


Abstract
The protoberberine alkaloid berberine carries methylenedioxy moiety and exerts a variety of pharmacological effects, such as anti-inflammation and lipid-lowering effects. Berberine causes potent CYP1B1 inhibition, whereas CYP1A2 shows resistance to the inhibition. To reveal the influence of oxidative metabolism on CYP1 inhibition by berberine, berberine oxidation and the metabolite-mediated inhibition were determined. After NADPH-fortified preincubation of berberine with P450, the inhibition of CYP1A1 and CYP1B1 variants (CYP1B1.1, CYP1B1.3, and CYP1B1.4) by berberine was not enhanced, and CYP1A2 remained resistant. Demethyleneberberine was identified as the most abundant metabolite of CYP1A1- and CYP1B1-catalyzed oxidations, and thalifendine was generated at a relatively low rate. CYP1A1-catalyzed berberine oxidation had the highest maximal velocity (Vmax) and exhibited positive cooperativity, suggesting the assistance of substrate binding when the first substrate was present. In contrast, the demethylenation by CYP1B1 showed the property of substrate inhibition. CYP1B1-catalyzed berberine oxidation had low Km values, but it had Vmax values less than 8% of those of CYP1A1. The dissociation constants generated from the binding spectrum and fluorescence quenching suggested that the low Km values of CYP1B1-catalyzed oxidation might include more than the rate constants describing berberine binding. The natural protoberberine/berberine fmetabolites with methylenedioxy ring-opening (palmatine, jatrorrhizine, and demethyleneberberine) and the demethylation (thalifendine and berberrubine) caused weak CYP1 inhibition. These results demonstrated that berberine was not efficiently oxidized by CYP1B1, and metabolism-dependent irreversible inactivation was minimal. Metabolites of berberine caused a relatively weak inhibition of CYP1.
Introduction
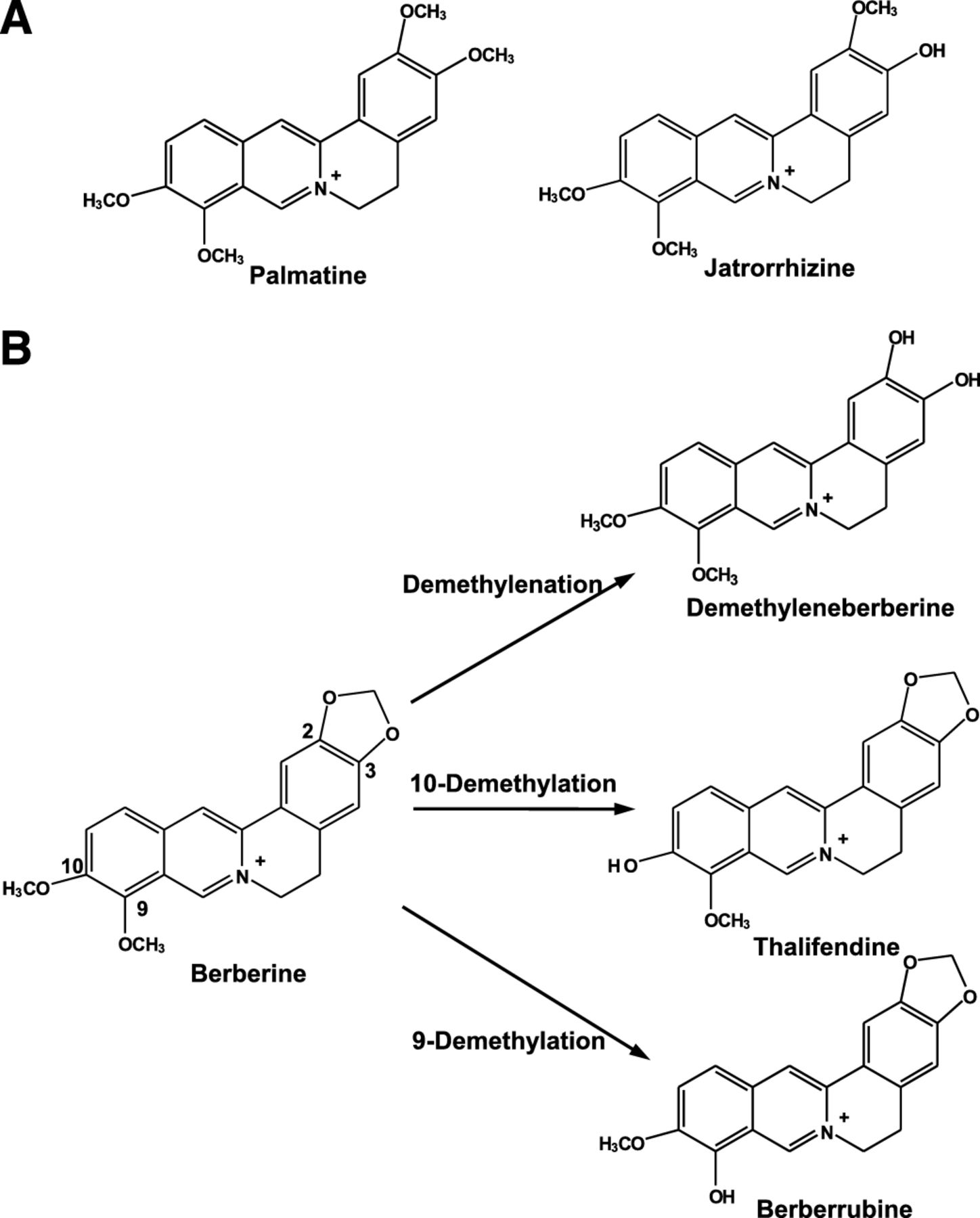
Protoberberine alkaloids, including berberine, palmatine, and jatrorrhizine, have been found in several medicinal plants, including Coptis chinensis (Huang-Lian), Berberis aristata (Indian barberry), and Hydrastis canadensis (goldenseal) (Grycova et al., 2007; Kulkarni and Dhir, 2010). Berberine appeared to be the most abundant protoberberines of Huang-Lian and goldenseal, and berberine and its metabolites were the main contributors to their pharmacological effects (Abourashed and Khan, 2001; Tang et al., 2009; Cao et al., 2013). In human urine, the glucuronide/sulfate conjugates of demethyleneberberine (demethylenation), thalifendine (10-demethylation), berberrubine (9-demethylation), and jatrorrhizine (ring breakage) have been identified (Liu et al., 2009) (Scheme 1). In HepG2 cells, the metabolites, demethyleneberberine and thalifendine, showed the inductive effect of berberine on the mRNA levels of either the insulin receptor or the low-density lipoprotein receptor, but to a lesser extent (Li et al., 2011). The 24-hour exposure to 15 μM berberrubine, jatrorrhizine, and thalifendine decreased the cellular triglyceride level in HepG2 cells (Cao et al., 2013). Unlike berberine, palmatine and jatrorrhizine do not have the methylenedioxyl substitute. Palmatine and jatrorrhizine were oxidized by liver microsomal enzymes or P450s to form the hydroxylation and/or demethylation products (Yang et al., 2009; Zhou et al., 2013). The clinical impact of their metabolites remained unclear.

