


Abstract
The measurement of the effect of new chemical entities on human UDP-glucuronosyltransferase (UGT) marker activities using in vitro experimentation represents an important experimental approach in drug development to guide clinical drug-interaction study designs or support claims that no in vivo interaction will occur. Selective high-performance liquid chromatography-tandem mass spectrometry functional assays of authentic glucuronides for five major hepatic UGT probe substrates were developed: β-estradiol-3-glucuronide (UGT1A1), trifluoperazine-N-glucuronide (UGT1A4), 5-hydroxytryptophol-O-glucuronide (UGT1A6), propofol-O-glucuronide (UGT1A9), and zidovudine-5′-glucuronide (UGT2B7). High analytical sensitivity permitted characterization of enzyme kinetic parameters at low human liver microsomal and recombinant UGT protein concentration (0.025 mg/ml), which led to a new recommended optimal universal alamethicin activation concentration of 10 μg/ml for microsomes. Alamethicin was not required for recombinant UGT incubations. Apparent enzyme kinetic parameters, particularly for UGT1A1 and UGT1A4, were affected by nonspecific binding. Unbound intrinsic clearance for UGT1A9 and UGT2B7 increased significantly after addition of 2% bovine serum albumin, with minimal changes for UGT1A1, UGT1A4, and UGT1A6. Eleven potential UGT and cytochrome P450 inhibitors were evaluated as UGT inhibitors, resulting in observation of nonselective UGT inhibition by chrysin, mefenamic acid, silibinin, tangeretin, ketoconazole, itraconazole, ritonavir, and verapamil. The pan-cytochrome P450 inhibitor, 1-aminobenzotriazole, minimally inhibited UGT activities and may be useful in reaction phenotyping of mixed UGT and cytochrome P450 substrates. These methods should prove useful in the routine assessments of the potential for new drug candidates to elicit pharmacokinetic drug interactions via inhibition of human UGT activities and the identification of UGT enzyme-selective chemical inhibitors.
Introduction
Drug-drug interactions (DDIs) due to the inhibition of UDP-glucuronosyltransferases (UGTs), both as perpetrator and victim, should be considered when developing new chemical entities and are important in drug discovery research or evaluation of patient safety, as noted by regulatory agencies (Bjornsson et al., 2003; Zhang et al., 2010). In general, the risk of significant clinical DDIs due to the inhibition of UGTs during drug coadministration is low compared with inhibition of cytochrome P450 (P450) activities (Williams et al., 2004; Kiang et al., 2005). Likewise, few clinically relevant examples that require dose adjustment for a poor UGT-metabolizer genotype exist, except for clinical reports on UGT1A1 substrates (Toffoli et al., 2006; Williams et al., 2008; Court, 2010). Nevertheless, a thorough evaluation throughout the discovery and development of new drugs is required to inform particular groups of the potential for a significant DDI liability. Accordingly, the development of reliable and robust assay conditions and analytical methods for measuring glucuronidation in vitro, which could be used to obtain relative activity factors (Venkatakrishnan et al., 2000) and to develop appropriate tools that are useful in identifying UGT enzyme-selective chemical inhibitors, are required to advance UGT reaction-phenotyping techniques (Miners et al., 2010a; Parkinson et al., 2010).
UGT1A1, -1A3, -1A4, -1A6, -1A9, -2B7, and -2B15 play a significant role in hepatic drug and xenobiotic metabolism (Miners et al., 2010a) of the 19 human UGTs currently identified (Mackenzie et al., 2005; Miners et al., 2010b). The triad of UGT reaction-phenotyping techniques are not equally advanced (Court, 2004). In particular, a limited number of UGT-selective probe substrates are available to correlate in vitro-glucuronidation activities of the major UGTs expressed in human liver with those of drugs or new chemical entities in human liver microsomes (HLM) or hepatocytes, or to evaluate UGT contribution in DDIs through either enzyme inhibition or induction (Court, 2005; Miners et al., 2010a). During the course of these studies, we developed selective liquid chromatography equipped with tandem mass spectrometry (LC-MS/MS) functional assays of authentic glucuronides for five major hepatic UGT probe substrates (Fig. 1). Selective UGT1A1 substrates include bilirubin and etoposide, but, due to challenging substrate assays, β-estradiol (ES) is mostly used to characterize UGT1A1 kinetics, with the understanding that other hepatic UGTs (e.g., UGT1A3) may contribute to β-estradiol-3-glucuronide (ES3-G) formation (Lépine et al., 2004; Itäaho et al., 2008). Trifluoperazine (TFP) is selective for measurement of UGT1A4 activity (Uchaipichat et al., 2006a), and other potentially selective substrates have been characterized (e.g., 1-hydroxymidazolam) (Miners et al., 2010a). In addition to serotonin (5-hydroxytryptamine), the metabolite 5-hydroxytryptophol (5HTOL) has been identified with LC/UV quantification as a marker substrate for UGT1A6 (Krishnaswamy et al., 2004). Literature reports have shown convincing evidence of propofol (PRO) selectivity for UGT1A9 activity (Soars et al., 2004; Court, 2005), and 3′-azido-3′-deoxythymidine or zidovudine (AZT) is an established UGT2B7 probe substrate (Court et al., 2003).

Major reactions catalyzed by human UDP-glucuronosyltransferase enzymes. Internal standards for LC-MS/MS analytical methods shown for each reaction.
Several experimental variables affect in vitro UGT enzyme activity and ultimately intrinsic clearance (CLint) estimates, including buffer type, pH, ionic strength, latency, organic solvent, glucuronide stability, atypical kinetics, and the albumin effect (Easterbrook et al., 2001; Fisher et al., 2001; Boase and Miners, 2002; Soars et al., 2003; Uchaipichat et al., 2004; Engtrakul et al., 2005; Rowland et al., 2007, 2008). The latter results from long-chain unsaturated fatty acids being released from membranes during the course of an incubation and acts as a potent inhibitor of UGT1A9, UGT2B7, and microsomal glucuronidation activity, which results in overestimation of the Km value (Tsoutsikos et al., 2004; Rowland et al., 2007, 2008). Inhibitory fatty acids are sequestered by addition of bovine serum albumin (BSA), and altered incubation conditions (in the presence and absence of BSA) should be evaluated to select appropriate substrate concentrations (≤Km) when used in an inhibitor screening assay. The objectives of this study are to develop improved analytical methods (Donato et al., 2010) for measuring the in vitro activities of five major human UGT enzymes that use authentic analytical glucuronide standards (Fig. 1) analogous to those used for P450 (Walsky and Obach, 2004). We also evaluated these methods in high-throughput UGT inhibitor screening of several potential inhibitors of these enzymes. Various attributes of incubation conditions frequently used to measure UGT activities (e.g., albumin, buffer, saccharolactone, and alamethicin) were explored to define optimal conditions for each enzyme at low protein concentration. In the course of these studies, the more precise identification of the glucuronide metabolite of 5-hydroxytryptophol was made to facilitate the development of the assay for UGT1A6. The findings are presented here, and these assay methods should be useful to investigators who are engaged in research on these enzymes and the xenobiotics they metabolize.
Materials and Methods
Materials.
Substrates, metabolite standards, internal standards (IS), and other materials were from the following sources: AZT-5′-glucuronide (AZT-G), [13C6]AZT-5′-glucuronide, 5-hydroxytryptophol, propofol-O-glucuronide (PRO-G), trifluoperazine-N-glucuronide, and [D3]trifluoperazine-N-glucuronide (Cerilliant, Austin, TX); magnesium chloride, potassium phosphate, Tris (base), and Tris-HCl (Mallinckrodt Baker, Inc., Phillipsburg, NJ); ammonium formate and formic acid, (Fluka, Buchs, Switzerland); alamethicin, AZT, bovine serum albumin (crude BSA), diclofenac, β-estradiol, β-estradiol-3-glucuronide, β-estradiol-17-glucuronide, propofol, d-saccharic acid 1,4-lactone (saccharolactone), and uridine-diphosphate-glucuronic acid trisodium salt (UDPGA) (Sigma-Aldrich, St. Louis, MO); and 1-naphthyl-glucuronide (Sequoia Research Products, Ltd., Pangbourne, UK). Inhibitors were obtained from Sigma-Aldrich or Sequoia Research Products. Pure (100%) stable-labeled dimethyl sulfoxide (DMSO)-d6 and methanol-d4 were obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). 5-Hydroxytryptophol-O-glucuronide (5HTOL-G) was obtained by biosynthesis as detailed under 5-Hydroxytryptophol-O-glucuronide Isolation, Quantitation, and Structural Elucidation. Other reagents and solvents used were from standard suppliers and were of reagent or high-performance liquid chromatography (HPLC) grade, with all purities as defined by the manufacturer.
Human liver microsomes were prepared from a mixed gender pool of 50 donors (provided by BD Biosciences, Woburn, MA). Recombinant UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7 Supersomes were heterologously expressed from human cDNA in a baculovirus expression system; protein concentrations and initial activity assessments were provided by BD Biosciences.
General UGT Assay Incubation Conditions.
Specific aspects of the incubation condition for each assay are defined in Table 1. In general, for the final optimized method, a premix containing HLM or expressed UGT enzyme (0.025 mg/ml) were mixed with 100 mM Tris-HCl buffer (pH 7.5 at 37°C), MgCl2 (5 mM), substrate, and alamethicin (10 μg/ml), with or without 2% BSA. The premix was placed on ice for 15 min to allow alamethicin pore formation, and then aliquots of this mixture (0.09 ml) were delivered to each well of a polypropylene 96-well polymerase chain reaction plate (maintained at 37°C) that contained the inhibitor or control solvent (DMSO), as applicable. Final solvent concentrations were 1% (v/v) or less. Incubations were commenced with the addition of UDPGA (5 mM) to a final incubation volume of 0.1 ml and incubated at 37°C for the period defined in Table 1. Incubations were typically terminated by addition of acidified organic solvent (acetonitrile) that contained the internal standard. The terminated incubation mixtures were centrifuged and either directly injected, evaporated under nitrogen and reconstituted, or filtered before transferring into a receiver 96-well, microtiter plate for LC-MS/MS analysis, as described for each assay below.
Incubation conditions and analytical parameters for human UDP-glucuronosyltransferase assays
Optimization of UGT Assay Incubation Conditions.
