


Abstract
DPC 681 (N-[(3-fluorophenyl)methyl]glycyl-N-{3-[((3-aminophenyl) sulfonyl)-2-(aminophenyl)amino]-(1S,2S)-2-hydroxy-1-(phenyl-methyl)propyl}-3-methyl-l-valinamide) is a potent peptide-like human immunodeficiency virus protease inhibitor that was evaluated in phase I clinical trials. In primary cultures of hepatocytes, DPC 681 significantly induced the testosterone 6β-hydroxylase activity of rat CYP3A, but not human CYP3A4. Western blot analysis, however, demonstrated a 3-fold increase in expression of CYP3A4 protein by 20 μM DPC 681 in primary cultures of human hepatocytes. Subsequent studies showed that DPC 681 was a potent inhibitor of human CYP3A4 (IC50 = 0.039 μM) and rat CYP3A (IC50 = 1.62 μM). Moreover, DPC 681 was a mechanism-based inactivator of CYP3A4 with KI and kinact of 0.24 μM and 0.22 min-1, respectively. Thus, DPC 681 is both a potent inhibitor and a strong inducer of CYP3A4. Induction of CYP3A4 by DPC 681 was masked in vitro by autoinactivation, similar to the protease inhibitor ritonavir. In pharmacokinetic studies in healthy human volunteers and rats, DPC 681 was found to highly autoinduce its metabolism. Human volunteers dosed with DPC 681 at 600 mg twice daily for 14 days had a 75% decrease in the mean area under the concentration-time curve and a more than 3-fold increase in apparent clearance as compared with that on day 1. Because the primary route of DPC 681 clearance is via CYP3A metabolism, the increased clearance observed in clinical studies is due to induction of human CYP3A4 expression.
DPC 6811 (N-[(3-fluorophenyl)methyl]glycyl-N-{3-[((3-aminophenyl)sulfonyl)-2-(aminophenyl)amino]-(1S,2S)-2-hydroxy-1-(phenyl-methyl)propyl}-3-methyl-l-valinamide) (Fig. 1) is a potent peptide-like human immunodeficiency virus (HIV) protease inhibitor. Clinically available protease inhibitors include amprenavir, indinavir, nelfinavir, ritonavir, and saquinavir (Molla et al., 1998; Barry et al., 1999). Combinations of a protease inhibitor with other antiretroviral drugs from different classes are very effective treatments for HIV infection. DPC 681 could potentially be combined with nucleoside reverse transcriptase inhibitors, including didanosine, lamivudine, stavudine, zalcitabine, and zidovudine (Barry et al., 1999), non-nucleoside reverse transcriptase inhibitors, including delavirdine, efavirenz, and nevirapine (Barry et al., 1999; Smith et al., 2001), or other HIV protease inhibitors such as ritonavir, for treatment of HIV infection. In addition, HIV-infected patients also receive other drugs for the treatment of opportunistic infections, concurrent symptoms, or the relief of side effects of antiretroviral drugs. Since clinical drug-drug interactions are a serious concern in the treatment of HIV infection, the drug-drug interaction potential of DPC 681 was studied.
Chemical structure of DPC 681.
Human CYP3A4 is an important drug-metabolizing enzyme, expressed in the small intestine and liver. The expression of CYP3A4 can be induced in vivo by a variety of drugs, including some antiretroviral agents such as ritonavir (Luo et al., 2002). Notably, all peptide-like HIV protease inhibitors and NNRTIs are primarily metabolized by CYP3A4 (Molla et al., 1998; Smith et al., 2001; Tran et al., 2001). Consequently, CYP3A4 induction or inhibition could affect oral bio-availability (first-pass effect) and enhance or inhibit hepatic metabolism of drugs themselves and concomitantly administrated drugs. Because of this, DPC 681 was tested for its potential to induce and inhibit CYP3A4.
In the present study, DPC 681 was demonstrated to be a strong inducer of human and rat CYP3A isozymes. However, induction of human CYP3A4 in primary culture of hepatocytes by DPC 681, as demonstrated by Western blot analysis, was masked by its potent CYP3A4 inhibition when testosterone 6β-hydroxylase activity was measured. Similar to ritonavir (Kumar et al., 1996, 1999; Eagling et al., 1997; Hsu et al., 1997; Koudriakova et al., 1998; Luo et al., 2002), DPC 681 appears to show concurrent mechanism-based inactivation and induction of CYP3A4, which makes it more difficult to predict clinical outcomes from in vitro data and understand underlying and overlapping mechanisms in vivo.
Materials and Methods
Materials. Dexamethasone and rifampin (rifampicin) were purchased from Sigma-Aldrich (St. Louis, MO). Ritonavir was obtained from Moravek Bio-chemicals (Brea, CA). Testosterone was purchased from Steraloids (Wilton, NH). Pooled human liver microsomes and Sprague-Dawley rat liver microsomes were purchased from BD Gentest (Woburn, MA). DPC 681 was prepared by Bristol-Myers Squibb Co. (Wilmington, DE).