Structures of palmatine and jatrorrhizine (A) and the metabolism of berberine to form demethyleneberberine, thalifendine, and berberrubine (B).
In humans and rodents, the bioavailability of berberine was low, and about half was eliminated in the form of metabolites (Ma and Ma, 2013; Spinozzi et al., 2014). In participants taking 15 mg/kg berberine chloride for 3 months, the mean steady-state plasma concentrations of the unconjugated forms of berberine, berberrubine, demethyleneberberine, and jatrorrhizine were 1.7–6.7 nM (Spinozzi et al., 2014). Zuo et al. (2006) reported that plasma concentrations of berberrubine, demethyleneberberine, jatrorrhizine, and thalifendine were generally higher than the plasma berberine concentration in rats treated orally with 40 mg/kg berberine. The area under the curve of the concentration versus time (AUC)0-t values of plasma berberine metabolites (in the unconjugated and conjugated forms) were 2–50 times greater than that of berberine. In rat livers, the AUC0-t value of demethyleneberberine was comparable to that of berberine in rats treated orally with 100 mg/kg berberine (Liu et al., 2010). At 4 hours after oral administration, the hepatic berberine and demethyleneberberine concentrations were about 0.30 μM. In the heart and kidney, the AUC0-t value of demethyleneberberine was also comparable to that of berberine. Besides the extensive hepatic metabolism of berberine, high tissue distribution might also be one of the factors leading to the low plasma concentration of berberine (Liu et al., 2010). The AUC0-t values of unconjugated berberine in the liver, kidney, lung, and spleen were 40–7530% greater than that in plasma. The AUC0-t values of the unconjugated metabolites, such as demethyleneberberine, were also higher in the liver and heart than in the plasma. The high tissue-distribution level may cause significant biochemical influence not only in the liver, but also in the extrahepatic tissues.
In the NADPH-dependent oxidation of berberine by human liver microsomal enzymes and the recombinant hepatic P450s, demthyleneberberine appeared to be the most abundant metabolite. To a lesser extent, thalifendine was also generated (Li et al., 2011) (Scheme 1). The formation of berberrubine and jatrorrhizine was below the detection limit. Among the main hepatic P450s, CYP2D6 had the highest berberine oxidation activities. Although CYP1A2 had berberine demethylenation and 10-demethylation activities of about 50–60% of those of CYP2D6, the contribution of CYP1A2, CYP2D6, and CYP3A4 to human liver microsomal demethylenation was estimated to be almost equal, and the contribution of CYP1A2 to the 10-demethylation was predominant (Li et al., 2011).
Although the liver has the most abundant P450 enzymes, extrahepatic CYP1A1 and CYP1B1 play important toxicological roles, such as the oxidation of carcinogen benzo(a)pyrene in target organs, including the breast, lung, and ovary (Shimada and Fujii-Kuriyama, 2004). Estradiol is metabolized by CYP1A1 to generate mainly the 2-hydroxylation metabolite. CYP1B1 is predominantly expressed in steroidogenic tissues and preferentially generates the genotoxic 4-hydroxyestradiol (Lee et al., 2003), which has been noticed as a marker of mammary tumors (Liehr and Ricci, 1996). Recently, CYP1A1 and CYP1B1 have also been identified as being involved in the oxidation of arachidonic acid (Choudhary et al., 2004). CYP1B1 was considered to be involved in angiotensin II-induced hypertension (Jennings et al., 2010). Using 7-ethoxyresorufin as a substrate, our previous report demonstrates that berberine potently inhibits CYP1B1, and CYP1A2 shows resistance to berberine-mediated inhibition (Lo et al., 2013). To elucidate the influence of metabolism on the berberine-mediated inhibition of human CYP1s, berberine oxidation and the CYP1 inhibition by oxidation metabolites were determined in recombinant enzyme systems.
Materials and Methods
Chemicals and Enzymes.