Linearity of product formation with respect to time and protein concentration were conducted with HLM and recombinant UGT (rUGT) for each assay. Multiple time-course experiments were conducted to evaluate the effects of the following: 100 mM Tris-HCl versus 100 mM phosphate buffers (pH 7.5), MgCl2 concentration (0, 1, 5, and 10 mM), and use of 5 mM saccharolactone. Incubations (1.0 ml) were conducted as described under General UGT Assay Incubation Conditions at four HLM or rUGT protein concentrations (0.01, 0.025, 0.05, 0.1 mg/ml) for each specific UGT assay at substrate concentrations approximating S50 or Km. Aliquots (0.1 ml, n = 2) were typically collected over a 90-min time course, terminated, and analyzed for metabolite formation as described for each assay below.
The optimal alamethicin concentration in incubation (Table 2) was determined using pooled HLM and rUGT for UGT1A1, UGT1A4, and UGT2B7 since initial incubations for UGT2B7 performed with published alamethicin concentrations (50 μg/mg protein) shown optimal for pore-formation (Fisher et al., 2000) were not linear for AZT-G formation with respect to protein. Alamethicin, which was dissolved in MeOH/water (50:50) as a 100× stock, was evaluated with four pooled HLM or rUGT protein concentrations (0.01, 0.05, 0.2, 0.5 mg/ml), and each was assessed without and with alamethicin (1–25 μg/ml, n = 3) pretreatment at the appropriate substrate S50 or Km.
Optimal alamethicin concentration for activation of β-estradiol (UGT1A1) and trifluoperazine (UGT1A4) glucuronidation in human liver microsomes
Determination of Nonspecific Protein Binding.
The fraction of unbound substrate in incubation matrices (fu,inc) was determined by equilibrium dialysis using the rapid equilibrium dialysis method (Waters et al., 2008). Substrates were spiked into the human liver microsomal sample (0.025 mg/ml, with and without 2% BSA; 2.0-ml incubation volume) at a concentration close to the Km or S50, and then dialyzed against incubation mixture on a shaker (450 rpm) within a humidified CO2 (5%) incubator at 37°C in the absence of protein (HLM or BSA) for 4 h. Aliquots (0.22 ml) were removed to assess recovery, and, at the end of the incubation, protein was precipitated with acetonitrile (0.18 ml containing 5% DMSO and IS) and analyzed by LC-MS/MS as described for each assay below.
UGT Enzyme Kinetic and Inhibition Experiments.
Substrate saturation experiments were conducted to generate rate data for determining the appropriate enzyme kinetic fit (see Data Analysis), and the apparent substrate Km and Vmax values were calculated for each assay in pooled HLM and rUGT enzymes (Tables 3 and 4). The experiments were performed using at least eight substrate concentrations that spanned the anticipated Km (n = 3) with and without addition of 2% BSA (w/v). Incubations were typically performed in a polypropylene 96-well polymerase chain reaction plate, which was incubated using a thermostatically controlled heater block or a shaking water bath at 37°C, as described above. In brief, aliquots (0.09 ml) of pooled HLM or rUGT enzyme premix were prewarmed at 37°C for 5 min before initiation with 0.01 ml of UDPGA (5 mM). The samples were incubated for the times indicated in Table 1, terminated by the addition of an internal standard as described for each assay, and analyzed to quantify the specific glucuronide products by LC-MS/MS, as detailed for each assay below.
Kinetic parameters for human UDP-glucuronosyltransferase activities in human liver microsomes and recombinant UGT enzymes
Unbound kinetic parameters for human UDP-glucuronosyltransferase activities in human liver microsomes
β-Estradiol-3-Glucuronide (UGT1A1) Assay.
Unless otherwise indicated, ES (3–1000 μM final) was incubated as described under General UGT Assay Incubation Conditions. Stock solutions of ES (100 mM), ES3-G (1 mM), and α-naphthylglucuronide (0.5 mM), as internal standard, were prepared in DMSO. Reactions (0.1 ml) were terminated at the indicated times (Table 1) by addition of 0.05 ml of 47:50:3 acetonitrile/water/formic acid containing α-naphthylglucuronide (5 μM final), then centrifuged at 500g for 10 min, and the supernatant was transferred to 96-well plates for LC-MS/MS analysis. ES3-G standard curve samples (0.02–2.5 μM final concentration) were serially diluted (1:100) in incubation matrix and treated identically to samples. The LC-MS/MS method used a binary gradient using water/0.1% formic acid (mobile phase A) and acetonitrile/0.1% formic acid (mobile phase B). A mobile phase composition of 29% B was held for 2.0 min, then ramped to 90% B over 2.1 min, returned to 29% B over 0.1 min, and held. ES and ES3-G were analytically separated on a Phenomenex Gemini 5μ C18 2.0 × 50 mm (Phenomenex, Torrance, CA) column. Under these HPLC conditions ES3-G, β-estradiol-17-glucuronide, and the IS (α-naphthylglucuronide) had elution times of 1.3, 1.9, and 1.1 min, respectively.
Trifluoperazine N-Glucuronide (UGT1A4) Assay.
Unless otherwise indicated, TFP (1–550 μM final concentration) was incubated as described under General UGT Assay Incubation Conditions. Stock solutions of TFP (55 mM) were prepared in DMSO. TFP-G (0.858 mM), and [D3]TFP-G (0.03 mM), as internal standard, were prepared in water/acetonitrile (50:50). Reactions (0.1 ml) were terminated at the indicated time (Table 1) by addition of 0.01 ml of 17:80:3 acetonitrile/water/formic acid that contained [D3]trifluoperazine-N-glucuronide ([D3]TFP-G, 3 μM final) as IS and centrifuged at 500g for 10 min, and the supernatant was transferred to 96-well plates for LC-MS/MS analysis as described below. TFP-G standard curve samples (0.017–8.6 μM final concentration) were serially diluted (1:100) in incubation mixture and treated identically to samples. The LC-MS/MS method used a binary gradient using water/0.1% formic acid (mobile phase A) and acetonitrile/0.1% formic acid (mobile phase B). A mobile phase composition of 10% B was held for 0.1 min, then ramped to 80% B over 1.9 min (2.0 min total), returned to 10% B over 0.1 min, and held (3.0 min total). Analytes were separated on a Phenomenex Synergi max-RP 4μ 2 × 30 mm column. Under these HPLC conditions, TFP-G and the IS ([D3]TFP-G) had elution times of 1.2 and 1.1 min, respectively.
5-Hydroxytryptophol-O-glucuronide (UGT1A6) Assay.
Unless otherwise indicated, 5HTOL (20–10,000 μM final) was incubated as described under General UGT Assay Incubation Conditions. Stock solutions of 5HTOL, 5HTOL-G (6.6 mM), and diclofenac (0.32 μM) were prepared in water, DMSO-d6, and acetonitrile, respectively. 5HTOL-G concentrations were determined by quantitative NMR using the artificial signal insertion for calculation of concentration observed (aSICCO) method described by Walker et al. (2011). Reactions (0.1 ml) were terminated at the indicated times (Table 1) by the addition of 0.3 ml of acetonitrile, which contained diclofenac (0.24 μM) as an internal standard, and centrifuged at 500g for 10 min. The supernatant (0.3 ml) was transferred to 96-well plates, evaporated to dryness under nitrogen at 40°C, and reconstituted in 5:95 acetonitrile/water (0.1 ml) containing 0.1% formic acid for LC-MS/MS analysis as described below. 5HTOL-G standard curve samples (0.01–20 μM) were serially diluted (1:10) in incubation matrix and treated identically to samples. The LC-MS/MS method used a binary gradient using 95:5 water/acetonitrile containing 0.1% formic acid (mobile phase A) and 95:5 acetonitrile/water containing 0.1% formic acid (mobile phase B). A mobile phase composition of 20% B was ramped to 95% B over 1.6 min and maintained for 0.2 min (1.8 min total), returned to 10% B over 0.1 min, and held (2.5 min total). Analytes were separated on a Waters XTerra MS 3.5μ C18 2.1 × 100 mm (Waters, Milford, MA) column. Under these conditions, 5HTOL-G and the IS (diclofenac) had elution times of 1.4 and 1.1 min, respectively.
Propofol-O-glucuronide (UGT1A9) Assay.
Unless otherwise indicated, PRO (1–1000 μM final) was incubated as described under General UGT Assay Incubation Conditions. Stock solutions of PRO (100 mM) and PRO-G (1.1 mM) were prepared in methanol/water (50:50) and diclofenac (0.629 μM), as internal standard, in acetonitrile. Reactions (0.2 ml) were terminated at the indicated times (Table 1) by the addition of 0.4 ml of acetonitrile containing 0.63 μM diclofenac (0.42 μM final concentration) as IS, centrifuged at 500g for 10 min, and the supernatant was transferred to 96-well plates for LC-MS/MS analysis as described below. PRO-G standard curve samples (0.14–28 μM final) were serially diluted (1:40) in incubation matrix and treated identically to samples. The LC-MS/MS method used a binary gradient using 95:5 water/acetonitrile containing 0.01% formic acid (mobile phase A) and acetonitrile/0.01% formic acid (mobile phase B). A mobile phase composition of 90% B was held for 1.2 min, then ramped to 97% B over 1.2 min (4.2 min total), returned to 90% B over 0.1 min, and held (5.0 min total). Analytes were separated with a Waters SunFire 3.5μ C18 2.1 × 30 mm column. Under these HPLC conditions, PRO-G and the IS (diclofenac) had elution times of 1.3 and 1.1 min, respectively. PRO quantitation for nonspecific protein binding was performed using the following: an AB Sciex (AB Sciex LLC, Foster City, CA) API5500 QTrap mass spectrometer equipped with an electrospray source (electrospray ionization) in negative detection mode; two Shimadzu LC-20AD pumps, a CBM20A controller, and DGU-20A solvent degasser (Shimadzu, Columbia, MD); a LEAP CTC HTS PAL autosampler (CTC Analytics, Carrboro, NC); and a Valco 2-position switching valve (Valco, Houston, TX). Separation was on a Kinetex 2.6μ C18 50 × 2 mm column and the LC-MS/MS method used a binary gradient using water/0.01% formic acid (mobile phase A) and acetonitrile (mobile phase B). A mobile phase composition of 5% B was held for 0.3 min, then ramped to 95% B over 2 min, held at 95% B for 0.3 min, returned to 5% B over 0.1 min, and held (3.0 min total).