Primary Culture of Human Hepatocytes. Human liver tissues were obtained as surgical wastes or rejected donor livers from the University of North Carolina at Chapel Hill School of Medicine (donors HL-N072 and HL-N075) or from the Department of Surgery at the University of Kansas Medical Center (Kansas City, KS) (donor H152). All tissues were obtained through qualified medical staff, with donor consent and with the approval of the appropriate hospital ethics committees. Human hepatocytes were isolated by a modification of the two-step collagenase digestion method (Li et al., 1992), with a minor modification (LeCluyse et al., 2000). The isolated human hepatocytes were cultured for CYP3A4 induction studies as described previously (Li et al., 1992; LeCluyse et al., 2000). Primary cultures of human hepatocytes were maintained for 2 days followed by three daily additions of DPC 681 at final concentrations of 2, 10, and 20 μM or rifampin (Pichard et al., 1990; LeCluyse et al., 2000) at a final concentration of 10 μM.
Primary Culture of Rat Hepatocytes. Rat hepatocytes were obtained from 12-week-old male and female Sprague-Dawley rats (250–300 g; Charles River Laboratories, Inc., Wilmington, MA) by a two-step perfusion with Krebs-Henseleit buffer and collagenase (LeCluyse et al., 1996). Isolated hepatocytes were cultured for CYP3A induction studies as described previously (LeCluyse et al., 1996). Primary cultures of rat hepatocytes were maintained for 2 days followed by three daily additions of DPC 681 at final concentrations of 2, 10, and 20 μM or dexamethasone (Ledirac et al., 2000) at a final concentration of 10 μM.
Inhibition of Testosterone 6β-Hydroxylase Activity. Inhibition experiments with ritonavir and DPC 681 on testosterone 6β-hydroxylase activity in human and rat liver microsomes were performed to obtain IC50 values. Reaction mixtures (final volume, 1 ml) contained (final concentrations) 100 mM phosphate buffer, pH 7.4, 0.2 mg/ml liver microsomes, 200 μM testosterone, 1% acetonitrile, 0.5% DMSO, and seven concentrations (0–12.5 μM) of DPC 681 or ritonavir and were preincubated at 37°C for 5 min. The reactions were initiated with addition of NADPH (final concentration, 1.0 mM), incubated at 37°C for 10 min, and stopped with addition of 1.0 ml of ice-cold acetonitrile containing 20 μM 6β-hydroxyprogesterone (internal standard). Mechanism-based inactivation experiments of CYP3A4 by DPC 681 or ritonavir were performed to obtain KI and kinact (Mayhew et al., 2000). The reaction mixtures (final volume, 0.5 ml) contained 100 mM phosphate buffer, pH 7.4, 4 mg/ml liver microsomes, 0.5% DMSO, and six concentrations (0–8 μM) of ritonavir or DPC 681 and were preincubated at 37°C for 5 min. The mechanism-based inactivations were initiated with addition of NADPH (final concentration, 1 mM) and incubated for 0, 3, 7, 13, and 25 min. At these time points, 50 μl of reaction mixture was transferred to the testosterone assay reaction mixture (final volume, 1 ml) which contained 100 mM phosphate buffer, pH 7.4, 200 μM testosterone, 1 mM NADPH, and 1% acetonitrile. The reactions were incubated at 37°C for 10 min and stopped as aforementioned. The formation of 6β-hydroxytestosterone was analyzed by HPLC (Pearce et al., 1996).
Pharmacokinetic Study in Humans. Multiple doses of DPC 681 were orally administered to six healthy male human volunteers between the ages of 18 and 50 years. A single 600 mg dose was given on the first day, 24 h after which DPC 681 was administered every 12 h for 13 days. The last dose was given in the evening of day 14. All morning dose administrations were in the fasted state and at least 1 h before the breakfast meal. All evening dose administrations were at least 2 h after the last meal and at least 1 h before any evening snack. Subjects were confined to the clinical study unit starting on day - 1 and remained confined until 48 h after the administration of the last dose of DPC 681. No other drugs or CYP3A4 inhibitors such as grapefruit juice were given to the subjects before and during the trial. Blood samples were collected from the volunteers using EDTA as the anticoagulant before dosing and 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h after doses 1, 14, 15, 26, and 27.
Pharmacokinetic Study and CYP3A Induction in Rats. Sprague-Dawley rats (250–300 g; Charles River Laboratories, Inc.) were dosed with DPC 681 as a formulation of 1% Tween 80 and 0.16% methanesulfonic acid in water. Rats were given morning doses and afternoon doses (6 h after the morning doses) at 0 and 100 mg/kg via oral gavage for 12 days. There were 12 male and 12 female rats at both levels. Blood samples were taken on days 1 and 12 before and at 1, 3, 6, 7, 8, 10, and 12 h after the morning doses. Whole blood (0.5 ml) collected from the orbital plexus under light ether anesthesia was placed on ice immediately and subsequently centrifuged to obtain plasma. The plasma samples were kept frozen at -70°C until analysis. Plasma samples obtained from three rats per time point per group were pooled for analysis. Liver samples (2 g) were obtained at necropsy on day 15 from four rats of each gender group and were snap-frozen in liquid nitrogen and stored at -70°C until CYP activity determinations were made.