Berberine chloride, glucose-6-phosphate, glucose-6-phosphate dehydrogenase (yeast), and β-NADP+ sodium salt were purchased from Sigma-Aldrich (St. Louis, MO). Palmatine and jatrorrhizine were isolated and purified from Fibraurea tinctoria by T. S. Wu (Department of Chemistry, National Chung-Kung University, Tainan, Taiwan, ROC) following the method as previously described (Su et al., 2007). Berberrubine, demethyleneberberine, and thalifendine were synthesized by C. C. Sheng (Division of Chinese Medicinal Chemistry, National Research Institute of Chinese Medicine, Taipei, Taiwan, ROC) following the methods reported by Liu et al. (2009, 2010). The structures of isolated natural products and synthetic metabolites were identified by 1H-NMR and 13C-NMR analyses (Su et al., 2007; Bodiwala et al., 2011). The purities of synthetic metabolites were ≥98% based on the NMR and high-performance liquid chromatography (HPLC) analyses.
Preparation of Bacterial Membranes Expressing Human CYP1 Enzymes and CYP1B1 Variants.
The expression constructs of CYP1A1*1, CYP1A2*1, and CYP1B1*3 with N-terminal modification were generously provided by F. Peter Guengerich (Nashville, TN) (Shimada et al., 1998b). The construct for expressing CYP1B1.1 (Leu432) was prepared as described before (Lo et al., 2013). Bicistronic human constructs consisting of the coding sequence of P450 followed by that of NADPH-P450 reductase were transformed to Escherichia coli DH5α by electroporation (Gene Pulser II; Bio-Rad, Hercules, CA). The construct of CYP1B1*4 was prepared by introducing the 4390A>G substitution to CYP1B1*1 using the primer-directed enzymatic amplification method, according to the manufacturer’s instructions for the QuickChange Lightning site-directed mutagenesis kit (Stratagene, an Agilent Technologies Company, La Jolla, CA). The oligonucleotide primer sets used for mutagenesis were described in a previous report (Lo et al., 2013). P450 expression and membrane preparation were performed following the methods of Shimada et al. (1998a) and Parikh et al. (1997), respectively. The P450 content of the bacterial membrane fraction was determined following the spectrophotometric method (Omura and Sato, 1964) using a spectral determination buffer for dilution (Parikh et al., 1997).
CYP1 Activity and the Inhibitory Effect Determinations.
The 7-ethoxyresorufin O-deethylation (EROD) activities of CYP1A1/1A2/1B1 were determined using 5 pmol P450/ml. The generation of the metabolite resorufin was determined by measuring the increase of the fluorescence intensity (excitation wavelength: 550 nm; emission wavelength: 585 nm) (Pohl and Fouts, 1980). For the determination of the inhibitory effect, P450 activity was determined in the incubation with or without NADPH-fortified preincubation. The NADPH-fortified preincubation consisted of P450, increasing concentrations of protoberberines, and a NADPH-generating system in 50 mM HEPES buffer, pH 7.85. The NADPH-generating system comprised 0.5 mM NADP+, 5 mM glucose-6-phosphate, and 1.0 U/ml glucose-6-phosphate dehydrogenase. After a 10-minute preincubation, the substrate 7-ethoxyresorufin (2 μM) was added into the reaction mixtures and the O-deethylation activity was determined, as described above.
Berberine Oxidation.
Berberine oxidation activity was determined using a method modified from the method reported by Li et al. (2011). Berberine chloride was dissolved in dimethyl sulfoxide. Berberine oxidation activity was determined in an incubation mixture (0.5 ml) consisting of 50 mM potassium phosphate buffer, pH 7.4, 20 pmol/ml P450, 0.5 mM NADP+, 5 mM glucose-6-phosphate, 10 mM MgCl2, and various concentrations of berberine chloride. The oxidation was initiated by the addition of 1.0 U/ml glucose-6-phosphate dehydrogenase and incubated at 37°C for 30 minutes. Reaction was terminated by the addition of 1 ml methanol. After centrifugation at 13,800g for 10 minutes at 4°C, the supernatant was subjected to HPLC analysis using a HPLC system (Hitachi High-Tech, Tokyo, Japan) equipped with a C18 column (Cosmosil, 4.6 mm × 25 cm, 5 μm; Nacalai Tesque, Kyoto, Japan) and an absorbance detector (Hitachi 7420, Tokyo, Japan). Metabolites were separated by 17.5% acetonitrile and 10% methanol in 20 mM sodium phosphate buffer, pH 3.0, at a flow rate of 1 ml/min. The production of demethyleneberberine and thalifendine was determined by measuring the absorbance at 346 nm, and the quantification was performed using the synthetic metabolite standards. Based on the high berberine oxidation activity of CYP1A1, the oxidation products generated by CYP1A1 were further identified using a liquid chromatography-mass spectrometry system. A HPLC system (Chromaster; Hitachi High-Tech) was equipped with an absorbance detector (SPD-20A; Shimadzu, Kyoto, Japan) and a mass spectrometer (ExpressionS CMS; Adivon, Ithaca, NY). The metabolites were separated with a mobile phase of 35% methanol containing 0.1% formic acid at a flow rate of 0.2 ml/min. Ions were detected using the positive ion mode with an electrospray voltage of 3500 V and gas temperature at 250°C. The generation of demethyleneberberine and thalifendine was identified by a selected ion mode with the m/z range of 324–325 and 322–323, respectively.
Intrinsic Fluorescence Measurements.
The binding of berberine to P450 in membrane fractions was monitored by determining the decrease of intrinsic fluorescence intensity (fluorescence quenching) using a method modified from the report of Liu et al. (2012). CYP1A2 and CYP1B1 variant-expressed bacterial membrane fractions were diluted to 0.2 μM P450 using 0.1 M potassium phosphate buffer (pH 7.4). The emission fluorescence spectra of P450 were measured between 280 and 400 nm (excitation wavelength at 295 nm). The emission wavelength with maximal fluorescence intensity was used for the following determination of fluorescence quenching. The fluorescence intensity of bacterial membrane fractions expressing P450 was measured at 336 nm. The reduction of fluorescence intensity by the addition of increasing concentrations of berberine (I) was calculated. The dissociation constants (Kd) were estimated by nonlinear regression analysis (SigmaPlot 11.0; Jandel Scientific, San Rafael, CA), according to the equation for a single binding site: F0 − F = ΔFmax × I/(Kd + I), where F0 and F are the fluorescence intensities before and after the addition of berberine, respectively. ΔFmax is the maximal value of (F0 − F) (Sampedro et al., 2007). The estimates of variance (denoted by ±) are presented from the analysis of individual sets of data.