AZT-5′-glucuronide (UGT2B7) Assay.
Unless otherwise indicated, AZT (180–4500 μM final concentration) was incubated as described under General UGT Assay Incubation Conditions. Stock solutions of AZT (1100 mM), AZT-G (0.5 mM), and [13C6]AZT-G (0.5 mM), as internal standard, were prepared in acetonitrile/water (50:50). Reactions (0.1 ml) were terminated at the indicated times (Table 1) by the addition of 0.01 ml of 5:92:3 acetonitrile/water/formic acid containing 10 μM [13C6]AZT-G (1 μM final concentration) as IS, filtered on a Millipore (Millipore Corporation, Billerica, MA) Multiscreen-HA 0.45 μm mixed cellulose ester 96-well membrane vacuum filtration module, and the filtrate was transferred to 96-well plates for LC-MS/MS analysis as described below. AZT-G standard curve samples (0.05–5 μM final concentration) were serially diluted (1:100) in incubation matrix and treated identically to samples. The LC-MS/MS method used a binary gradient using 5 mM ammonium formate/0.05% formic acid (mobile phase A) and 95:5 acetonitrile/methanol containing 0.05% formic acid (mobile phase B). A mobile phase composition of 2% B was held for 0.5 min, then ramped to 35% B over 2.1 min (2.6 min total), returned to 2% B over 0.1 min, and held (4.0 min). Analytes were separated on a Phenomenex Luna 5μ C18(2) 3.0 × 30 mm column. Under these HPLC conditions, AZT-G and the IS ([13C6]AZT-G) had elution times of 2.0 min.
UGT Inhibition Assays.
In experiments where inhibition of glucuronide formation was investigated (Table 5), 50 and 500 μM inhibitors (final concentration) in DMSO (or solvent control) were added to the 96-well plate (n = 2) before addition of the rUGT enzyme mixture and initiated with UDPGA (5 mM) as described above. For IC50 determination, HLM or rUGT were incubated with increasing inhibitor concentrations (0.1–100 μM) as described above. UGT substrate concentrations were at or below Km; ES (10 or 100 μM with BSA), TFP (40 μM HLM or 67 μM with BSA; 10 μM rUGT or 140 μM with BSA), 5HTOL (350 μM), PRO (100 μM HLM, 200 μM rUGT, or 40 μM with BSA), and AZT (842 μM HLM, 1080 μM rUGT, 374 μM HLM with BS, or 596 μM rUGT with BSA).
Inhibition of human UDP-glucuronosyltransferase activities in recombinant UGT enzymes
Recombinant UGTs (0.025 mg/ml) were incubated with alamethicin (10 μg/ml), 50 or 500 μM inhibitor (or DMSO solvent control) in 100 mM Tris-HCl buffer (pH = 7.5) containing 5 mM MgCl2, 5 mM UDPGA, without 2% BSA as described under Materials and Methods. Percentage of activity remaining (relative to solvent control) represents the mean of two experiments. UGT substrate concentrations were at or below the respective Km; 10 μM ES, 10 μM TFP, 350 μM 5HTOL, 200 μM PRO, 842 μM AZT.
Instrumentation.
Analytical quantification was conducted by LC-MS/MS using three systems as identified in Table 1: the LC-MS/MS systems comprised an 1) AB Sciex LLC 4000 QTrap mass spectrometer equipped with an electrospray source, two Shimadzu LC-20AD pumps, a CBM20A controller and DGU-20A solvent degasser, a LEAP CTC HTS PAL autosampler using 20:80 acetonitrile/water and 80:20 acetonitrile/water (both containing 0.1% formic acid) as the wash solvents (CTC Analytics), and a Valco 2-position switching valve; 2) an AB Sciex LLC 4000 triple quadrupole mass spectrometer equipped with an electrospray source, two Jasco X-LCT 3080PU pumps, and a LC-NetII ADC controller (Jasco Analytical Instruments, Easton, MD), a LEAP CTC HTS PAL autosampler using 5:95 water/acetonitrile and methanol (both containing 0.1% formic acid) as the wash solvents (CTC Analytics), and a Valco 2-position switching valve; and 3) a Waters Micromass Quattro Ultima triple quadrupole mass spectrometer equipped with an electrospray ionization source (Micromass, Beverly, MA), two LC-10ADvp pumps with a SCL-10ADvp controller and DGU-14 solvent degasser (Shimadzu), a LEAP CTC HTS PAL autosampler with a multisolvent peristaltic self-washing system using 5:95 water/acetonitrile and 95:5 acetonitrile/water (both containing 0.1% formic acid) as the wash solvents (CTC Analytics), and a LabPro switching valve (Reodyne LLC, Rohnert Park, CA). In all three systems, flow was diverted from the mass spectrometer to waste for the first 0.5 min of the gradient to remove nonvolatile salts.
Quantitative and qualitative NMR was performed using a Bruker (Billerica, MA) Avance 600 MHz system controlled by TOPSPIN V2.0, equipped with a 5-mm TCI cryoprobe. Semipreparative HPLC was performed using a Shimadzu SiL-HTC autosampler, two LC-20AD solvent pumps, an SPD-M20A diode array detector, and a FRC-10A fraction collector.
5-Hydroxytryptophol-O-glucuronide Isolation, Quantitation, and Structural Elucidation.
A pure sample of 5HTOL-G was prepared by incubating 5HTOL (200 μM) with pooled HLM (1.0 mg/ml), UDPGA (1.0 mM), alamethicin (10 μg/ml), and MgCl2 (5 mM) in 100 mM potassium phosphate buffer (pH 7.5) containing 1% BSA (4–10-ml incubations). Incubations were conducted in a shaking water bath at 37°C for 3 h, protein was precipitated by addition of acetonitrile (30 ml), and supernatants were combined and concentrated using an evaporative centrifuge set to 37°C. The residue was reconstituted in 10:90 acetonitrile/water (2 ml) and purified using a semipreparative HPLC system in five injections (0.4 ml) using a binary gradient of 5 mM ammonium acetate/0.1% formic acid (mobile phase A) and acetonitrile (mobile phase B). A mobile phase composition of 10% B was held for 10 min, then ramped to 30% B over 30 min. Separation was performed on a Phenomenex Gemini 5μ C18 10 × 250 mm (Phenomenex), semipreparative HPLC column with a flow rate of 4 ml/min. Aliquots (0.025 ml) of fractions at retention times of UV peaks believed to be the metabolite and the parent were taken for HPLC/MS/UV analysis for verification. Fractions containing 5HTOL-G were combined and concentrated to dryness using an evaporative centrifuge set to 37°C (Genevac, Valley Cottage, NY). Under these HPLC conditions, 5HTOL-G had an elution time of 13.4 min. The isolate was reconstituted in DMSO-d6 (200 μl) and placed in 3-mm diameter tubes before NMR analysis. One-dimensional spectra were recorded using a sweep width of 12,000 Hz and a total recycle time of 7.2 s. The resulting time-averaged free-induction decays were transformed using an exponential line broadening of 1.0 Hz to enhance signal to noise. All spectra were referenced using residual DMSO-d6 (1H δ = 2.5 ppm and 13C δ = 39.5 relative to tetramethylsilane, δ = 0.00). Phasing, baseline correction, and integration were all performed manually. If needed, the BIAS and SLOPE functions for the integral calculation were adjusted manually. The correlation spectroscopy (COSY), multiplicity-edited heteronuclear single quantum coherence, and heteronuclear multiple bond coherence (HMBC) data were recorded using the standard pulse sequence provided by Bruker. Two-dimensional experiments were typically acquired using a 1K × 128 data with 16 dummy scans and a spectral width of 8000 Hz in the f2 dimension. The data were zero-filled to a size of 1K × 1K. A relaxation delay of 1.5 s was used between transients. Quantitation of the prepared 5HTOL-G sample was performed using the aSICCO method, as described previously (Walker et al., 2011).
Data Analysis.
Standard curve fitting was accomplished with AB Sciex Analyst software (version 1.4.2; AB Sciex LLC) or MassLynx QuanLynx software (version 4.1; Micromass) as described above and indicated in Table 1. Data were typically fit to quadratic curves using 1/x2 weighting, and standard curves were run for each experiment.
Substrate concentration [S] and velocity (V) data were fitted to the appropriate enzyme kinetic model by nonlinear least-squares regression analysis (SigmaPlot version 12; Systat Software, Inc., San Jose, CA) to derive the apparent enzyme kinetic parameters Vmax (maximal velocity) and Km or S50 (substrate concentration at half-maximal velocity). The Michaelis-Menten model (eq. 1), the substrate inhibition model (eq. 2), and the substrate activation model (eq. 3), which incorporates the Hill coefficient (n), were used:
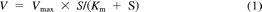
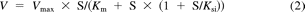
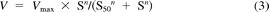 where Vmax is the maximal velocity, Km or S50 is the substrate concentration at half-maximal velocity, n is an exponent indicative of the degree of curve sigmoidicity, and Ksi is an inhibition constant. The best fit was based on a number of criteria, including visual inspection of the data plots (Michaelis-Menten and Eadie-Hofstee), distribution of the residuals, size of the sum of the squared residuals, and the S.E. of the estimates. Selection of models other than Michaelis-Menten was based on the F-test (P < 0.05) and the Akaike Information Criterion (AIC). The CLint was calculated as the Vmax/Km for Michaelis-Menten and substrate inhibition kinetics, and unbound intrinsic clearance (CLint,u) was corrected for the fraction of unbound substrate in incubation (fu,inc) as CLint/fu,inc. Because Km and S50 are not equivalent, the maximum clearance (CLmax) is suggested as an appropriate alternate clearance parameter for substrates exhibiting substrate activation kinetics or positive cooperativity (Houston and Kenworthy, 2000; Uchaipichat et al., 2004) and was calculated from (eq. 4):
where Vmax is the maximal velocity, Km or S50 is the substrate concentration at half-maximal velocity, n is an exponent indicative of the degree of curve sigmoidicity, and Ksi is an inhibition constant. The best fit was based on a number of criteria, including visual inspection of the data plots (Michaelis-Menten and Eadie-Hofstee), distribution of the residuals, size of the sum of the squared residuals, and the S.E. of the estimates. Selection of models other than Michaelis-Menten was based on the F-test (P < 0.05) and the Akaike Information Criterion (AIC). The CLint was calculated as the Vmax/Km for Michaelis-Menten and substrate inhibition kinetics, and unbound intrinsic clearance (CLint,u) was corrected for the fraction of unbound substrate in incubation (fu,inc) as CLint/fu,inc. Because Km and S50 are not equivalent, the maximum clearance (CLmax) is suggested as an appropriate alternate clearance parameter for substrates exhibiting substrate activation kinetics or positive cooperativity (Houston and Kenworthy, 2000; Uchaipichat et al., 2004) and was calculated from (eq. 4):
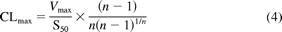
IC50 estimates for inhibition of glucuronidation were determined by nonlinear curve fitting with SigmaPlot (version 12; Systat Software, Inc.) and were defined as the concentration of inhibitor required to inhibit control glucuronidation reactions by 50%.