Testosterone Oxidation Assay. Microsomes were prepared from cultured human or rat hepatocytes 24 h after the final drug treatment as described previously (Li et al., 1992; LeCluyse et al., 2000). The concentration of protein was determined using a bicinchoninic acid protein assay (Pierce Chemical, Rockford, IL). Microsomal CYP3A4 activity was determined by measuring the 6β-hydroxylation of testosterone (Pearce et al., 1996). CYP3A4 induction was expressed as fold induction over the vehicle control.
Immunoblot Analysis. Equal amounts of microsomal protein (10 μg) were loaded onto polyacrylamide gels and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Levels of immunoreactive CYP3A4 protein in human hepatocyte microsomes were determined by Western immunoblotting analysis with CYP3A4-specific antibodies at a 1:2,000 dilution (Chemicon International, Temecula, CA) as described previously (LeCluyse et al., 2000; Luo et al., 2002). The relative amounts of CYP3A4 protein were estimated from densitometric analysis of the blot after scanning.
Analyses of DPC 681 in Rat and Human Plasma. A mixture of 0.2 ml of pooled rat plasma samples and 0.3 ml of 2 mM ammonium acetate solution was extracted with 5 ml of methyl t-butyl ether, followed by the addition of 0.2 ml of diluted sulfuric acid (pH ∼1) to the organic layer. HPLC fluorescence (excitation, 254 nm; emission, 377 nm) was performed using a MetaChem Basic C18 HPLC column (3 × 150 mm; ANSYS Technologies, Inc., Lake Forest, CA) heated at 30°C and an isocratic acetonitrile/water mobile phase that was run over 10 min at a flow rate of 0.7 ml/min. DPC IJ368, a structural analog of DPC 681, was used as an internal standard. Retention times of DPC 681 and the internal standard were 6.6 and 7.6 min. This assay was validated as precise, accurate, and specific in the range of 50 to 10,000 nM (Solon et al., 2002). The human plasma samples were analyzed with an LC/MS-MS method that was validated in the range of 5 to 5,000 nM of DPC 681 using 0.2 ml of plasma samples.
Statistics. Noncompartmental pharmacokinetics parameters were calculated using Kinetica (InnaPhase Corp., Philadelphia, PA).
Results