Binding Spectral Analysis.
The spectral changes due to the binding of berberine to recombinant P450 were determined by the titration of CYP1 enzymes with increasing concentrations of berberine at ambient temperature. The absorbance was measured in a wavelength scan mode using a spectrophotometer with a double beam (U3000; Hitachi, Tokyo, Japan). To prevent the absorbance interference of berberine, tandem (tandem/divided chambers) cuvettes were used. Both reference (without binding) and sample (with binding) cuvettes contained P450 and berberine in the buffer solution. Each side of a tandem cuvette contained 1 μM P450 (in 0.1 M potassium phosphate buffer, pH 7.4) and various concentrations of berberine diluted in 0.1 M potassium phosphate buffer, pH 7.4. Baseline correction was carried out in the absence of berberine. After the addition of berberine in the buffer chamber, solutions in the two chambers remained separate in the reference cuvette. In the sample cuvette, berberine and P450 solutions on each chamber were completely mixed, and the spectrum was recorded. The absorbance difference ∆A372–425 nm and ∆A370–420 nm was determined for the binding of berberine to CYP1A2 and CYP1B1, respectively. The spectral dissociation constant Ks was determined by nonolinear least-squares regression without weight according to the equation: ∆A = ∆Amax × S/(Ks + S), where S is the concentration of berberine or demethyleneberberine.
Data Analyses.
The concentration of an inhibitor required for causing a 50% decrease of catalytic activities (IC50) was calculated by curve fitting (Grafit; Erithacus Software, Staines, UK). The statistical significance of the differences between the two groups of IC50 values was evaluated using Student’s t test. Kinetic analysis of the pattern with positive cooperativity was performed following the nonlinear least-squares regression (SigmaPlot software) based on the equation: v = (Vmax × Sn)/(S50n + Sn), where S50 is the substrate concentration showing half of the maximal velocity (Vmax) and the Hill coefficient n is a measure of cooperativity. For substrate inhibition, the kinetics can be regarded as a form of uncompetitive inhibition by the substrate (Houston and Kenworthy, 2000). Values of velocities (v) at various substrate concentrations (S) were fitted by nonlinear least-squares regression without weight according to the equation: v = Vmax × S/{Km + S[1 + (S/Ki)]}, where Ki was the inhibitor constant. The 10-demethylation activity of CYP1B1 was too low to be determined at a berberine concentration less than 1 μM. Thus, the hyperbolic plot was analyzed following the Michaelis-Menten equation: v = Vmax × S/(Km + S). Estimates of variance (denoted by ±) are presented from the analysis of individual sets of data.
Results
The Inhibition after NADPH-Fortified Preincubation of CYP1 with Protoberberines.
Recombinant CYP1A1, CYP1A2, and CYP1B1.1 had EROD activities of 77.9 ± 5.5, 4.7 ± 1.2, and 9.0 ± 2.7 nmol/min/nmol P450, respectively. NADPH-fortified preincubation provides an incubation of the CYP1 system with protoberberines in the presence of the NADPH-generating system. This preincubation allows the preoxidation of protoberberines by CYP1 to be investigated regarding the influence of oxidative metabolism on the CYP1 inhibition by themselves. The inhibition of CYP1A1 by berberine, palmatine, and jatrorrhizine was decreased by the preoxidation (Table 1). The inhibition of CYP1B1 by berberine and jatrorrhizine was also decreased by the preoxidation. The IC50 values were increased by 51–157%, probably due to the metabolism of protoberberines forming metabolites with fewer inhibitory effects than the parent chemicals. With the preoxidation for the same time period (10 minutes), berberine caused more potent inhibition of CYP1B1 than palmatine and jatrorrhizine did. Even with preoxidation, CYP1A2-catalyzed EROD activity was still resistant to the inhibition by protoberberines. Among CYP1 enzymes, berberine preferentially inhibited CYP1B1 activity in the enzyme assay with and without the NADPH-fortified preincubation of P450 with berberine (Table 1). Unlike berberine, palmatine and jatrorrhizine with the methylenedioxy ring opening did not show preference in CYP1B1 inhibition. Because berberine caused potent inhibition of CYP1B1, the influence of preoxidation on the inhibition of CYP1B1 variants by berberine was further studied. CYP1B1.3 (10.3 ± 1.5 nmol/min/nmol P450) and CYP1B1.4 (16.7 ± 2.1 nmol/min/nmol P450) had EROD activities similar to CYP1B1.1. The preoxidation with berberine did not cause obvious decreases in the IC50 values for the inhibition of CYP1B1.3 and CYP1B1.4 activities (Table 1).
Effect of preoxidation on the inhibition of 7-ethoxyresorufin O-deethylation activity of CYP1A1, CYP1A2, and CYP1B1 variants by berberine, palmatine, and jatrorrhizine
IC50 values were calculated by graph-fitting software. Estimates of variance generated from the curve fitting of individual sets of data were less than 15%. Data represent the mean ± S.E. of three determinations.
Berberine Demethylenation and 10-Demethylation Catalyzed by CYP1 Enzymes.