Results
General UGT Assay Incubation Conditions.
In this report, we describe optimized LC-MS/MS analytical methods for five human UDP-glucuronosyltransferase assays. Although efforts were made to control any potential analytical method variability, a potentially greater source of variability resides with the incubation method. Linear conditions for each assay were established by conducting the incubations at four protein concentrations and measuring the formation of glucuronide metabolite over time. The incubation times were selected such that all reactions were linear with time (<10% substrate consumption), and the lowest protein concentration was selected such that the amount of glucuronide metabolite formed were within the dynamic range of the analytical assays (Table 1).
Optimization of UGT Incubation Conditions.
Because experiments were performed at significantly lower protein concentrations (0.025 mg/ml) than typically described previously (0.25–2.5 mg/ml) (Fisher et al., 2000; Court, 2004; Donato et al., 2010), and considering that enzyme kinetic parameters are affected by incubation conditions (Boase and Miners, 2002), a limited number of incubation conditions known to affect UGT enzyme activity were evaluated. The overall goal was to select universal incubation conditions for all five UGT assays developed, and this applies to the protein concentration of 0.025 mg/ml used in this study.
Tris versus Phosphate Buffers.
Activity comparisons in human liver microsomes and recombinant UGT enzymes for UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7 substrates were conducted in 100 mM Tris-HCl (pH 7.5 at 37°C) and 100 mM potassium phosphate (pH 7.5) buffers to evaluate which provided the greatest overall glucuronide metabolite formation. Incubations in Tris buffer with HLM showed marked increases in metabolite formation for UGT1A4 (2-fold) and UGT1A9 (∼1.5-fold), whereas the other UGTs activities were relatively unaffected (Fig. 2). In rUGT incubations, Tris buffer resulted in increased product formation for all UGTs studied, except UGT1A6. Based on these results, a 100 mM Tris-HCl buffer was selected for all subsequent experiments.
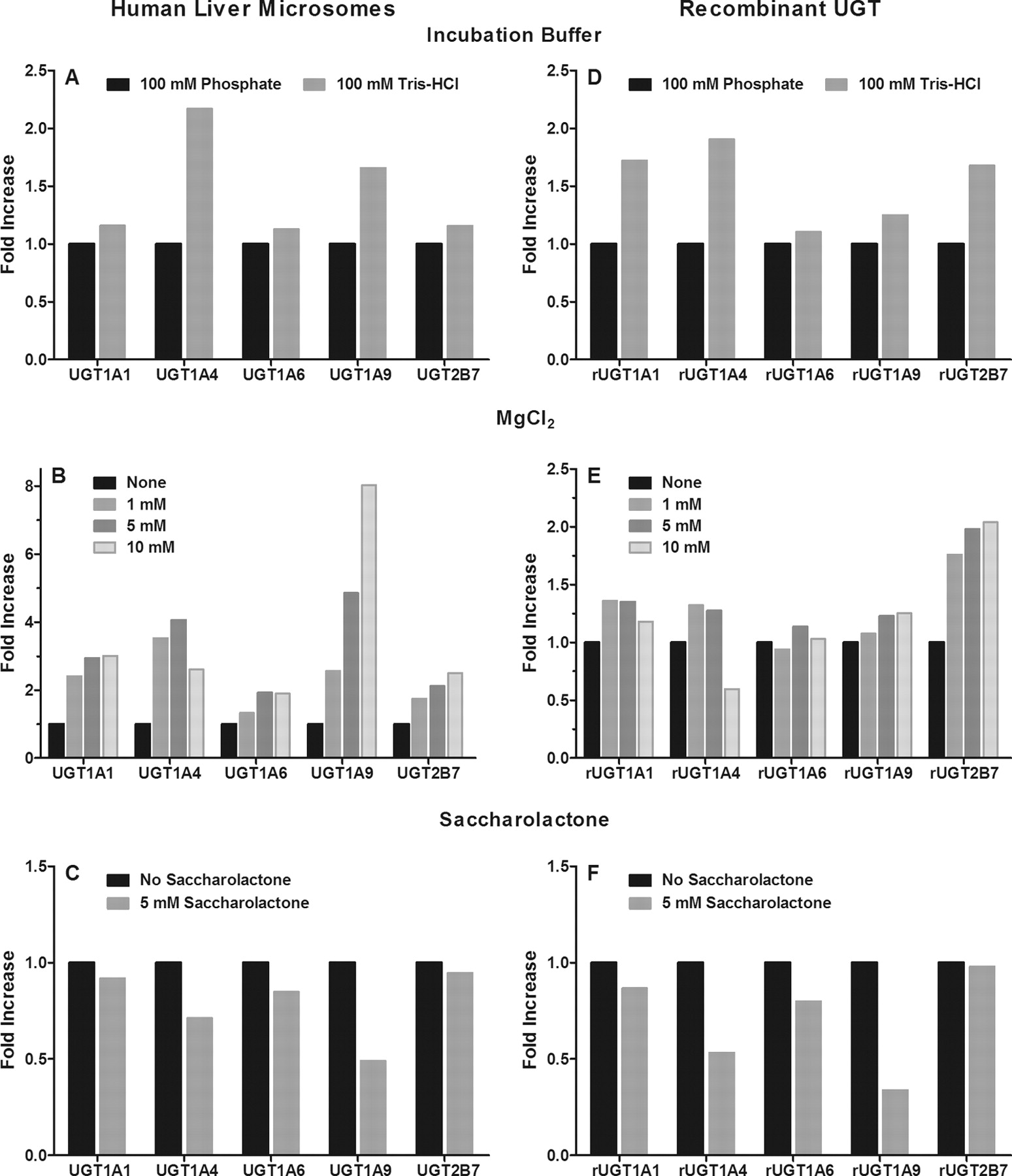
Effect of incubation conditions on major human hepatic UGT activities in pooled human liver microsomes (A–C), and recombinantly expressed UGT enzymes (D–F). Values expressed as fold increase above activity in presence of phosphate buffer (A and D), absence of MgCl2 (B and E), or absence of saccharolactone (C and F).
MgCl2 Concentration.
To determine a common magnesium chloride concentration for use in all UGT assays, glucuronide metabolite formation was measured in HLM and rUGT enzymes for all UGT assays at 0, 1, 5, and 10 mM MgCl2. The effect of MgCl2 concentration on UGT activities in HLM and recombinant UGT enzymes are shown in Fig. 2. The inclusion of MgCl2 in HLM resulted in increased glucuronidation activity, ranging from 2- to 4-fold for UGT1A1, UGT1A4, UGT1A6, and UGT2B7, and up to 8-fold for UGT1A9. Similar trends with more modest activation were observed in rUGTs with a 10 mM MgCl2 concentration, resulting in decreased activity for rUGT1A4. Based on these findings, a 5 mM MgCl2 concentration was selected as general UGT incubation condition.
Inclusion of Saccharolactone.
The effect of saccharolactone, an inhibitor of β-glucuronidase, was assessed at 5 mM to determine whether its use was required with HLM, and/or rUGTs for all assays developed. The fold-change in metabolite formation in the presence and absence of saccharolactone for incubations with HLM and rUGTs are shown in Fig. 2. Saccharolactone inclusion did not significantly impact the activities of UGT1A1, UGT1A6, and UGT2B7, whereas decreases in metabolite formation were observed for UGT1A4 and UGT1A9. Based on these results, saccharolactone was excluded from the optimized UGT incubation conditions.
Alamethicin Concentration in UGT Incubations.
After observing a lack of linearity between AZT-G formation and protein concentration in HLM, which had been pretreated with alamethicin at the previously reported optimal concentration for activation (50 μg/mg HLM) (Fisher et al., 2000), the use of alamethicin at low protein concentrations was evaluated further. Four HLM protein concentrations were assessed at eight alamethicin concentrations using the optimized incubation conditions for UGT2B7 (Fig. 3), UGT1A1, and UGT1A4 (Table 2). Similar results were observed for all three enzymes studied. In general, we observed initial activation of UGT activity at a universal alamethicin concentration of 5 μg/ml and apparent maximal activation at 8.55 μg/ml. The corresponding alamethicin concentrations for 8.55 μg/ml expressed as microgram per milligram HLM are 855, 171, 42.8, and 17.1 μg/mg for HLM concentrations of 0.01, 0.05, 0.2, and 0.5 mg/ml HLM, respectively (Table 2). Therefore, maximal activation would be observed with 50 μg alamethicin/mg HLM only when protein concentrations exceed 0.17 mg/ml (8.55 μg/ml divided by 50 μg/mg). Accordingly, it is the alamethicin solution concentration (microgram per milliter) that is relevant to increasing metabolite formation at the microsomal protein concentrations examined (0.01–0.5 mg/ml), irrespective of protein concentration in incubation. Based on these results, an alamethicin concentration of 10 μg/ml is suggested as optimal for activation of UGT activity regardless of the protein concentration used; therefore, corresponding alamethicin concentrations required for activation at protein concentrations of 0.01, 0.05, 0.2, and 0.5 mg/ml are 1000, 200, 50, and 20 μg/mg HLM, which indicates that a significantly higher alamethicin/protein ratio is required for UGT activation at low protein concentrations. At microsomal protein concentrations below 0.2 mg/ml, no activation is realized at an alamethicin concentration of 50 μg/mg (see Fig. 3B). AZT-G formation (UGT2B7) with HLM is plotted as log alamethicin concentration in microgram per milliliter versus metabolite formed for each protein concentration (Fig. 3A). The same data were plotted as the log of alamethicin concentration in microgram per milligram microsomal protein versus metabolite formed for each protein concentration to illustrate the lack of effect at low microsomal protein concentrations (Fig. 3B). An alamethicin solution concentration of at least 10 μg/ml (400 μg alamethicin/mg HLM at 0.025 mg/ml HLM) would result in optimal increases in metabolite formation in all three UGT assays and was used for all optimized incubations. Similar experiments were conducted with rUGT2B7 and showed no benefit from alamethicin addition at concentrations up to 25 μg/ml (data not shown).