CYP3A4 Induction in Cultured Human Hepatocytes. Microsomal testosterone 6β-hydroxylase activity from cultured human hepatocytes (three donors) was determined to assess CYP3A4 induction after incubation with DPC 681 at final concentrations of 2, 10, and 20 μM for 3 days. Although the prototypical CYP3A4 inducer rifampin (10 μM) significantly induced CYP3A4 activity 3.51-fold, DPC 681 did not markedly induce CYP3A4 activity over the vehicle control 0.1% DMSO (approximately 1.4-fold, n = 3, Fig. 2 and Table 1). However, Western blot analyses of microsomes prepared from those three donors showed that DPC 681 as well as rifampin induced CYP3A4 expression markedly (Fig. 3 and Table 1). The mean increases in expression (n = 3) were 2.2-, 2.8-, and 3.3-fold over vehicle control by DPC 681 at the concentrations of 2, 10, and 20 μM, respectively, whereas rifampin induced the CYP3A4 protein by 4.98-fold at a final concentration of 10 μM (Table 1). The results of CYP3A4 activity and Western blotting were consistent for rifampin, but not for DPC 681 at high concentrations (10 and 20 μM). This suggests that the CYP3A4 activity (testosterone 6β-hydroxylation) of microsomes prepared from cultured human hepatocytes did not truly reflect the CYP3A4 induction by DPC 681.
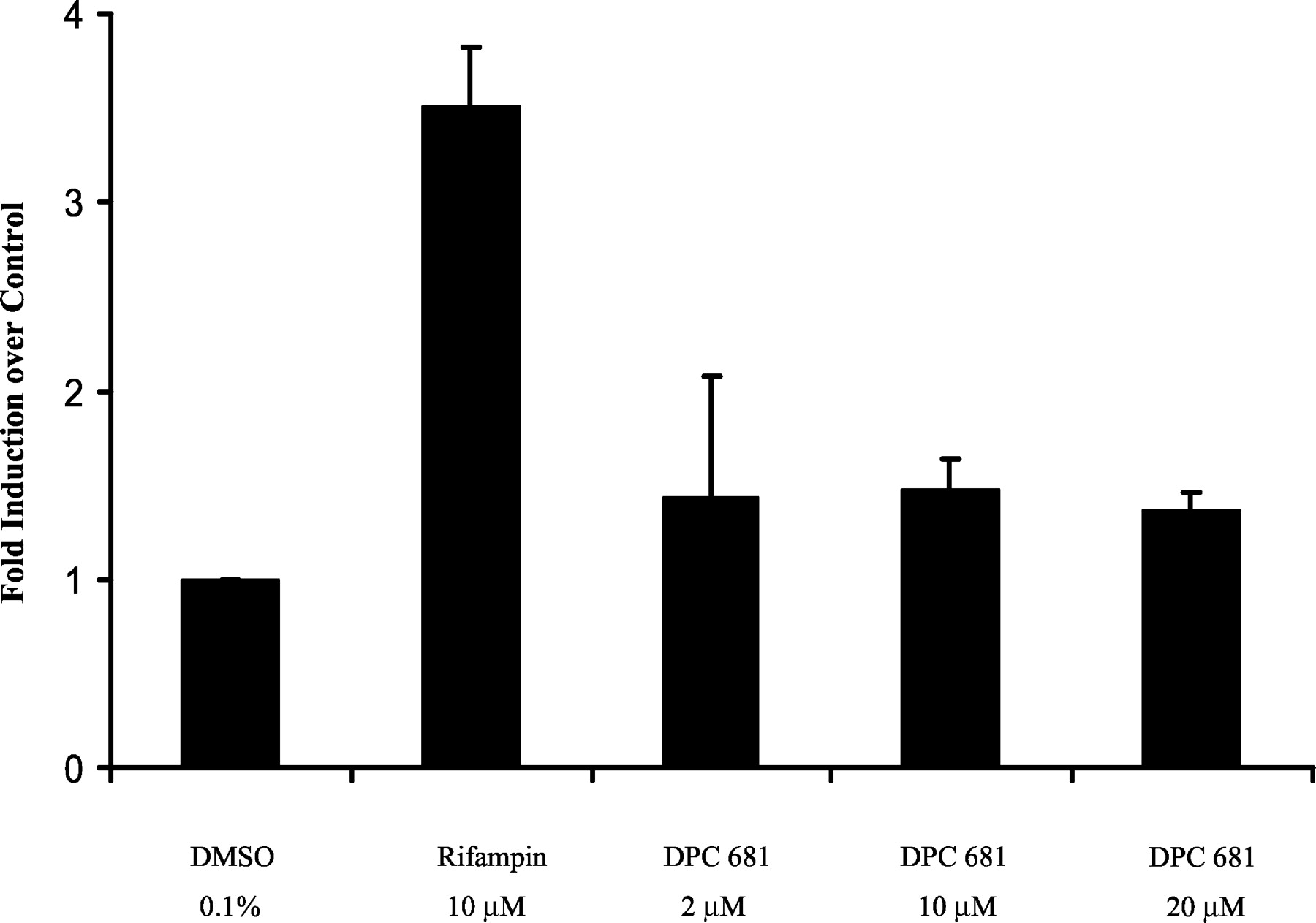
Induction of microsomal CYP3A4 activity by DPC 681 in primary culture of human hepatocytes.
Hepatocyte cultures were treated for 3 days with DPC 681 (2, 10, and 20 μM) or rifampin (10 μM). DMSO at 0.1% (v/v) was used as a vehicle control. Microsomes were prepared 24 h after the last treatment, and the CYP3A4 activity was determined by measuring testosterone 6β-hydroxylase activity (pmol/mg/min). The results are the mean ± standard deviations of three experiments from three donors as described under Experimental Procedures.
CYP3A4 induction by DPC 681 reflected by testosterone 6β-hydroxylase activity and Western blotting

Induction of immunoreactive CYP3A4 levels in cultured human hepatocytes.
Primary human hepatocytes were cultured and treated as described in Fig. 2. Equal amounts of microsomal protein (10 μg) from donor HL-N075 were used for Western blot analyses with CYP3A4-specific antibodies. The blots were scanned by densitometry.
