segments, 3; parallel threads, 1; and resampling iterations, 10,000).
External Validation of Computational Models and Statistical Analysis. The 98 molecules used to generate percent-inhibition data for both CYP2D6 and CYP3A4 in our laboratories were input as an .sdf file into the Chemtree software, and predictions were made with the appropriate set of 20 models coinciding with the same enzyme to generate computational predictions. The in vitro observed data and computationally predicted data for the average of the 20 tree models were compared and assessed using a number of statistical approaches, including the correlation of Spearman's ρ (Ekins et al., 2002), available in JMP 4.5 (SAS Institute Inc., Cary, NC). This test represents a correlation coefficient on the relative rank order of the data and not on the values themselves. This test also provides a statistical significance result, which can be expressed as the p value, where an observed value of <0.05 is meaningful. In addition, the observed and predicted data can be considered as binary values using a cutoff of less than 40% inhibition to represent desirable molecules and greater than 40% inhibition to indicate undesirable molecules. The molecules could be ranked from lowest- to highest-predicted percentage of inhibition, and then the rate of finding these poor inhibitors was assessed. This rate was compared with the random and the ideal rate of finding all the poor inhibitors at five data-point intervals and plotted accordingly. Such an approach represents selection of the molecules with the lowest percentage of inhibition first. The standard deviations for replicate empirical determinations of percent-inhibition data were also determined.
Results and Discussion
In recent years, there have been a number of studies published that describe computational models for various absorption, distribution, metabolism, excretion, and toxicity properties, including drug-drug interactions. In the majority of cases, the validation of these models has been performed using a relatively small data set of empirical data that is sometimes confined to a low chemical diversity or similar structural series (Ekins et al., 2000, 2001). In the present study, we have described models using a commercially available software package for molecular descriptor generation and computational QSAR model building. We have coupled this software with a large set of experimentally determined CYP2D6 and CYP3A4 inhibition data from a commercial source to generate models. In addition, we have tested these computational models with data generated by our own laboratory but not included in model generation or resembling those molecules in the model training sets.
The average of the 20 CYP2D6 Chemtree models resulted in an observed versus predicted correlation of r2 = 0.88 for the training set (Fig. 1a). The average of the 20 CYP3A4 Chemtree models resulted in an observed versus predicted correlation of r2 = 0.82 for the training set (Fig. 1b). Percent-inhibition data were generated for 98 diverse commercially available molecules with both CYP2D6 and CYP3A4 and were compared with the predictions generated with the respective Chemtree models. The average of the 20 CYP2D6 Chemtree models was able to generate a statistically significant rank ordering of the percent-inhibition data based on the Spearman's ρ rank of 0.61, p = 0.0001. This represents a significant improvement over the random identification of the molecules with less than 40% inhibition. When ranked in order of increasing percentage of inhibition of CYP2D6, virtually all of the first 50 compounds were predicted correctly (Fig. 2a). The average of the 20 CYP3A4 Chemtree models was to a lesser extent also able to generate a statistically significant rank ordering of the percent-inhibition data based on the Spearman's ρ rank of 0.49, p = 0.0001. This was also an improvement over random identification of the molecules with less than 40% inhibition (Fig. 2b) but was not of the same magnitude as the predictions for CYP2D6 (Fig. 2a). A ranking of such inhibition data may be all that is required for early drug discovery in order to select the best molecules with the greatest odds of progressing without failure due to interactions with these enzymes.

CYP2D6 (a) and CYP3A4 (b) Chemtree training sets.
a, CYP2D6 Chemtree training set for the average of 20 models showing the observed versus the predicted percent inhibition of 5 μM AMMC metabolism after incubation for 75 min at 37°C in the presence of the test compound (10 μM), as described under Materials and Methods; b, CYP3A4 Chemtree training set for the average of 20 models showing the observed versus predicted percent inhibition of 10 μM BFC metabolism for 20 min at 37°C in the presence of the test compound (10 μM) as described under Materials and Methods. Both figures also show the 95% confidence interval for the data.
