


Abstract
Drug-drug interactions (DDIs) that occur via mechanism-based inactivation of cytochrome P450 are of serious concern. Although several predictive models have been published, early risk assessment of MBIs is still challenging. For reversible inhibitors, the DDI risk categorization using [I]/Ki ([I], the inhibitor concentration; Ki, the inhibition constant) is widely used in drug discovery and development. Although a simple and reliable methodology such as [I]/Ki categorization for reversible inhibitors would be useful for mechanism-based inhibitors (MBIs), comprehensive analysis of an analogous measure reflecting in vitro potency for inactivation has not been reported. The aim of this study was to evaluate whether the term λ/kdeg (λ, first-order inactivation rate at a given MBI concentration; kdeg, enzyme degradation rate constant) would be useful in the prediction of the in vivo DDI risk of MBIs. Twenty-one MBIs with both in vivo area under the curve (AUC) change of marker substrates and in vitro inactivation parameters were identified in the literature and analyzed. The results of this analysis show that in vivo DDIs with >2-fold change of object drug AUC can be identified with the cutoff value of λ/kdeg = 1, where unbound steady-state Cmax is used for inhibitor concentration. However, the use of total Cmax led to great overprediction of DDI risk. The risk assessment using λ/kdeg coupled with unbound Cmax can be useful for the DDI risk evaluation of MBIs in drug discovery and development.
Introduction
Inhibitory drug-drug interactions (DDIs) are of serious concern in drug development because they can lead to restricted use or withdrawal of drugs from the market (Huang and Lesko, 2004; Wienkers and Heath, 2005). The clinical relevance of mechanism-based inactivators (MBIs) is illustrated by the fact that 24 (19%) of the identified 129 cytochrome P450 (P450) inhibitors on the U.S. market and 38% of the known strong inhibitors are MBIs of P450 enzymes (Isoherranen et al., 2009). Eight (33%) of the 24 MBIs caused strong interactions in vivo. The Pharmaceutical Research and Manufacturers of America (PhRMA) recently summarized the industry practices used in inactivation measurements and recommended practical methods for in vitro inactivation assays and for prediction of in vivo DDIs using in vitro data (Grimm et al., 2009). However, as described in the article, although several mathematical models for MBI predictions have been presented, quantitative prediction of in vivo DDIs is still challenging. For reversible inhibitors, the DDI risk categorization using [I]/Ki ([I], the inhibitor concentration; Ki, the inhibition constant) is widely used and accepted in drug discovery and development. For irreversible inhibitors, the U.S. Food and Drug Administration (FDA) draft guidance for industry of drug interactions studies (released in February 2012; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf) recommends calculating an R-value equal to {kinact × [I]/(KI + [I]) + kdeg}/kdeg to assess the in vivo DDI risk of MBIs. In this equation, [I] is the inactivator concentration calculated from the total (free and bound) systemic inhibitor concentration, kinact is the maximal inactivation rate, KI is the inactivator concentration when the rate of inactivation reaches half of kinact, and kdeg is the rate constant for enzyme degradation in vivo. If this R-value is >1.1 (or 11 for CYP3A inhibition in the gut), the investigational drug is considered to be a possible P450 inhibitor in vivo and further evaluation is necessary. How well this R-value reflects the magnitude of in vivo DDI risk and whether false positives and false negatives are common has not been reported.
A recent review showed that 13 (42%) of 31 in vitro MBIs were neither moderate nor potent inhibitors in vivo (VandenBrink and Isoherranen, 2010). This suggests that drugs classified as in vitro MBIs do not always cause clinically significant DDIs. In another report of subset of MBIs, relatively accurate predictions of in vivo DDIs were reported (Fahmi et al., 2009). The published methods for quantitative prediction of in vivo DDIs for MBIs are shown in eqs. 1 and 2:
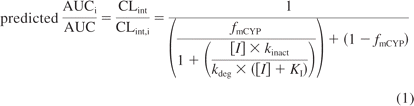
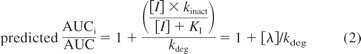 where AUCi/AUC is the fold increase in probe AUC, CLint/CLint,i is the fold decrease in probe CLint, and λ is the apparent first-order inactivation rate at a given inhibitor concentration (Mayhew et al., 2000; Grimm et al., 2009). Equation 2 assumes that the probe is entirely cleared by a single, inhibited pathway (fm = 1) in the liver, and it is mathematically equal to the R-value used in the FDA draft guidance for predicting inactivation risk for systemic clearance. Equation 2 can also be written as AUCi/AUC = 1 + λ/kdeg, which is similar to the AUCi/AUC = 1 + [I]/Ki used in risk assessment of reversible inhibitors. The aim of this study was to determine whether the λ/kdeg value obtained from in vitro data could be reliably used to identify DDI risk of in vitro MBIs. The correlation between the magnitude of in vivo DDI and the predicted λ/kdeg was determined for known in vitro MBIs and the false-positive and false-negative rates were evaluated.