CYP3A Induction in Cultured Rat Hepatocytes. DPC 681 significantly induced rat CYP3A activity (testosterone 6β-hydroxylation) in primary cultures of both male and female rat hepatocytes (Fig. 4). The induction was approximately 3-fold over the vehicle control (n = 3), whereas the prototypical rodent CYP3A inducer dexamethasone showed approximately 5-fold induction of CYP3A activity.
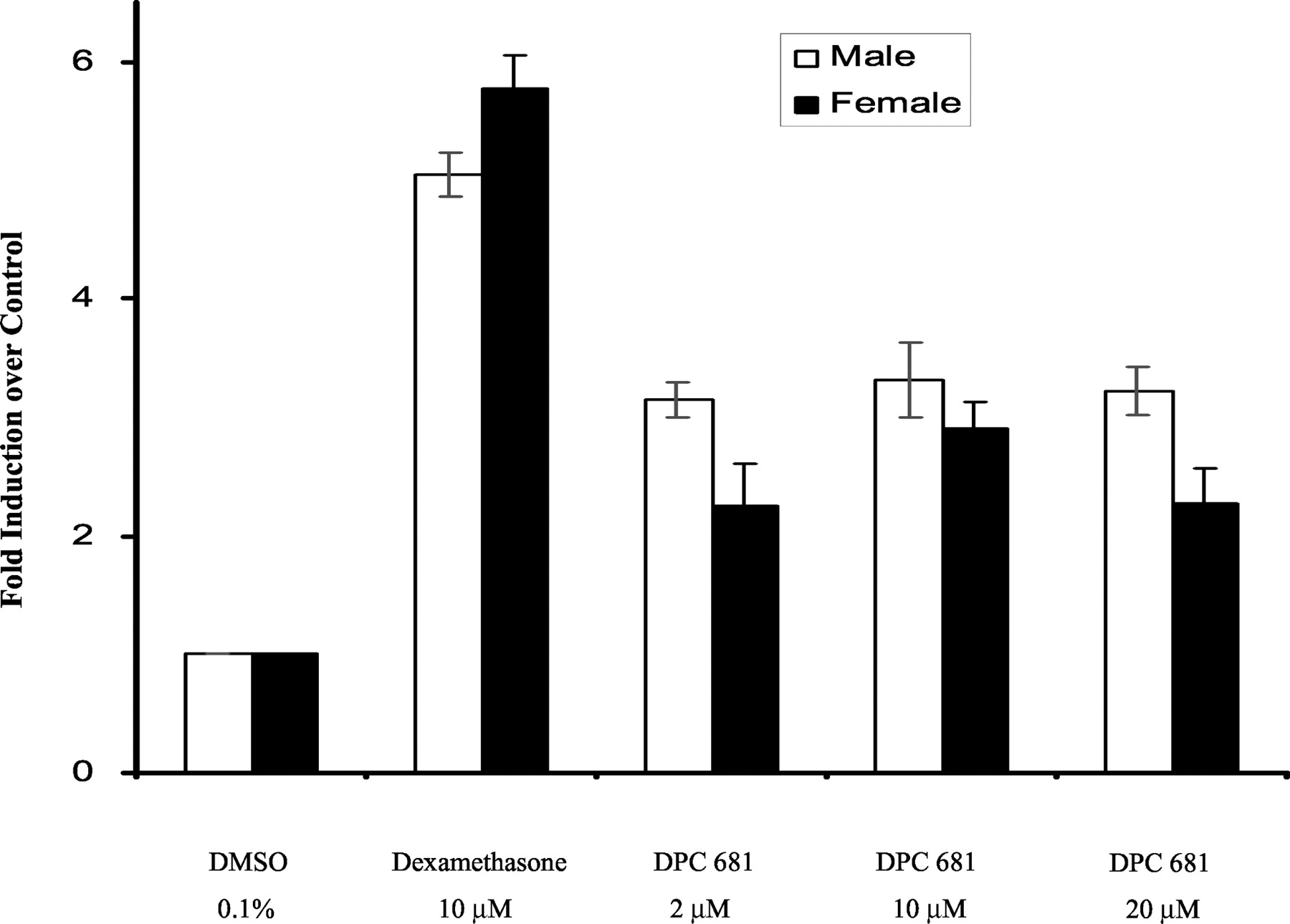
Induction of microsomal CYP3A4 activity by DPC 681 in primary culture of rat hepatocytes.
Male and female hepatocyte cultures were treated for 3 days with DPC 681 (2, 10, and 20 μM) or dexamethasone (10 μM). DMSO at 0.1% (v/v) was used as a vehicle control. Microsomes were prepared 24 h after the last treatment, and the CYP3A activity was determined by measuring testosterone 6β-hydroxylase activity (pmol/mg/min). The results are the mean ± standard deviations of three determinants.
Inhibition of Human CYP3A4 and Rat CYP3A Enzymes. Experiments were further conducted to evaluate the potential inhibition of CYP3A activities by DPC 681. As presented in Table 2, DPC 681 potently inhibited the human CYP3A4, measured as testosterone 6β-hydroxylase activity, with an IC50 of 0.039 μM. DPC 681 also inhibited rat CYP3A enzymes; however, the IC50 (1.62 μM) was approximately 40-fold greater than that observed in human liver microsomes. The results also indicated that DPC 681 was a mechanism-based inactivator of CYP3A4, with KI and kinact being 0.24 μM and 0.22 min-1, respectively. Ritonavir was also examined for its inhibition of human and rat CYP3A enzymes. Ritonavir showed more potent inhibition of both human CYP3A4 and rat CYP3A enzymes than DPC 681.
Inhibition of CYP3A activity by DPC 681 and ritonavir in human and rat liver microsomes as measured by testosterone 6β-hydroxylation
Pharmacokinetics in Rats. As shown in Fig. 5, after oral administration of DPC 681 at 100 mg/kg, twice daily, the AUC0–6h after dose 2 on day 1 in male and female rats was 175 and 220 μM · h, respectively, whereas the AUC0–6h after dose 24 on day 12 was 28 and 37 μM · h, respectively. The AUC of DPC 681 after dose 24 was less than 20% of that after dose 2 in both male and female rats, indicating the autoinduction of rat hepatic and/or intestinal CYP3A expression by DPC 681.