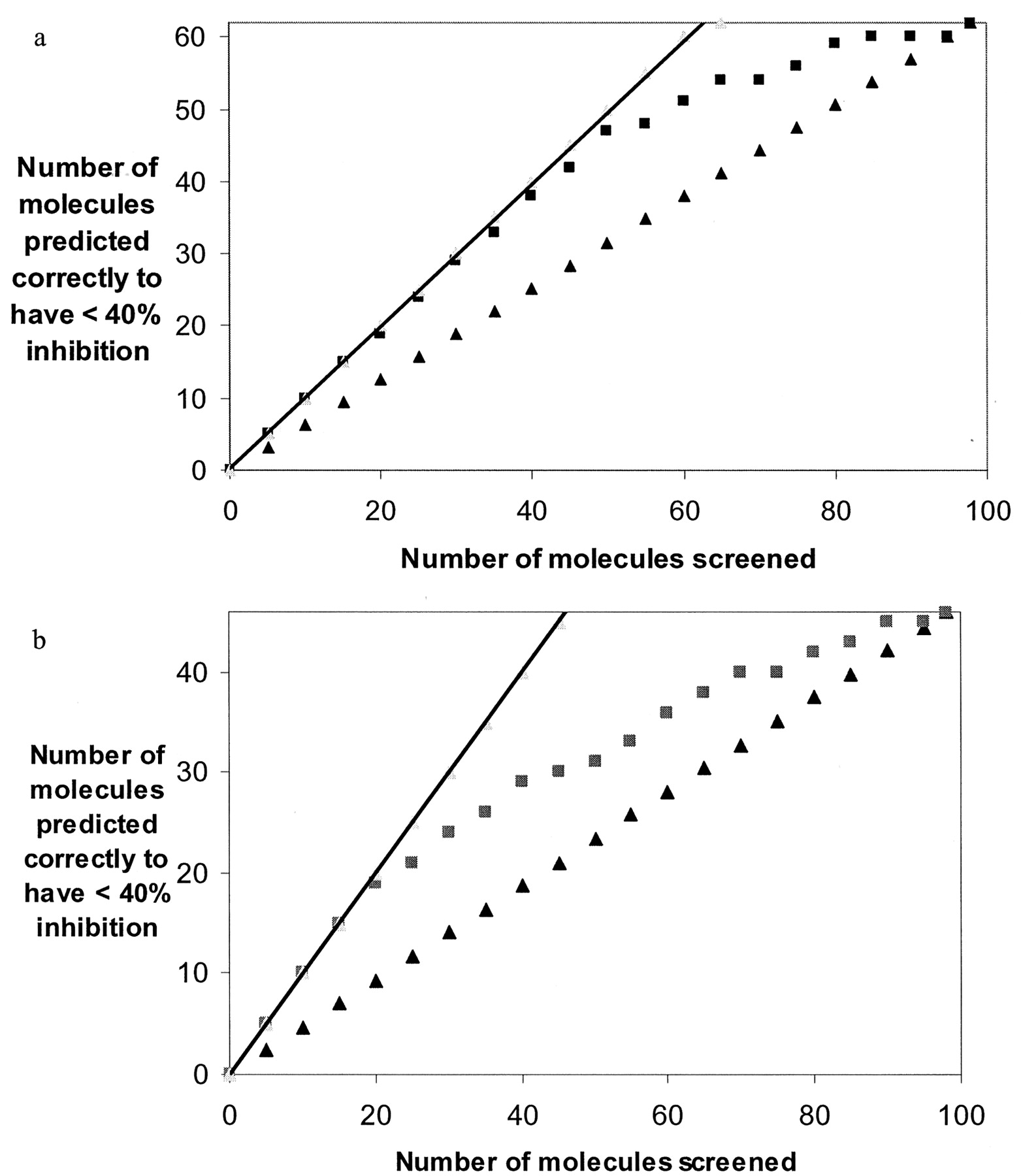
Evaluation of the 20 CYP2D6 Chemtree models using the 98-molecule test set showing the random rate (triangles) and ideal rate (line) of finding those molecules with less than 40% inhibition (62 molecules) (a) and evaluation of the 20 CYP3A4 Chemtree models using the 98-molecule test set showing the random rate (triangles) and ideal rate (line) of finding those molecules with less than 40% inhibition (46 molecules) (b).
In a and b, the rank order of the absolute predicted inhibition values (squares) was used to rank the molecules from low to high percentage of inhibition and to identify the molecules with less than 40% inhibition.
The current models generated are only as good as the data used in terms of both training and testing. The experimental error of 10% for the empirical data must be considered when the predictions are assessed and compared with experimental values, as this suggests that a cutoff for inhibition used as a go/no go decision (for example, less than 40% in this example) should have some degree of flexibility. Typically, results with greater than 20% standard deviation were observed with compounds that were noted as having solubility issues. Further limitations with the predictive powers of our generated models appear to be related to the training data set. These sets were inadvertently heavily skewed toward the low-percent inhibition molecules (Fig. 1, a and b), and it is likely that this is representative of the actual case for available drug-like molecules. This might be different in laboratories where some project areas may be inadvertently synthesizing potent inhibitors for P450s as a consequence of combinatorial chemistry or parallel synthesis. As more empirical data become available, its incorporation into the training set should enhance the future predictive power of our models. Continuous improvements in throughput for in vitro drug-drug interaction studies could be viewed as a means to obviate the need for computational predictions. However, in a number of companies, large virtual libraries of molecules must be evaluated and refined prior to synthesis or purchase, which argues in favor of rapid computational filters for drug-drug interactions and other important properties. Chemtree and algorithm technologies developed to handle nonlinear data clearly will play a valuable role in this regard to aid in filtering and compound selection.
Footnotes
-
↵1 Abbreviations used are: DDI, drug-drug interaction; QSAR, quantitative structure-activity relationship; AMMC, 3-[2-N,N-diethyl-N-methylammonium)-ethyl]-7-methoxy-4-methylcoumarin; AHMC, 3-[2-N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin; BFC, 7-benzyloxy-4-trifluoromethylcoumarin; DMSO, dimethyl sulfoxide; HFC, 7-hydroxy-4-trifluoromethylcoumarin.
- Received March 27, 2003.
- Accepted May 15, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}