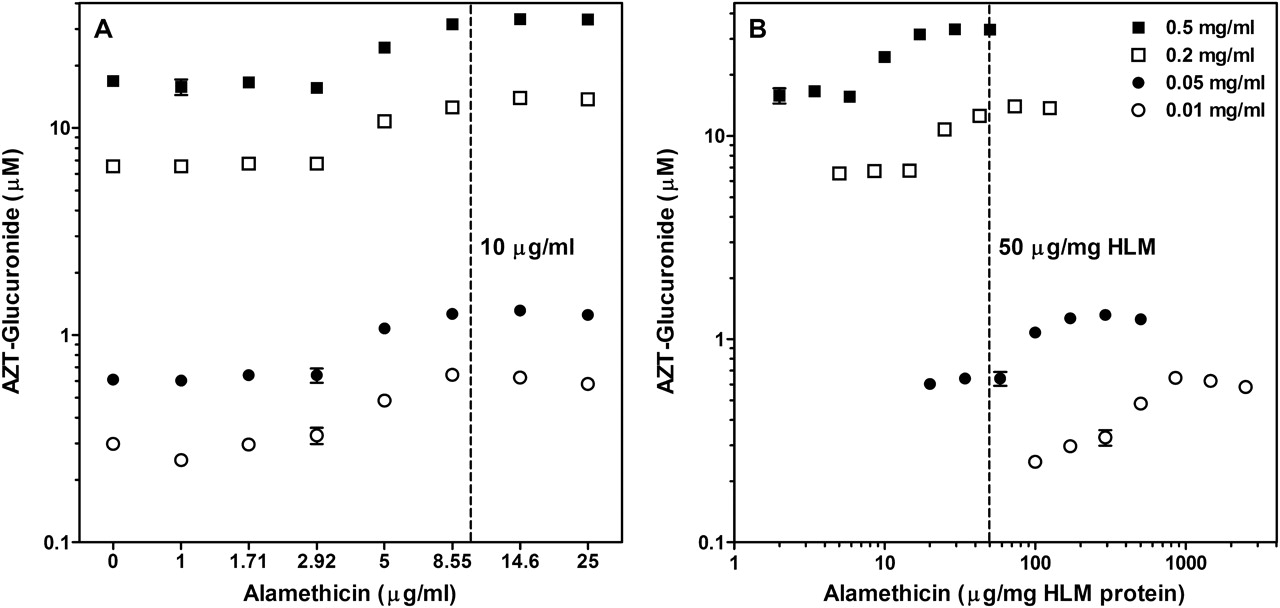
Optimal alamethicin concentration for activation of AZT glucuronidation in human liver microsomes. A, AZT-G formation shown at four concentrations (0.01–0.5 mg/ml) of pooled HLM versus alamethicin concentration expressed as microgram of alamethicin per milliliter of incubate. B, the same data were also plotted expressing alamethicin concentration as microgram of alamethicin per milligram of microsomal protein. Dashed lines signify the current recommended optimal alamethicin activation concentration (10 μg/ml) (A) and the previously used alamethicin activation concentration (50 μg/mg) (B). Corresponding data for β-estradiol and trifluoperazine glucuronidation are shown in Table 2.
Structural Interpretation of 5-Hydroxytryptophol Glucuronide.
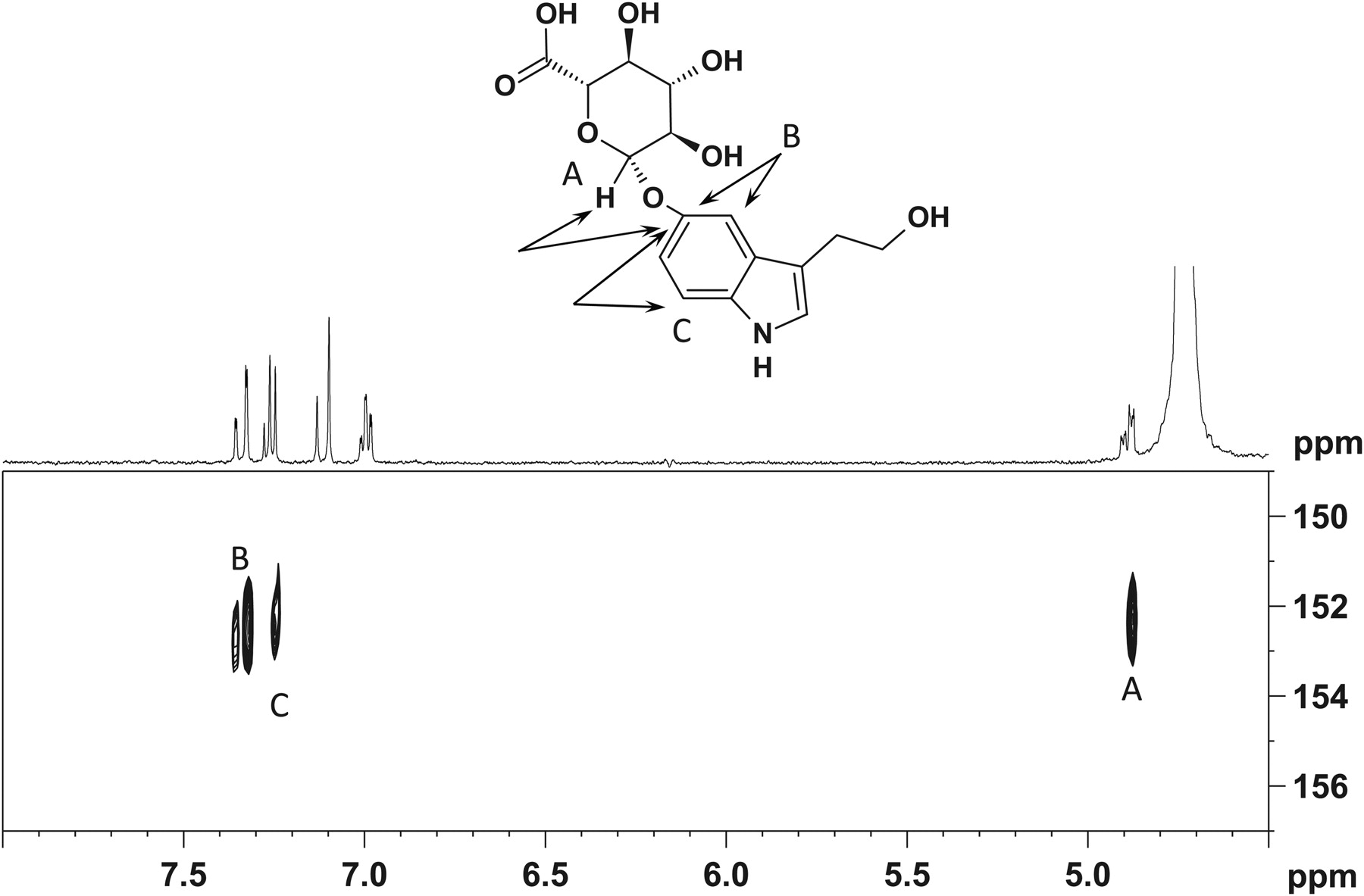
HPLC analysis of the isolated fraction of purified 5HTOL-G indicated a single UV peak at a retention time of 13.4 min, using the semipreparative HPLC system. Using an analytical column (Phenomenex HydroRP 4μ C18, 4.6 × 150 mm), the isolate also appeared as a single peak at 5.3 min, suggesting a single isolated component. There are three biologically plausible positions for glucuronidation of 5HTOL; 1) the C5 (phenolic) hydroxyl of the indole, 2) the aliphatic hydroxyl, and 3) the amine group of the indole (Fig. 4). The 1H-spectrum of the isolate, when dissolved in DMSO-d6, contains two resonances at δ 10.78 and 10.67 that together integrate to a single hydrogen. The COSY spectrum contains cross-peaks, which indicates that these resonances are coupled to resonances at δ 7.18 and 7.11 (Fig. 4). When the sample is diluted in methanol-d4, the δ 10.78 and 10.67 resonances are absent from both the 1H and COSY spectrum (Supplemental Fig. 1). Based on these data, the δ 10.78 and 10.67 are assigned as the NH of the indole and eliminates this site as a potential site for glucuronidation.