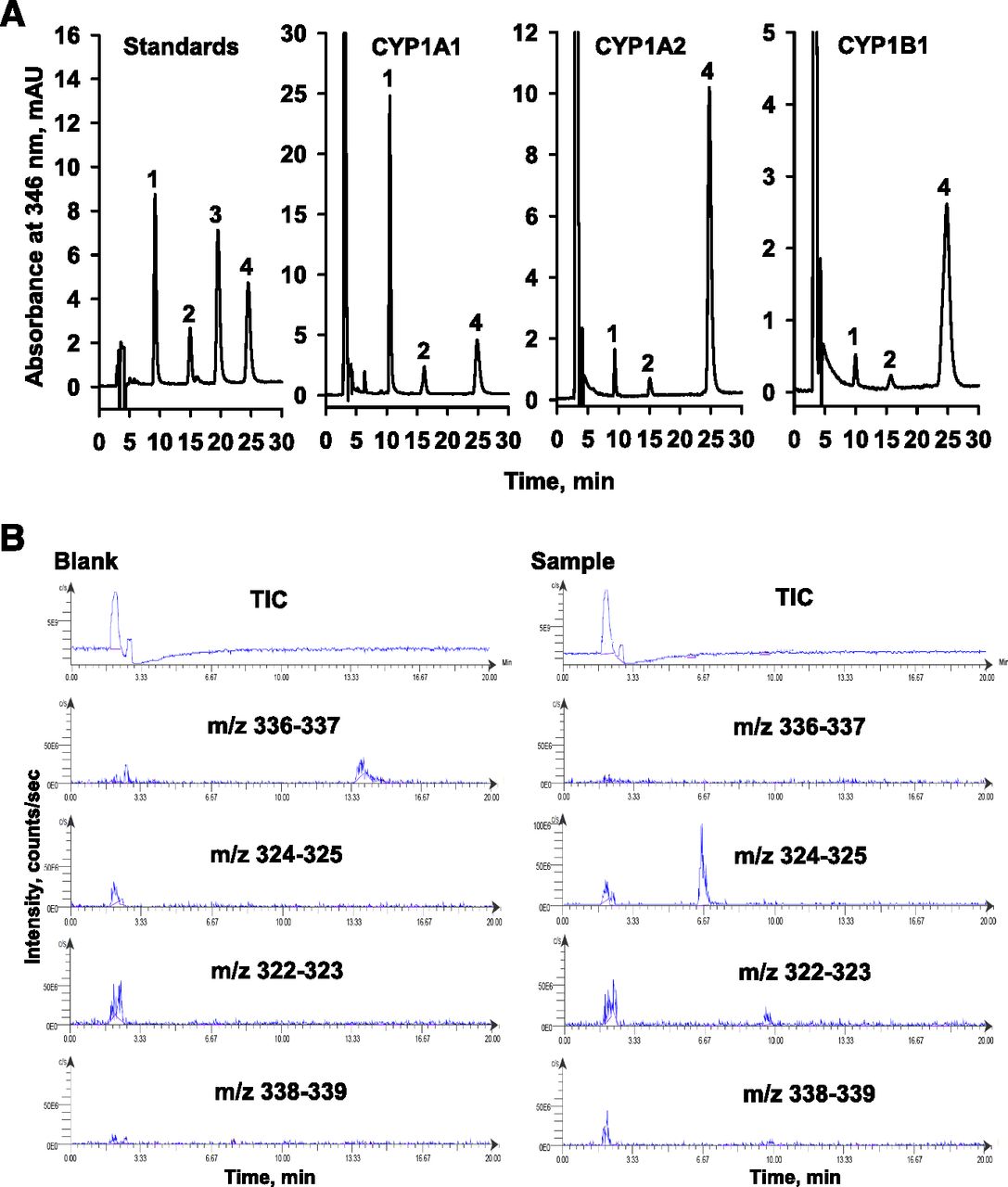
HPLC analysis revealed that CYP1A1-catalyzed berberine oxidation generated demethyleneberberine (demethylenation) and thalifendine (10-demethylation), whereas berberrubine was not detected (Fig. 1A). The formation of demethyleneberberine and thalifendine has been further identified by liquid chromatography-mass spectrometry analysis (Fig. 1B). In CYP1A1-catalyzed berberine demethylenation and 10-demethylation, the plots of velocity versus berberine concentration were sigmoidal (Fig. 2, upper panel). The Lineweaver-Burk plots showed a nonlinear pattern and the Eadie-Hofstee plot showed a convex curve, indicating positive cooperativity. Nonlinear regression analysis of the plot of velocity versus berberine concentration generated kinetic parameters for demethylenation and 10-demethylation with an r value of 0.99 (Table 2). In CYP1B1-catalyzed demethylenation, the velocity of CYP1B1 decreased at berberine concentrations higher than 7.5 μM, suggesting a substrate inhibition pattern (Fig. 2, bottom panel). Nonlinear regression analysis according to substrate inhibition generated the parameters with r = 0.99 (Table 2). The plot of CYP1B1-catalyzed 10-demethylation velocity versus berberine concentration was hyperbolic, and nonlinear regression analysis following the Michaelis-Meten equation generated the parameters with r = 0.99. The maximal demethylenation and 10-demethylation activities (Vmax) of CYP1B1 were only 5% and 8% of those of CYP1A1, respectively. The Km values of demethylenation and 10-demethylation by CYP1B1 were 4% and 48% of those by CYP1A1, respectively.