where AUCi/AUC is the fold increase in probe AUC, CLint/CLint,i is the fold decrease in probe CLint, and λ is the apparent first-order inactivation rate at a given inhibitor concentration (Mayhew et al., 2000; Grimm et al., 2009). Equation 2 assumes that the probe is entirely cleared by a single, inhibited pathway (fm = 1) in the liver, and it is mathematically equal to the R-value used in the FDA draft guidance for predicting inactivation risk for systemic clearance. Equation 2 can also be written as AUCi/AUC = 1 + λ/kdeg, which is similar to the AUCi/AUC = 1 + [I]/Ki used in risk assessment of reversible inhibitors. The aim of this study was to determine whether the λ/kdeg value obtained from in vitro data could be reliably used to identify DDI risk of in vitro MBIs. The correlation between the magnitude of in vivo DDI and the predicted λ/kdeg was determined for known in vitro MBIs and the false-positive and false-negative rates were evaluated.
Materials and Methods
Data Collection.
The University of Washington Metabolism and Transport Drug Interaction Database (http://www.druginteractioninfo.org) was queried to identify known P450 mechanism-based inactivators with KI and kinact values determined using human liver microsomes or recombinant systems, and to retrieve all reported in vivo interactions for the mechanism-based inactivators extracted. From the resulting list of in vivo interaction studies, those conducted with a known marker substrate (FDA Draft Guidance for Industry, 2012) were selected, and the change in object AUC was recorded. The inhibitor concentrations measured in the interaction study were used if available. For studies that did not measure the plasma concentrations of the inhibitor, literature data using the same dosing regimen were used to obtain steady-state Cmax values for the inactivator. If data for the inactivator were not available at the dose level used in the in vivo interaction studies, the concentrations were dose-normalized to obtain predicted Cmax values for the inactivator. The plasma or serum protein binding data for the inactivators were also collected from the literature. If multiple KI and kinact values for the inactivator were available, the value used for analysis was chosen according to the following criteria: 1) the KI and kinact were tested using the same probe as that used in the in vivo DDI study, and 2) the study with the lowest microsomal protein concentration was used.
Assessment of the Evaluation Methodology to Predict In Vivo DDI Risk.
The steady-state inactivator concentrations and in vitro kinact, KI, and kdeg values were used to compute the λ/kdeg values. Both total and unbound Cmax at steady state were used for inactivator concentrations because total Cmax is used in the [I]/Ki risk assessment for reversible inhibitors, whereas use of the unbound systemic Cmax rather than total systemic Cmax or estimated unbound portal Cmax yielded the most accurate DDI predictions for MBIs in a previous study (Obach et al., 2007). The reported turnover half-life (t1/2) of 36 to 51 h for CYP1A2, 32 h for CYP2B6, 104 h for CYP2C9, 26 h for CYP2C19, 70 h for CYP2D6, and 26 to 79 h for CYP3A4 were used to calculate kdeg (1/min) values (Yang et al., 2008). The median values were used for CYP1A2 and CYP3A4. Based on the λ/kdeg from eq. (2), the likelihood that a drug will cause in vivo interactions was classified as likely (λ/kdeg > 1), possible (1 > λ/kdeg > 0.1), or remote (0.1 > λ/kdeg), then compared with actual AUC change of object drug.
Results and Discussion
Twenty-one inactivators with complete in vivo and in vitro data were identified, and the λ/kdeg values were calculated. Because several inhibitors had multiple DDI studies, a total of 160 in vivo studies were analyzed. Figure 1 shows the correlation between predicted risk (λ/kdeg) and the in vivo AUC change for all DDI studies analyzed. The analysis using unbound inhibitor concentrations and all reported in vivo DDI studies is shown in Fig. 1, A and B. The relationship between greatest observed in vivo DDI (the maximal in vivo DDI risk) with a given inhibitor and the predicted λ/kdeg with accepted P450 marker probes is shown in Fig. 1, C and D. The in vivo studies and in vitro parameters used are summarized in Table 1. The DDIs with >2-fold AUC change of object drugs could be identified using a λ/kdeg cutoff value of 1 and unbound inactivator Cmax (Fig. 1, C and D). The use of total Cmax in the λ/kdeg calculation resulted in exaggerated risk prediction and an increase in the number of false positives (Fig. 1E). These results suggest that unbound Cmax rather than total Cmax would be appropriate for DDI risk assessment with MBIs.

DDI risk assessment for the identified MBIs. A, all 160 in vivo DDI studies for the 21 inactivators were analyzed using calculated λ/kdeg coupled with unbound steady-state Cmax. B, the relationship between λ/kdeg and AUC change for individual inactivators are categorized as shown. The numbers after each MBI indicate the number of in vivo studies conducted within that category. In vivo studies with the most sensitive probes for the inactivated P450 and highest in vivo interactions for the 21 inactivators were analyzed using λ/kdeg coupled with unbound steady-state Cmax (C), and the relationship between λ/kdeg and AUC change for individual inactivators is shown for different risk categories in D. E, all 160 in vivo DDI studies for the 21 inactivators were analyzed using λ/kdeg coupled with total steady-state Cmax. A λ/kdeg of 1 predicts a 2-fold AUC increase in vivo for an ideal probe substrate.