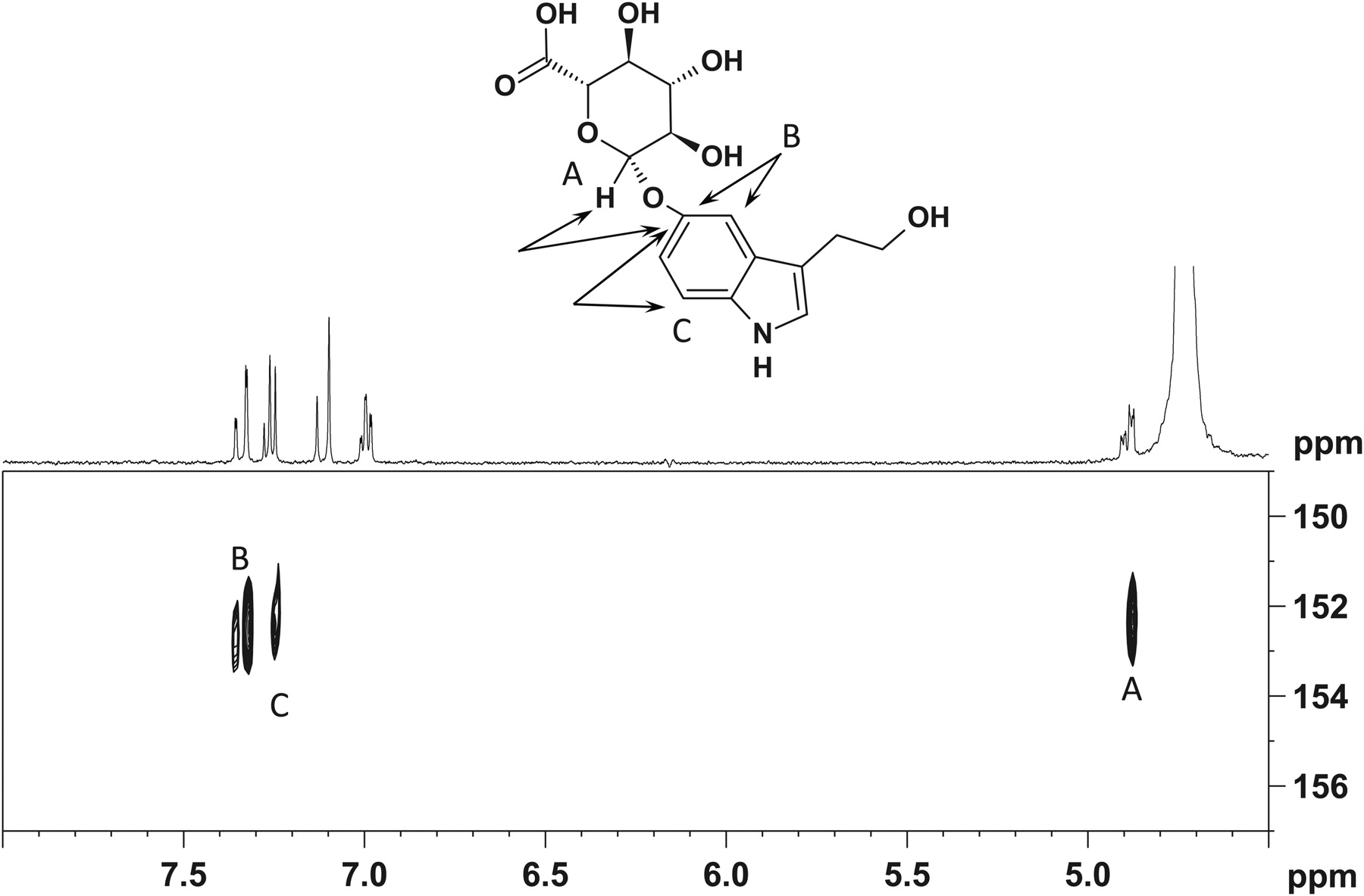
Structure of 5-hydroxytryptophol-O-glucuronide showing NMR HMBC coupling at positions A, B, and C, which confirm glucuronidation at the phenolic 5-hydroxytryptophol position.
The HMBC spectrum of the isolated sample diluted in methanol-d4 contains cross-peaks that indicate long-range coupling from the anomeric proton (δ 4.89 ppm) to a carbon with a chemical shift of 152.7 ppm. The HMBC data also contains cross-peaks from two aromatic proton resonances at 7.33 and 7.25 ppm to the carbon at 152.7 ppm (Supplemental Fig. 2). This strongly indicates that the 5HTOL is glucuronidated at the C5 (phenolic) position of the indole. The HMBC spectrum (diluted in methanol-d4) contains no evidence for any coupling of the anomeric proton to any aliphatic carbon. These combined data suggest a single site of glucuronidation of 5HTOL in human liver microsomes at the phenolic oxygen (Fig. 4). These experiments confirmed that of the three likely glucuronidation sites, the analogous serotonin 5-hydroxyl moiety was glucuronidated (Krishnaswamy et al., 2003).
Nonspecific Binding.
The fraction of unbound substrate in incubation (fu,inc) is shown in Table 4 and was determined at substrate concentrations close to Km or S50 for ES (170 μM), TFP (67 μM), AZT (373 μM), or a concentration range for 5HTOL (3, 30, or 300 μM) and PRO (4 or 40 μM). Substrate recovery for all compounds met acceptance criteria and ranged from 73 to 134%. ES is moderately bound (14% free) to HLM at 0.025 mg/ml and is highly bound (3.9% free) with the addition of 2% BSA. These values reflect significant microsomal binding, even at low protein concentrations, and are in agreement with reported ES binding to BSA (9–11% free with 0.5% BSA) (Rowland et al., 2009) and high plasma protein binding (∼2% free). TFP was moderately bound to HLM (28% free) and highly bound with 2% BSA (6.1% free), in agreement with previously reported microsomal binding at the concentration tested (Uchaipichat et al., 2006a). The results of previous studies indicated a degree of saturable protein binding for TFP in HLM between a concentration range of 10 to 200 μM, with fu,inc increasing from 0.21 to 0.59 (Uchaipichat et al., 2006a). In this study, unbound TFP enzyme kinetic parameters (Table 4) did not account for the possibility of saturable binding. 5HTOL was poorly bound to HLM (93% free) and HLM with BSA (90% free), and mean fu,inc is reported in Table 4. The fu,inc in HLM at 3, 30, or 300 μM were 0.85, 0.98, and 0.95, respectively, and for HLM with BSA 0.79, 0.94, and 0.97, respectively. PRO at 4 and 40 μM was not bound to HLM (100% free) and moderately bound with BSA (17% free), in agreement with previous HLM (70% free at 0.5 mg/ml) and BSA-binding studies (∼20–60% free) (Rowland et al., 2008, 2009). AZT exhibited moderate binding to HLM and HLM with BSA (69% free), in agreement with microsomal (60% free) and BSA binding (49% free) reported by Kilford et al. (2009), and reported AZT plasma protein binding of 62 to 66% free (product label) or 72 to 82% free (Luzier and Morse, 1993). A lower degree of AZT microsomal or BSA binding (≥90% free) was reported by other authors (Court et al., 2003; Rowland et al., 2007, 2009).
β-Estradiol-3-glucuronide Glucuronidation (UGT1A1).
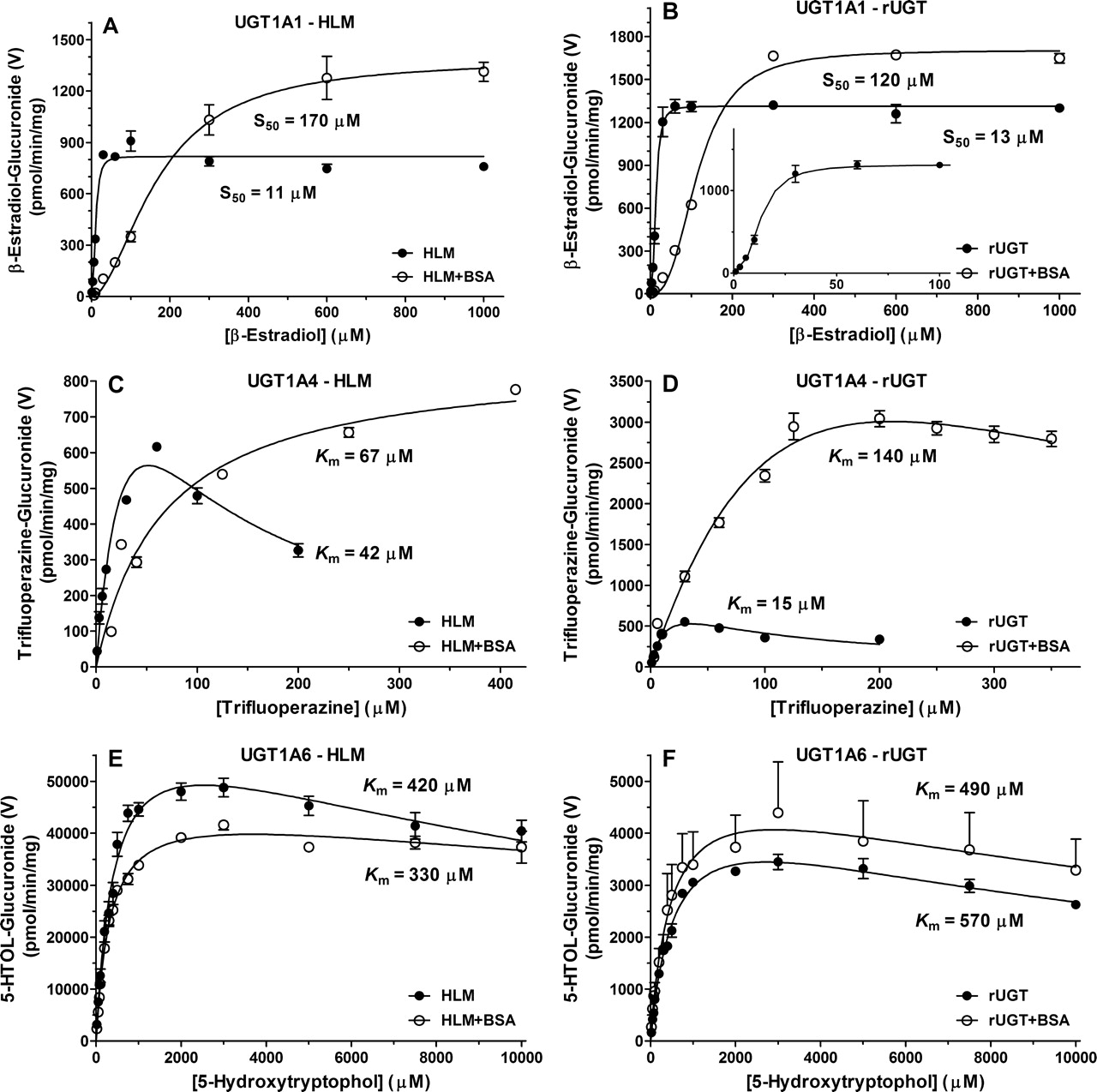
An LC-MS/MS assay for β-estradiol-3-glucuronosyltransferase, a probe substrate for UGT1A1 activity (Williams et al., 2002), was adopted and optimized for use at low protein concentrations. The resulting ES3-G product formation displayed atypical kinetics (curved Eadie-Hofstee plot) and best fit the Hill equation, which is consistent with previous literature (Fisher et al., 2000). The mean Hill coefficient value (n), which gives an indication of the degree of sigmoidicity of the curve, ranged from 1.8 to 2.7 for all incubation conditions (Table 3; Fig. 5), which is indicative of homotropic activation or positive cooperativity. The mean HLM and rUGT S50 values of 11 and 13 μM, respectively, is in agreement with apparent S50 values previously reported for HLM ranging from 17 to 61 μM (Fisher et al., 2000; Alkharfy and Frye, 2002; Williams et al., 2002; Soars et al., 2003; Itäaho et al., 2008) and 8.7 to 23 μM for rUGT1A1 (Soars et al., 2003; Lépine et al., 2004; Fujiwara et al., 2007; Zhou et al., 2011).