The chromatrograms of HPLC (A) and liquid chromatography-mass spectrometry (B) analyses of berberine oxidation products generated by CYP1A1, CYP1A2, and CYP1B1. In a 0.5 ml assay without NADPH, the incubation mixture containing 2.7 μM demethyleneberberine (1), 2.4 μM thalifendine (2), 2.4 μM berberrubine (3), and 4.8 μM berberine (4) was prepared following the assay method and used as the external standards for peak identification and quantification. The chromatograms of P450-generated berberine oxidation products show the chromatograms obtained from the reaction using 20 nM P450 and 10 μM berberine. The berberine oxidation products generated by CYP1A1 were further analyzed by liquid chromatography-mass spectrometry. Berberine oxidation was performed using 50 nM P450 and 10 μM berberine. After incubation for 30 minutes, the reaction product was subjected to liquid chromatography-mass spectrometry analysis. The blank incubation was carried out in the absence of NADPH. A selected ion mode was used to find the ionized berberine (m/z = 336–337), demethyleneberberine (m/z = 324–325), thalifendine (m/z = 322–323), and jatrorrhizine (m/z = 338–339).

Kinetic analyses of berberine demethylenation and 10-demethylation by human CYP1A1 and CYP1B1. The plots of velocity (v) versus berberine concentration (S) of CYP1-catalyzed berberine demethylenation, the 1/v versus 1/S (Lineweaver-Burk plot), and v versus v/S (Eadie-Hofstee plot) are shown in the right, middle, and left panels, respectively. Lines show the best fit as determined by nonlinear least squares regression according to the cooperative oxidation and substrate inhibition for CYP1A1 and CYP1B1, respectively. Data represent the mean ± S.E. of three to four determinations.
Kinetic parameters of berberine demethylenation and 10-demethylation by CYP1A1 and CYP1B1
Kinetic parameters were determined by the nonlinear regression analysis according to each kinetic pattern, as described in Materials and Methods. Data represent the values and the estimated variances (denoted by ±).
Inhibition of the 7-Eoxyresorufin O-Deethylation Activity of CYP1 by Demethyleneberberine, Thalifendine, and Berberrubine.
In the assays of CYP1 activity, the presence of demethyleneberberine, thalifendine, and berberrubine all resulted in a preferential inhibition of CYP1B1 with IC50 values between 8.0 and 26.7 μM (Table 3). Thalifendine inhibited CYP1A1 activity with an IC50 value of 30.4 ± 1.2 μM, whereas demethyleneberberine and berberrubine had IC50 values higher than 60 μM. Compared with the results with the parent compound berberine (Table 1), inhibition of CYP1A1/CYP1B1 by metabolites of berberine was relatively weak. The EROD activity of CYP1A2 was not inhibited by demethyleneberberine, thalifendine, and berberrubine at the concentration up to 60 μM.
Inhibition of the 7-ethoxyresorufin O-deethylation activity of CYP1 by demethyleneberberine, thalifendine, and berberrubine
Inhibition of CYP1-catalyzed 7-ethoxyresorufin O-deethylation activity was studied in the assays without NADPH-fortified preincubation. IC50 value was calculated, as described in Materials and Methods. Data show the estimates and estimates of variance (denoted by ±) determined from the curve fit analysis of individual sets of data.
Quenching of the Fluorescence of CYP1A2 and CYP1B1 Variants with Berberine.
To examine the interaction of CYP1A2 and CYP1B1 variants with berberine, the quenching of tryptophan fluorescence of the apoprotein of P450 by berberine was determined. CYP1B1.3 and CYP1B1.4 carry mutations at positions away from the berberine-binding site in the apoprotein of P450. Upon excitation at 295 nm, bacterial membranes expressing recombinant CYP1B1 variants had intrinsic fluorescence emission peaks at 336–343 nm. The dissociation constant (Kd) was calculated by determining the fluorescence quenching, presumably reflecting the binding of berberine to P450. The plots of fluorescence quenching versus berberine concentrations were hyperbolic (plot not shown). The apparent Kd values for the interaction of berberine with CYP1B1 variants were 14.6–15.2 μM (Table 4). The differences between the binding affinities of berberine to CYP1B1 variants were minimal. However, the Kd value for the interaction of berberine with CYP1A2 was 18.2 ± 1.3 μM, which was 20–25% higher than the values for CYP1B1 variants.
The fluorescence quenching of CYP1A2 and CYP1B1 variants by the binding of berberine in the recombinant enzyme systems
Data show the estimates and estimates of variance (denoted by ±) determined from the curve fit analysis of individual sets of data.
The Spectral Changes Due to the Binding of Berberine to CYP1A2 and CYP1B1.
In contrast to the inhibition resistance and high oxidation rate of CYP1A2 when a high concentration of berberine was present (Li et al., 2011), CYP1B1 was potently inhibited by berberine and had a low berberine oxidation rate. To examine the spin state changes in hemoproteins of CYP1A2 and CYP1B1.1 (L432), spectral changes due to the binding of berberine to P450 were studied. The absorbance spectrum as seen with berberine was analyzed first. Berberine had a maximal absorbance at 420 nm, and this spectral property interfered with the binding spectral analysis (Fig. 3A). By using a set of tandem cuvettes to measure the binding spectra, spectral analyses revealed that CYP1A2 and CYP1B1 generated a difference spectrum with the maximal absorbance at 372 and 370 nm, respectively (Fig. 3, B and C). The spectral troughs were at 425 and 420 nm for the binding to CYP1A2 and CYP1B1, respectively. Unexpectedly, the binding of berberine to CYP1A2 and CYP1B1 caused the same type of spectral change, and the binding spectra showed the characteristics of type I spectrum. However, another absorbance peak was observed at ∼470 nm, which was not present in a typical type I spectrum. The dissociation constant (Ks) values were calculated from the absorbance difference of the ∆A372–425 nm and ∆A370–420 nm for the binding to CYP1A2 and CYP1B1, respectively (Fig. 3, B and C). The Ks values for the binding of berberine to CYP1A2 and CYP1B1 were 10.1 ± 2.5 and 7.4 ± 1.3 μM, respectively. CYP1B1 had a spectral dissociation constant 27% lower than the value of CYP1A2.