In vivo and in vitro data used for risk assessment
This analysis shows the effect of probe sensitivity in observed DDI risk. When all 160 in vivo studies were included, 58 (36%) were categorized into the zone between AUC change <2-fold and λ/kdeg > 1. This demonstrates an overprediction of the in vivo risk with many substrates. Some inactivators distributed between the different zones from low to high DDI risk mainly due to different probes used and variable probe sensitivity (Fig. 1B). This is not unexpected, because it is known that the fm of object drugs as well as the Fg of CYP3A4 substrates are important factors that affect the magnitude of in vivo DDIs. For example, for diltiazem, a 1.5- and 3.8-fold increase in AUC of quinidine (fmCYP3A4 = 0.76) and midazolam (fmCYP3A4 = 0.94) (Brown et al., 2005), respectively, were observed, despite the fact that dosing regimens in both studies were similar (Backman et al., 1994; Laganière et al., 1996). For paroxetine, a 1.7- and 5.2-fold increase in AUC of imipramine (fmCYP2D6 = 0.46) and desipramine (fmCYP2D6 = 0.88) (Brown et al., 2005), respectively, were observed (Albers et al., 1996; Alderman et al., 1997). This demonstrates that simple risk analysis does not take into account the effect of multiple clearance pathways, genetic polymorphisms, and polytherapy on the magnitude of the DDIs observed in individual patients.
When the in vivo data were analyzed for individual MBIs using only the largest observed in vivo interactions (Fig. 1, C and D), the portion of data points falling in the zone between AUC change <2-fold and λ/kdeg > 1 was decreased [5 of 21 (24%)] but still showed a significant false-positive rate. Using the R-value of 1.1, which is equivalent to a λ/kdeg cutoff of >0.1, the false-positive rate was 38%, demonstrating a significant overprediction of DDI risk even when unbound Cmax was used. The false positives included the two CYP2B6 MBIs, clopidogrel and ticlopidine. The overprediction with these two MBIs is likely due to low fm of the probe bupropion by CYP2B6 and the contribution of alternative elimination pathways (reduction to threohydrobupropion and erythrohydrobupropion) (Faucette et al., 2000; Reese et al., 2008). Hence, clopidogrel and ticlopidine are expected to cause more potent DDIs with a higher fmCYP2B6 substrate. Indeed, ticlopidine was a weaker MBI for CYP2C19 than CYP2B6 in vitro, but in vivo a 6-fold increase in the AUC of omeprazole (CYP2C19 probe) was observed. Zileuton, which had a λ/kdeg value >1, suggesting a significant DDI risk in vivo, resulted in a weak interaction in vivo (1.92-fold increase in AUC of theophylline). In addition to CYP1A2, theophylline is also cleared by CYP3A4, CYP2E1, and renal clearance, suggesting that use of a higher fm probe such as caffeine would result in a correct risk categorization. The fact that tadalafil induces CYP3A in vitro (Ring et al., 2005), and most likely in vivo, is a likely reason for overprediction of CYP3A4 inhibition in vivo. The reasons for the overprediction of CYP3A4 DDI risk by amiodarone and fluoxetine are unknown.
In conclusion, the results show that the use of λ/kdeg with unbound steady-state Cmax can be useful for identifying high DDI risk compounds, but this method is not applicable for accurate quantitative prediction. The presented approach does not account for probe fm by inhibited pathway, gut extraction of the probe, possible simultaneous induction, and competitive inhibition by the inhibitor. Therefore, it is less accurate for predicting DDI magnitude than other existing static methods (Fahmi et al., 2009) or physiologically based modeling. However, the presented method is expected to provide the highest risk estimate, in comparison with other methods, for a potential MBI unless significant gut extraction of the object drug exists. More compounds with weak in vivo interactions and known in vitro MBIs need to be identified to determine the false-positive or false-negative rates of this method. Because this dataset includes the currently known in vivo inhibitors that are MBIs, it would be especially important to obtain data from in vivo DDI studies of compounds in development that are MBIs to further demonstrate the utility of this method. This evaluation methodology is simple and could be used in drug discovery and development for risk assessment of MBIs without accounting for probe-specific values such as fm and gut metabolism.
Authorship Contributions
Participated in research design: Fujioka, Kunze, and Isoherranen.
Performed data analysis: Fujioka, Kunze, and Isoherranen.
Wrote or contributed to the writing of the manuscript: Fujioka, Kunze, and Isoherranen.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant P01-GM32165].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- DDI
- drug-drug interaction
- MBI
- mechanism-based inactivator
- P450
- cytochrome P450
- FDA
- U.S. Food and Drug Administration
- AUC
- area under the curve
- CLint
- intrinsic clearance
- fm
- the fraction of total clearance of the drug to which the affected P450 enzyme contributes.
- Received May 6, 2012.
- Accepted June 8, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}