Enzyme kinetics of β-estradiol-3-glucuronide (UGT1A1), trifluoperazine-N-glucuronide (UGT1A4), and 5-hydroxytryptophol-O-glucuronide (UGT1A6) formation in pooled HLM (A, C, and E) or recombinant UGTs (B, D, and F) in the absence and presence of 2% BSA. ES3-G formation displayed atypical kinetics, as elucidated by the insert at low substrate concentrations in B. TFP-G and 5HTOL-G formation displayed substrate inhibition kinetics, except TFP-G formation with HLM in presence of 2% BSA, which displayed Michaelis-Menten kinetics. Enzyme kinetic parameters and incubation conditions are summarized in Tables 3 and 4.
In the presence of 2% BSA, total S50 increased (9–15-fold) with minor increases (1.3–1.7-fold) in Vmax, resulting in decreased apparent CLmax (Table 3). However, when corrected for unbound kinetics (Table 4), S50 values were significantly lower (∼8–26-fold) than total S50 and more comparable between HLM and rUGT (1.4–6.6 μM) due to a high degree of microsomal and BSA binding (Table 4). The resultant HLM unbound maximal clearance (CLmax,u) decreased slightly with BSA addition, but it was relatively similar (within 2.6-fold) to values obtained in the absence of BSA.
Trifluoperazine N-Glucuronidation (UGT1A4).
A trifluoperazine-N-glucuronosyltransferase LC-MS/MS assay, which has been shown to be selective for measurement of UGT1A4 activity (Uchaipichat et al., 2006a) as previously quantified by LC/UV based on aglycone absorbance (Uchaipichat et al., 2006b), was developed. The structural identity of TFP-G isolated from in vitro HLM incubations were in agreement with the TFP-G analytical standard compared by NMR spectroscopy (data not shown). The TFP-G product formation data in HLM and rUGT exhibited substrate inhibition kinetics (Table 3; Fig. 5), whereas HLM in the presence of BSA displayed Michaelis-Menten kinetics. Substrate concentrations higher than 200 μM failed to remain in solution in the absence of BSA, accordingly the 300 and 550 μM TFP concentrations were not used for determining apparent enzyme kinetic parameters; however, solubility did not appear to be affected in the presence of BSA. The mean total and unbound Km values in HLM were 42 and 11 μM, respectively, in agreement with Km values reported in HLM that ranged from 35 to 84 μM or 4.7 to 7.6 μM when corrected for nonspecific binding (Uchaipichat et al., 2006a). In rUGT1A4, total and unbound Km values were 15 and 4.1 μM, respectively, in agreement with reported rUGT1A4 values (binding corrected) of 20 to 44 (4.1) μM (Uchaipichat et al., 2006a; Fujiwara et al., 2007; Kubota et al., 2007; Kerdpin et al., 2009). When corrected for nonspecific binding, HLM unbound CLint increased slightly (1.6-fold) due to a relatively higher degree of BSA binding (Table 4). Although unbound HLM Km was comparable with previous reports, Vmax was higher with a pronounced effect of BSA on rUGT1A4 Vmax, comparable with observations with lamotrigine as UGT1A4 substrate (Rowland et al., 2006).
5-Hydroxytryptophol-O-glucuronidation (UGT1A6).
A 5-hydroxytryptophol-O-glucuronosyltransferase LC-MS/MS assay for UGT1A6 was developed and optimized. The 5HTOL-G formation displayed weak substrate inhibition kinetics (Table 3; Fig. 5). The mean Km values in HLM and rUGT were 420 and 570 μM, respectively. Values cited for Km in HLM have ranged from 134 to 156 μM, and the value for rUGT1A6 was 135 μM (Krishnaswamy et al., 2004). In the presence of 2% BSA, unbound kinetic parameters (Table 4) did not change significantly, due to a low degree of nonspecific binding.
Propofol-O-glucuronidation (UGT1A9).
A propofol-O-glucuronosyltransferase LC/MS-MS assay was developed to evaluate UGT1A9 activities in HLM and rUGT. The PRO-G product formation data displayed substrate inhibition kinetics except for data collected using HLM in the presence of 2% BSA, which displayed Michaelis-Menten kinetics (Table 3; Fig. 6). The optimized method yielded mean Km values of 98 and 200 μM for HLM and rUGT1A9, respectively. Previously reported Km values obtained in HLM ranged from 64 to 280 μM (Soars et al., 2003; Shimizu et al., 2007; Rowland et al., 2008) and from 28 to 111 μM for rUGT1A9 (Soars et al., 2003; Rowland et al., 2008; Takahashi et al., 2008; Fujiwara et al., 2010). In the presence of 2% BSA, total Km values for HLM and rUGT1A9 were 46 and 63 μM for HLM and rUGT1A9, respectively. Previously reported Km values in the presence of BSA were 15.5 and 7.2 μM for HLM and rUGT, respectively (Rowland et al., 2008). As shown in Table 4, the addition of BSA resulted in significant increases in unbound CLint (7.1-fold).

Enzyme kinetics of propofol-O-glucuronide and AZT-5′-glucuronide formation in pooled HLM (A and C) or recombinant UGTs (B and D) in the absence and presence of 2% BSA. PRO-G formation displayed substrate inhibition kinetics, except with HLM in the presence of 2% BSA, which displayed Michaelis-Menten kinetics. AZT-G formation displayed Michaelis-Menten kinetics. Enzyme kinetic parameters and incubation conditions are summarized in Tables 2 and 3.
AZT-5′-glucuronidation (UGT2B7).
An AZT-5′-glucuronosyltransferase assay with LC-MS/MS detection (Engtrakul et al., 2005) was optimized to evaluate UGT2B7 activities in HLM and rUGT as an established UGT2B7 probe substrate (Court et al., 2003). The AZT-G product formation data displayed Michaelis-Menten kinetics (Table 3; Fig. 6). Statistical evaluation (F-test) indicated a statistically better enzyme kinetic fit (p < 0.05) with a substrate inhibition model for incubation conditions, except for rUGT in the presence of BSA. However, visual inspection of the fit did not justify use of a more complex model, and conclusive evidence would require incubation with substrate concentrations >4500 μM. The mean Km values for HLM (840 μM), HLM with BSA (160 μM), and rUGT (1100 μM) estimated with the substrate inhibition model were comparable with those reported in Table 3, where the mean total Km values for HLM and rUGT2B7 were 610 and 900 μM, respectively. Values are in agreement with published Km values in HLM ranging from 518 to 1600 μM, and for rUGT2B7 values of 478 to 770 μM have been reported (Court et al., 2003; Engtrakul et al., 2005; Uchaipichat et al., 2006b; Peterkin et al., 2007; Rowland et al., 2009). In the presence of 2% BSA, mean Km values for HLM and rUGT2B7 were 150 and 320 μM, respectively. Previously reported Km values in the presence of BSA were 69 to 105 μM and 40 to 70 μM for HLM and rUGT, respectively (Uchaipichat et al., 2006b; Rowland et al., 2007, 2009). Corrections for unbound concentrations in microsomes resulted in slight decreases in Km, whereas addition of BSA decreased Km (Table 4). Unbound HLM Km (100 μM) with BSA was comparable with globulin-free BSA (188 μM) and reported values in the presence of crude BSA (87 μM) (Rowland et al., 2007). The decreases in unbound Km (4.2-fold) and increases in Vmax (2.2-fold) in the presence of BSA resulted in a significantly increased unbound HLM CLint (9.2-fold) (Table 4).
UGT Inhibition.
Eleven previously reported UGT inhibitors and known P450 inhibitors were screened at two concentrations (50 and 500 μM) for their ability to inhibit the five hepatic UGTs studied, to evaluate a high-throughput UGT inhibition screening scenario (Table 5). Activities were compared with solvent control. Chrysin significantly inhibited UGT1A1, UGT1A6, UGT1A9, and UGT2B7; diflunisal appeared to be selective as UGT1A9 inhibitor; mefenamic acid mostly inhibited UGT1A9 and UGT2B7; silibinin was most potent as UGT1A1 inhibitor; and tangeretin inhibited UGT1A1 and UGT1A9. Valproic acid exhibited minimal UGT inhibition at 500 μM. The P450 inhibitor 1-aminobenzotriazole did not inhibit UGTs up to 500 μM, whereas the other P450 inhibitors tested appeared to be nonselective inhibitors of UGT1A1 and UGT1A4 (itraconazole); UGT1A1, UGT1A4, and UGT2B7 (ketoconazole and ritonavir); and weak UGT1A1 inhibition by verapamil at 50 μM with nonselective inhibition at the higher concentration tested.
IC50 experiments (Table 6) with chrysin confirmed potent inhibition of UGT1A1 with less than 10-fold selectivity over UGT1A6, UGT1A9, and UGT2B7. Itraconazole is a potent inhibitor (IC50 < 1 μM) of UGT1A1 and UGT1A4, whereas activities of UGT1A6, UGT1A9, and UGT2B7 were unaffected up to 100 μM. The addition of BSA typically increased IC50 values, probably due to an increase in nonspecific inhibitor binding (not specifically measured in these studies), reflecting the importance of correcting IC50 or Ki values for nonspecific binding when attempting DDI predictions.
Inhibition of human UDP-glucuronosyltransferase activities in human liver microsomes and recombinant UGT enzymes
Discussion
Over the past couple of decades, major advances were made in the characterization of UGT enzyme substrate and inhibitor selectivities, highlighting the importance of glucuronidation in drug metabolism, in vitro-in vivo extrapolation of drug clearance, and prediction of DDIs (Miners et al., 2010a). The UGT reaction-phenotyping techniques have progressed significantly with the identification and characterization of selective probe substrates, availability of recombinant UGTs, and optimization of in vitro incubation conditions required to measure drug glucuronidation. However, a serious shortcoming in the UGT reaction-phenotyping process is the lack of isoform-selective inhibitors that could be used in liver microsomal incubations, analogous to those available for P450 enzymes (Zhang et al., 2007). Selective UGT inhibitor probes are limited to hecogenin (UGT1A4), niflumic acid (UGT1A9), and fluconazole (UGT2B7) (Miners et al., 2010a). Thus, the confidence in UGT reaction phenotyping could benefit significantly from the identification of a greater number of enzyme-selective inhibitors for UGTs that contribute to hepatic drug metabolism. Accordingly, highly selective, robust, and sensitive LC-MS/MS analytical techniques would assist high-throughput screening efforts to increase future success in this endeavor.