The spectral difference due to interactions of berberine with CYP1A2 and CYP1B1. (A) shows the absorbance spectrum of berberine. To prevent the interference of the absorbance spectrum of berberine, tandem cuvettes were used for the detection of the difference spectrum resulting from the interaction of berberine with P450. The left panels of (B) and (C) show the spectra recorded at room temperature after the addition of increasing concentrations of berberine to diluted bacterial membrane fractions containing 0.5 μM CYP1A2 and CYP1B1, respectively. The same amount of berberine was added to the buffer chamber in the reference cuvette. The right panels of (B) and (C) show the plot of absorbance versus berberine concentration, and the solid line shows the best fit estimated from the curve fitting.
Discussion
The methylenedioxy phenyl derivatives have been reported to generate a complex of carbene metabolite intermediate and P450, resulting in a potent and irreversible inhibition after preoxidation (Murray, 2000). However, in human liver microsomes, results of spectral analysis indicated that no P450-MI complex was formed after NADPH-fortified microsomal incubation with berberine (Chatterjee and Franklin, 2003). Our findings revealed that the IC50 values were increased when berberine was preoxidized by CYP1A1/1B1, indicating that berberine was not a mechanism-based inhibitor of CYP1A1/1B1. Thus, carbene intermediate may not be generated during the demethylenation of berberine. Berberine oxidation by CYP1A1 and CYP1B1 generated demethyleneberberine and thalifendine. Demethyleneberberine and thalifendine inhibited CYP1B1 with a potency less than berberine did. The increased IC50 values after preoxidation of inhibitors can be attributed at least in part to the oxidation forming inactivated metabolites. Palmatine and jatrorrhizine do not carry the methylenedioxy moiety. Among the primary hepatic P450 forms, CYP1A2 showed the highest demethylation activity toward jatrorrhizine at a high concentration of 500 μM (Zhou et al., 2013). However, our findings revealed that jatrorrhizine at a concentration up to 60 μM could not compete with a substrate 7-ethoxyresorufin to cause CYP1A2 inhibition. Like berberine, the preoxidation reduced the CYP1A1 and CYP1B1 inhibition by jatrorrhizine and CYP1A1 inhibition by palmatine. The CYP1B1 inhibition by palmatine was less affected by the preoxidation. Palmatine and jatrorrhizine were not a mechanism-based inhibitor of CYP1A1/1B1, either.
The ratios (in parentheses) of IC50 values for CYP1B1 to CYP1A1 inhibition by the derivatives without the methylenedioxy ring were in the order of palmatine (5.4) > jatrorrhizine (1.3) > demethyleneberberine (< 0.13). The number (in parentheses) of methoxyl substitute added was in the order of palmatine (2) > jatrorrhizine (1) > demethyleneberberine (0). A greater methoxyl addition to the dihydroisoquinolino moiety seems to diminish the preference in CYP1B1 inhibition. With the methylenedioxyl substitute at C2 and C3, the monodemethylation at 9-(thalifendine) and 10-(berberubin) positions suppressed the CYP1A1 and CYP1B1 inhibitory effects, but remained the CYP1B1 preferential inhibition. Interestingly, none of the protoberberines or berberine metabolites inhibited CYP1A2. Based on our previous report of the docking of berberine to the putative CYP1B1 active site (Lo et al., 2013), the binding of derivatives/metabolites of berberine to CYP1B1 was predicted. Results of the best docking showed that the two hydroxyl side chains at C2 and C3 of demethyleneberberine were expected to be rotated more freely than the methylenedioxy ring (Supplemental Fig. 1, A and B). Among the derivatives and metabolites of berberine, palmatine caused the least inhibition of CYP1B1 activity. Using the ASP scoring (Korb et al., 2009) function of the ligand-docking software, the docking of palmatine to the putative binding site of CYP1B1 had the lowest score of 26.4. The lowest score of palmatine was consistent with its poor CYP1B1-inhibitory effect. The docking scores of berberine, jatrorrhizin, demethyleneberberine, thalifendine, and berberrubine were 38.2, 38.6, 37.8, 37.7, and 36.6, respectively. The docking scores were in the trend consistent with their IC50 values for CYP1B1 inhibition (berberine < jatrorrhizin < demethyleneberberine ≈ thalifendine < berberrubine). The presence of a bump or clash between heme of the P450 and the methoxyl substitution at C3 was predicted by docking monitoring (Supplemental Fig. 1D). This interaction may be associated with the relatively weak inhibition by palmatine. Thus, besides the important hydrogen bond formation of P450 with methoxyl substitutes at C9 and C10, the methylenedioxyl moiety of berberine provides the potential hydrophobic interaction (Lo et al., 2013) and a nonfree-rotating planar structure, resulting in the potent inhibition of CYP1B1 by berberine. These criteria can be important in the further drug development of a CYP1B1 selective inhibitor for the potential hypotensive and chemopreventive effects (Shimada and Fujii-Kuriyama, 2004; Jennings et al., 2010).