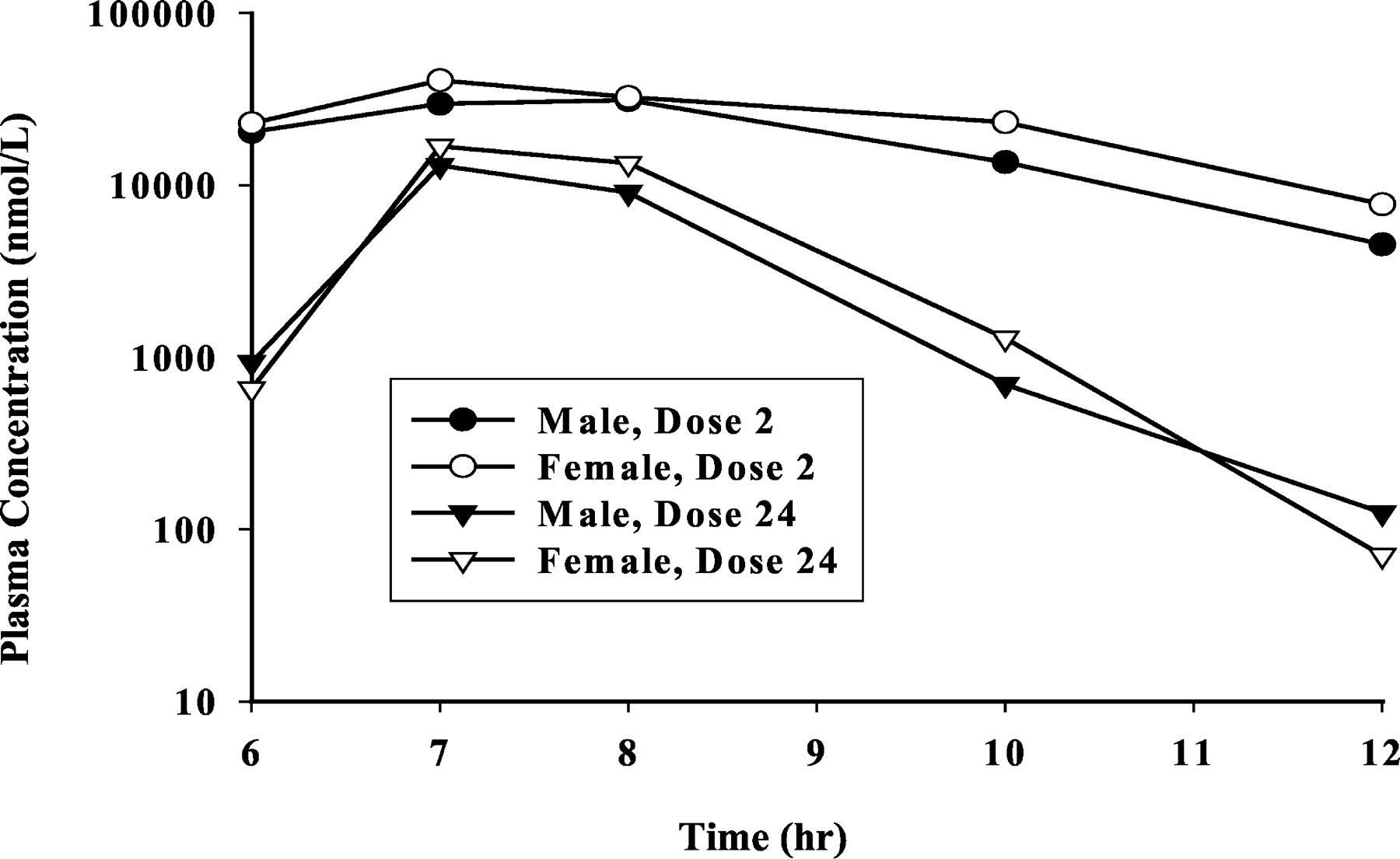
Pharmacokinetics of DPC 681 in rats.
Twelve (12) male and 12 female Sprague-Dawley rats were dosed with DPC 681 (100 mg/kg, twice daily, 6 h apart) via oral gavage for 12 days. The rats were bled before dosing and at 1, 3, 6, 7, 8, 10, and 12 h after the first daily dose on days 1 and 12. Plasma samples were analyzed by HPLC with fluorescence detection. Only concentration-time course curves after doses 2 and 24 are presented.
CYP3A Induction in Rats. Hepatic microsomes were prepared from livers of rats treated with DPC 681 at 0 or 100 mg/kg, twice daily, for 12 days (the same rats used for the pharmacokinetic study) and CYP3A activity (testosterone 6β-hydroxylation) was determined to measure CYP3A induction. As shown in Table 3, DPC 681 induced CYP3A activity 4- and 21-fold in male and female rats, respectively. A much higher fold induction over the vehicle control was seen in the female than in the male, possibly due to a much lower basal CYP3A activity in female rats (Johnson et al., 2000).
Induction of rat hepatic microsomal CYP3A activity by DPC 681
Pharmacokinetics in Healthy Human Volunteers. The pharmacokinetic parameters for DPC 681 orally dosed at 600 mg every 12 h for 14 days in six healthy human volunteers are shown in Table 4, and the DPC 681 concentration-time curves after doses 1, 14, and 26 are represented in Fig. 6. Compared with the parameters obtained after dose 1, after doses 14, 15, 26, and 27, AUC0–12h of DPC 681 was reduced 75 to 83% (P < 0.001), and Cmax was reduced 70 to 88% (P < 0.001), whereas the apparent clearance increased 3.4- to 5.9-fold (P < 0.001), and half-life (t1/2 h) was reduced only marginally. The marked reduction of AUC and Cmax and significant increase of apparent clearance after oral administration of DPC 681 for 7 to 14 days strongly suggested that DPC 681 induced its own metabolism and/or excretion after multiple doses. Because the primary metabolic route of DPC 681 in humans is CYP3A4 (Solon et al., 2002), this autoinduction may represent the combined induction of human hepatic and intestinal CYP3A4 expression by DPC 681.