Glucuronidation reactions have been described to be impacted significantly by in vitro incubation conditions such as buffer type and ionic strength, latency, glucuronide stability, atypical kinetics, and the albumin effect (refer to Introduction). Some standardization occurred over time, but universally accepted incubation conditions are not generally available with phosphate and Tris buffers used interchangeably for in vitro UGT reactions; however, some authors noted highest AZT glucuronidation activity with a physiologically relevant carbonate buffer or Williams E medium (Engtrakul et al., 2005). In this study, Tris buffer provided greater activity for UGT1A4 and UGT1A9 in HLM, whereas all rUGT activities, except rUGT1A6, were increased relative to phosphate buffer. Similar findings were observed for acetaminophen glucuronidation with rUGT1A9 (Mutlib et al., 2006) and negligible impact of Tris on UGT1A1 (ES) (Soars et al., 2003) or UGT2B7 (AZT) (Boase and Miners, 2002; Engtrakul et al., 2005) activities. Boase and Miners (2002) demonstrated a trend of higher apparent Vmax for AZT glucuronidation in Tris compared with phosphate buffers and, interestingly, a significant decrease in CLint at higher phosphate ionic strength (20–100 mM), primarily because of an increase in apparent Km. The mechanistic effect or impact of phosphate on UGT activity is not completely clear, although altered substrate selectivity and activity have been reported due to UGT phosphorylation (Basu et al., 2005).
Divalent metal ions increase UGT activity, and MgCl2 (2–10 mM) is typically included in in vitro UGT incubations, with some using MgCl2 concentrations closer the endoplasmic reticulum interior (1 mM) or as high as 50 mM (Fisher et al., 2001). Inclusion of MgCl2 increased UGT activity by ∼2- to 8-fold in HLM and to a lesser extent in rUGTs with 5 mM generally resulting in maximal stimulation of glucuronidation. A final MgCl2 concentration of 5 mM for the optimized incubation conditions is in agreement with general practice (4–5 mM) (Boase and Miners, 2002; Court, 2004). Saccharolactone, an inhibitor of endogenous β-glucuronidase-catalyzed hydrolysis of the glucuronide conjugate, is often added to UGT incubations, and in some cases it is required to preserve glucuronide stability (Bauman et al., 2005). In this study, saccharolactone did not increase glucuronide formation, and it occasionally decreased glucuronidation (e.g., UGT1A4 and UGT1A9). These findings are in agreement with reports indicating limited saccharolactone benefit or an inhibitory effect (Kaivosaari et al., 2008; Oleson and Court, 2008).
Alamethicin, a pore-forming peptide, is currently the preferred agent to activate microsomal UGT activity, presumably by increasing access of substrate and cofactor to the luminal orientation of UGT proteins (Fisher et al., 2001). A standard alamethicin concentration of 50 μg/mg microsomal protein is routinely used based on optimization (50–100 μg alamethicin/mg HLM) with microsomal protein concentrations ranging from 0.5 to 2.5 mg/ml (Fisher et al., 2000) and other supportive studies (Kaivosaari et al., 2008). Activation in hepatocytes required higher alamethicin concentrations (≥200 μg/ml) (Bánhegyi et al., 1993). Development of LC-MS/MS analytical methods allows use of lower protein concentrations in incubation, and initial optimization (0.01–0.1 mg/ml HLM) indicated a lack of linearity in glucuronide product formation using standard alamethicin activation conditions (50 μg/mg). Further evaluation of optimal alamethicin concentration (2–2500 μg/mg HLM) indicated dependence of the critical alamethicin activation concentration on microsomal protein concentration and loss of activation at low protein concentrations (Table 2; Fig. 3), even with alamethicin levels as high as 292 μg/mg microsomal protein. Accordingly, maximal activation could occur with as little as 17.1 μg/mg HLM at high HLM protein concentration, whereas lower HLM protein incubations may require as much as 855 μg alamethicin/mg protein. Hence, to standardize, it is more appropriate to express optimal alamethicin activation concentrations as microgram per milliliter in the incubation, especially when using HLM protein concentrations <0.2 mg/ml (Fig. 3). The exact reason for this phenomenon is not clear, but it is apparent that a critical alamethicin concentration in solution is required for optimal activation, independent of protein concentration. The current recommendation is to use a standard alamethicin activation concentration of at least 10 μg/ml incubate for microsomal protein concentrations between 0.01 and 0.5 mg/ml. In contrast, although alamethicin is often included in incubations of insect cell baculoviral-expressed UGTs, no activation was observed in this study, which suggests little benefit and is consistent with similar reports (Kaivosaari et al., 2008).
Enzyme kinetic parameters for UGT1A9 and UGT2B7 were most significantly affected by the addition of 2% BSA, whereas UGT1A1, UGT1A4, and UGT1A6 were affected to a lesser degree. In general, total Km or S50 increased for UGT1A1 and UGT1A4, remained unchanged for UGT1A6, and decreased for UGT1A9 and UGT2B7, whereas apparent Vmax remained relatively unaffected (<2-fold change). When corrected for nonspecific binding in the incubation, the unbound CLint in the presence of BSA increased most significantly for UGT1A9 and UGT2B7 and remained relatively unaffected for other UGTs investigated. Previous studies indicated that BSA decreased PRO Km without affecting Vmax in HLM and rUGT, and similar observations were found for AZT in HLM (Rowland et al., 2008, 2009). A more recent study indicated decreases in Km, but significant increases in Vmax for entacapone (UGT1A9), and only decreases in the Km for AZT in HLM and rUGT (Manevski et al., 2011). These findings indicate more complex mechanisms, other that competitive inhibition by fatty acids may be involved in the inhibition of enzyme activity released from membranes. It has been postulated that the albumin effect is affected by enzyme source or different fatty acid compositions released from membranes (Rowland et al., 2008, 2009), which could result in differences in apparent kinetic parameter estimates for HLM or rUGT; however, comparisons between the human embryonic kidney (HEK293) and Spodoptera frugiperda (Sf9) expression systems does not explain the observations for entacapone (Manevski et al., 2011). It should also be noted that in vitro CLint could differ depending on microsomal enzyme source due to significant interindividual variation of AZT glucuronidation rates (Peterkin et al., 2007), and apparent Vmax estimates were determined without an authentic glucuronide standard (Rowland et al., 2007).
The S50 for UGT1A1-catalyzed ES glucuronidation reported in this study (1.4–6.6 μM) is significantly lower than previous observations for apparent ES S50 (17–61 μM) (Fisher et al., 2000; Itäaho et al., 2008) because of considerable microsomal binding even when using a low protein concentration in incubation, reiterating the importance of correcting for nonspecific binding in kinetic parameter estimates. Although glucuronidation is often considered to be a low-affinity process (Williams et al., 2004), high micromolar affinity for ES and TFP (4.1–11 μM) was observed, similar to other reports for UGT1A substrates (Goosen et al., 2007; Liu et al., 2010).
Inhibition of UGT activities was confirmed with potential UGT and characterized P450 inhibitors, which indicated nonselective inhibition by chrysin, mefenamic acid, silibinin, and tangeretin. Diflunisal may be a potent and selective UGT1A9 inhibitor, but requires it further characterization because weak UGT2B7 inhibition (IC50 = 370 μM) was reported previously (Knights et al., 2009). Nonselective UGT inhibition was apparent with ketoconazole, itraconazole, ritonavir, and verapamil, in agreement with previous reports on ketoconazole (Liu et al., 2011; Zhou et al., 2011). Of the P450 inhibitors investigated, minimal inhibition was observed with the pan-P450 inhibitor 1-aminobenzotriazole, which indicated that it may be useful when phenotyping mixed UGT and P450 substrates and estimating fractional metabolism (fm) by P450 versus UGT (Kilford et al., 2009).
In summary, we describe optimized in vitro incubation and alamethicin activation conditions, LC-MS/MS analytical methods using authentic glucuronide standards, and kinetic parameters for five hepatic UGTs. These methods should prove useful in the routine assessments of the potential for new drug candidates to elicit pharmacokinetic drug interactions via inhibition of human UGT activities. The methods should also be advantageous when screening larger compound libraries to identify UGT enzyme-selective chemical inhibitors.
Authorship Contributions
Participated in research design: Walsky, Bauman, Lapham, Bourcier, Giddens, Obach, Hyland, and Goosen.
Conducted experiments: Walsky, Bauman, Lapham, Bourcier, Giddens, Negahban, Hyland, and Ryder.
Contributed new reagents or analytic tools: Walsky, Bauman, Lapham, Bourcier, Giddens, Negahban, and Ryder.
Performed data analysis: Walsky, Bauman, Lapham, Bourcier, Giddens, Negahban, Ryder, Hyland, and Goosen.
Wrote or contributed to the writing of the manuscript: Walsky, Bauman, Lapham, Bourcier, Giddens, Negahban, Ryder, Obach, Hyland, and Goosen.
Acknowledgments
We acknowledge Dr. Brian Ethell for helpful scientific discussions and thank Howard Miller and Mark Snyder for experimental assistance with the high-throughput experimental automation.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- DDI
- drug-drug interaction
- UGT
- UDP-glucuronosyltransferase
- P450
- cytochrome P450
- HLM
- human liver microsomes
- LC-MS/MS
- liquid chromatography equipped with tandem mass spectrometry
- ES
- β-estradiol
- ES3-G
- β-estradiol-3-glucuronide
- TFP
- trifluoperazine
- 5HTOL
- 5-hydroxytryptophol
- PRO
- propofol
- AZT
- 3′-azido-3′-deoxythymidine or zidovudine
- CLint
- intrinsic clearance
- BSA
- bovine serum albumin
- AZT-G
- AZT-5′-glucuronide
- PRO-G
- propofol-O-glucuronide
- TFP-G
- trifluoperazine-N-glucuronide
- UDPGA
- UDP-glucuronic acid
- DMSO
- dimethyl sulfoxide
- 5HTOL-G
- 5-hydroxytryptophol-O-glucuronide
- HPLC
- high-performance liquid chromatography
- rUGT
- recombinant UGT
- IS
- internal standard
- aSICCO
- artificial signal insertion for calculation of concentration observed
- COSY
- correlation spectroscopy
- HMBC
- heteronuclear multiple bond coherence
- CLmax
- maximal clearance.

- Received September 30, 2011.
- Accepted February 22, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}