In a study of supersomes, CYP1A2-catalyzed berberine demethylenation had Km and Vmax of 126 μM and 4.8 nmol/min/nmol P450, respectively (Li et al., 2011). The 10-demethylation had Km and Vmax values of 100 μM and 5.4 nmol/min/nmol P450, respectively. Compared with the velocities of CYP1A1 and CYP1A2, the Vmax of CYP1B1-mediated berberine oxidation was very low. Due to the opposite position of methylenedioxyl and the 10-methoxyl groups of berberine, the generation of demethyleneberberine and thalifendine requires two binding orientations of berberine. Results of docking analysis consistently supported the orientation preference in the demethylenation by CYP1B1. Although CYP1B1 exhibited a low Km value for berberine oxidation and was potently inhibited by berberine, berberine was a poorly oxidized substrate of CYP1B1. CYP1A1 had a high Vmax and a Km value within a range between those of CYP1B1 and CYP1A2. Unexpectedly, the Vmax value of CYP1A1 was greater than that of CYP2D6 reported by Li et al. (2011). Although the difference in the alternative expression systems can be an influencing factor of the Vmax value, CYP2D6 and CYP1A1 may play crucial roles in the berberine oxidation in respective high-expression tissues due to their differential tissue distribution. Although CYP1A2 exhibited a relatively high Km value in berberine oxidation and showed resistance to the inhibition by berberine (Li et al., 2011; Lo et al., 2013), the maximal oxidation velocity by CYP1A2 was about five-fold greater than that of CYP1B1. However, the intrinsic clearance (Kcat/Km) of berberine by CYP1A2 was relatively low. Thus, upon a high-exposure concentration of berberine in the liver, CYP1A2 may appear to efficiently metabolize berberine. However, upon a low-exposure concentration of berberine, CYP1A1- and CYP1B1-catalyzed oxidations may become important, especially in the extrahepatic tissues. The plots of velocity versus berberine concentration of CYP1A1-catalyzed berberine demethylenation and 10-demethylation activities showed a sigmoidal pattern, and the Eadie-Hofstee plots of both activities showed a convex property, suggesting that there may be two substrate binding sites or that the presence of a substrate caused a conformational change of CYP1A1. The binding of the first substrate may assist the binding of a second substrate to CYP1A1. However, to date, there was no report supporting that there were two substrate binding sites in CYP1A1. In CYP1B1-catalyzed berberine demethylenation, the activity decreased with the increased berberine concentration. Together with the potent inhibition of CYP1B1-catalyzed EROD activity by berberine (Lo et al., 2013), these results reveal that the second substrate binds to the inhibitor site, which is nonproductive. Consistent with the results reported by Li et al. (2011), our determination of CYP1A2-catalyzed berberine oxidation showed the hyperbolic property of Michaelis-Menten kinetics (data not shown). These results demonstrate the differential kinetic properties of berberine oxidation by CYP1 isoforms, which can be crucial in the assessment of berberine oxidation in vivo.
Due to the difference of CYP1A2 and CYP1B1 being substrate and inhibitor, the interactions of berberine with heme and the apoprotein of CYP1A2/1B1 were analyzed with regard to the binding spectrum and fluorescence quenching, respectively. As a methylenedioxy phenyl moiety-carrying natural product, gomisin caused a mechanism-based inhibition of CYP3A4 and showed a marker type III binding spectrum with the characteristic absorbance peaks at 455 and 430 nm (Iwata et al., 2004). However, the amine oxidation of methylenedioxymethamphetamine caused a mechanism-based inhibition of CYP2D and showed the type I binding spectrum, in which the peak and through of the spectrum appeared at wavelengths of about 390 and 420 nm, respectively (Delaforge et al., 1999). Structurally diverse methylenedioxyl derivatives caused differential binding spectra. Berberine was not a mechanism-based inhibitor of CYP1B1, and it appeared to be an inhibitor rather than an efficiently oxidized substrate. A ligand showing the type II binding spectrum was suggested to be an inhibitor and exhibited low turnover rates (Chiba et al., 2001). The type II binding spectrum was suggested to result from the potent coordination to the heme, which leads to the shift of hemoprotein to a low-spin dominant form. This spin state change may increase the redox potential and lead to the relatively low oxidation rate of the ligand. Both the spectral change due to the binding of berberine to inhibition-resistant CYP1A2 and sensitive CYP1B1 elicited the same pattern of the type I spectrum. The type I spectrum indicated that the binding of berberine to CYP1A2 and CYP1B1 caused the shift in equilibrium to a high-spin hemoprotein and that berberine tends to be the substrate of CYP1A2 and CYP1B1. Compared with the 20–27% differences in the dissociation constants (Kd, Ks) from the binding analysis, the difference in Km values of berberine oxidation by CYP1B1 and CYP1A2 was large, especially in the demethylenation. The big difference in Km may be attributed to factors beyond the rate constants describing berberine binding, in which the formation of activated Michaelis complex and product release may be involved (Yun et al., 2000).
In conclusion, our findings reveal that berberine is not a mechanism-based inhibitor of human CYP1 and it is poorly oxidized by CYP1B1. Among CYP1 isoforms, CYP1A1 had the highest berberine oxidation activity comparable to that of hepatic CYP2D6. CYP1A1-mediated oxidation can be important for the biologic effect of berberine, especially in extrahepatic tissues. None of the berberine metabolites caused CYP1A1 and CYP1B1 inhibition as potently as berberine did, indicating that berberine-mediated inhibition can be eliminated through metabolism.
Acknowledgments
We sincerely appreciate Dr. F. Peter Guengerich (Vanderbilt University, Nashville, TN) for generously providing the expression constructs of human P450s.
Authorship Contributions
Participated in research design: Ueng.
Conducted experiments: Lo, Shen, Chang, Huang, Wu, Ueng.
Performed data analysis: Lo, Shen, Chang, Tsai, Huang, Wu, Ueng.
Wrote or contributed to the writing of the manuscript: Ueng, Tsai.
Footnotes
- Received February 24, 2015.
- Accepted May 7, 2015.
S.-N.L. and C.-C.S. contributed equally to this work.
This work was supported by Ministry of Science and Technology, Taiwan [Grants NSC101-2320-B-077-001-MY3 and MM10211-0226] and National Research Institute of Chinese Medicine, Taiwan [Grants D123101].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- EROD
- 7-ethoxyresorufin O-deethylation
- HPLC
- high-performance liquid chromatography
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}