Pharmacokinetic parameters for DPC 681 dosed at 600 mg every 12 h for 14 days (except day 1 with a single 600 mg dose) in six healthy human volunteers

Pharmacokinetics of DPC 681 in healthy human volunteers.
DPC 681 was orally administrated (600 mg, twice daily, 12 h apart except day 1 with a single 600 mg dose) to six healthy male human volunteers for 14 days. Blood samples were collected after doses 1, 14, and 26, and analyzed with a LC/MS-MS method. The results are the mean ± standard errors of six determinants.
Discussion
The present study demonstrated that DPC 681 is a potent human CYP3A4 inducer. The marked reduction of AUC and Cmax, and significant increase of apparent clearance of DPC 681 in healthy human volunteers after multiple doses of DPC 681 indicates that this compound induces CYP3A4, since its primary metabolic route in vivo is via CYP3A4 metabolism (Solon et al., 2002). The Western blot analysis clearly showed that DPC 681 (20 μM) increased the level of CYP3A4 protein approximately 3.3-fold over the vehicle control in primary culture of human hepatocytes from three donors. However, when the activity of CYP3A4 in the microsomes prepared from the cultured human hepatocytes was measured, little increase in CYP3A4 activity was observed. The discrepancy was probably due to potent inhibition, in particular the mechanism-based inactivation of CYP3A4 by DPC 681. The present study demonstrated that DPC 681 inhibited CYP3A4 and also inactivated CYP3A4, leading to the loss of enzyme activity. Therefore, the activity of CYP3A4 in the microsomes prepared from human hepatocytes treated with DPC 681 did not accurately reflect the CYP3A4 induction potential of DPC 681.
DPC 681 also induced rat CYP3A isozymes, as demonstrated by the increased apparent clearance and liver CYP3A activity after multiple oral administration for 12 days and the increase of the CYP3A activity in cultured rat hepatocytes in vitro. It has been demonstrated that the expression of rat CYP3A2, 3A9, and 3A18 is age- and sex-dependent. In the livers of adult male rats, CYP3A2 and CYP3A18 are predominantly expressed, whereas only CYP3A9 is predominantly expressed in livers of adult female rats (Mahnke et al., 1997; Robertson et al., 1998; Johnson et al., 2000). The inducible isoforms CYP3A1 and CYP3A23 (Huss and Kasper, 1998; Hoen et al., 2000), however, are expressed in both male and female rats. Interestingly, DPC 681 showed species differences in CYP3A induction. DPC 681 did not induce canine CYP3A activity, as demonstrated by no change in the exposure of DPC 681 and no increase in the microsomal CYP3A activity in the livers obtained from beagle dogs treated with DPC 681 at 100 mg/kg, twice daily for 13 days (data not shown). This is probably due to the fact that the ligand binding domain of canine pregnane X receptor (PXR) is 87 and 75% identical to that of human and rat PXRs, respectively (Moore et al., 2002). PXR, also called steroid and xenobiotic receptor or pregnane X receptor, was found to mediate the drug-induced expression of CYP3A4 (Bertilsson et al., 1998; Lehmann et al., 1998; Xie et al., 2000).
In primary cultures of human hepatocytes, CYP3A4 expression could be determined by analyzing CYP3A4 activity (such as testosterone 6β-hydroxylation or 1′-OH midazolam hydroxylation), or levels of CYP3A4 protein and mRNA (Czerwinski et al., 2002; Luo et al., 2002). Care should be taken when CYP3A4 activity is determined to reflect CYP3A4 induction of a test compound that is a potent mechanism-based CYP3A4 inhibitor, because its inactivation may mask the induction effect. Ritonavir and troleandomycin, as demonstrated in a previous study (Luo et al., 2002), and DPC 681, as shown in the present study, are examples of this type of compound.
DPC 681 induced both rat and human CYP3A; however, when the activity of CYP3A was measured, approximately 3-fold induction was seen in cultured rat hepatocytes, but only 1.4-fold induction was observed in cultured human hepatocytes. This may be due to different effects of DPC 681 on human and rat CYP3A activity. First, the IC50 of DPC 681 for CYP3A was 1.62 μM when assaying the testosterone 6β-hydroxylation with rat liver microsomes, whereas it was 0.039 μM when using human liver microsomes. Second, DPC 681 was demonstrated to be a mechanism-based inactivator of human CYP3A4.
In a previous study, CYP3A4 activity from cultured human hepatocytes treated with ritonavir was lower than the control activities and became undetectable at 20 μM, whereas the respective CYP3A4 protein level assessed by Western blot analysis was 1.8-fold of the control (Luo et al., 2002). This discrepancy between activity and protein expression was found to be due to the potent CYP3A4 inhibition by ritonavir, in particular, the mechanism inactivation of the CYP3A4 activity (Kumar et al., 1996, 1999; Eagling et al., 1997; Koudriakova et al., 1998). In the present study, the CYP3A4 activity from cultured human hepatocytes treated with DPC 681 at 20 μM was 1.4-fold of the control activity, whereas the respective CYP3A4 protein level was approximately 3-fold of the control level. Comparison of ritonavir and DPC 681 in CYP3A4 inhibition may provide some explanation to the above observation. Under the same experimental conditions, the IC50 of DPC 681 on CYP3A4 activity was 0.039 μM, approximately 4-fold higher than with ritonavir (0.010 μM). Furthermore, ritonavir was shown to be more potent than DPC 681 in mechanism-based inactivation of CYP3A4. The KI of DPC 681 is 0.24 μM, which is 6-fold higher than that of ritonavir (0.038 μM).
Induction of CYP3As appears to be a major factor in the enhanced metabolism and/or excretion of DPC 681 in humans and rats. However, induction of P-glycoprotein (PGP) also may partially contribute to the reduced absorption and the enhanced excretion of DPC 681. PGP is highly expressed in intestine, brain, kidney, testes, and canalicular membrane. As an efflux pump, it could reduce intestinal absorption, diminish distribution to certain tissues (such as brain and testes), and increase intestinal, biliary, and renal excretion of a drug molecule (Troutman et al., 2001). PXR is believed to be a key transcriptional factor of MDR1, because a PXR response element (direct repeat separated by 4-base pair motif) was identified at about -8 kilobase pairs upstream from the MDR1 encoding region (Geick et al., 2001), and PXR activators including rifampin, ritonavir, and St. John's wort were shown to increase PGP expression (Greiner et al., 1999; Hennessy et al., 2001; Perloff et al., 2001). In a previous study, it has been demonstrated that DPC 681 is a substrate as well as a competitive inhibitor of PGP (Solon et al., 2002). In the present study, DPC 681 was shown to potently induce CYP3A4, very possibly through PXR activation. Thus, DPC 681 has the potential to induce PGP expression in humans, and consequently reduces the absorption while enhancing the excretion of DPC 681.
Inhibition and induction of the CYP3A4 activity have been shown to reduce and enhance the metabolism of CYP3A4 substrates, respectively, and consequently alter the pharmacokinetics of many drugs. In fact, inhibition and induction of CYP3A4 have raised many drug-drug interaction issues in the area of drug discovery and development. Similarly, inhibition and induction of PGP could have an opposite effect on the fate of coadministrated substrates. Notably, DPC 681 was shown to be a substrate, an inhibitor, and an inducer of human CYP3A4. In addition, it is a substrate, an inhibitor, and possibly an inducer of human PGP. Because of these effects, it may become difficult to predict a drug-drug interaction when DPC 681 is administrated with other CYP3A4 or PGP substrates. One might predict that after a single dose or in the early stage of multiple dosing, DPC 681 may show an inhibitory effect on CYP3A4 substrates; however, after prolonged treatment, CYP3A4 induction will occur and could even become the predominant effect. In addition, interindividual differences in the basal CYP3A4 levels will contribute to the variability of inhibition or induction response. In other words, DPC 681 may show inhibition and induction of CYP3A4 at the same time or transition from predominant inhibition to predominant induction.
DPC 681 and ritonavir resemble one another in many aspects. Both of them are potent peptide-like HIV protease inhibitors (Molla et al., 1998; Solon et al., 2002); CYP3A4 substrates, inducers, inhibitors, and mechanism-based inactivators (Kumar et al., 1996, 1999; Eagling et al., 1997; Hsu et al., 1997; Koudriakova et al., 1998; Luo et al., 2002; Solon et al., 2002); and PGP substrates, inhibitors, and possibly inducers (Perloff et al., 2001; Solon et al., 2002). However, compared with ritonavir, DPC 681 demonstrated much higher CYP3A4 induction in humans. In the present study, after administration of DPC 681 to humans (600 mg, twice daily) for more than 7 days, its AUC was decreased 75% compared with that on day 1. In contrast, after administration of ritonavir to humans (500 mg, twice daily), its AUC was not significantly decreased, although lower predose plasma concentrations were observed (Hsu et al., 1997). In addition, ritonavir shows more potent CYP3A4 inhibition in vivo as well as in vitro.
In summary, DPC 681 was found to concurrently induce and inhibit human and rat CYP3A isozymes. Induction of CYP3A4 and possibly PGP led to the enhanced metabolism and excretion of DPC 681 in humans. The CYP3A4 induction by DPC 681 was masked by mechanism-based inactivation in primary culture of human hepatocytes when CYP3A4 activity was determined to assess the induction response. Furthermore, concurrent induction and mechanism-based inactivation of CYP3A4 could complicate predictions of drug-drug interactions in vivo by DPC 681 and concomitant drugs.
Acknowledgments
We thank Dr. Scott Grossman for his review and comments and XenoTech LLC (Kansas City, Kansas) for CYP3A4 induction (donor H152).
Footnotes
-
↵1 Abbreviations used are: DPC 681, N-[(3-fluorophenyl)methyl]glycyl-N-{3-[((3-aminophenyl) sulfonyl)-2-(aminophenyl)amino]-(1S,2S)-2-hydroxy-1-(phenyl-methyl)propyl}-3-methyl-l-valinamide; AUC, area under the plasma concentration-time curve; CYP, cytochrome P450; DMSO, dimethylsulfoxide; HIV, human immunodeficiency virus; HPLC, high-performance liquid chromatography; LC/ MS-MS, liquid chromatography/tandem mass spectrometry; PGP, P-glycoprotein; PXR, pregnane X receptor.
- Received March 14, 2003.
- Accepted June 10, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}