


Abstract
AZD7325 [4-amino-8-(2-fluoro-6-methoxyphenyl)-N-propylcinnoline-3-carboxamide] is a selective GABAAα2,3 receptor modulator intended for the treatment of anxiety disorders through oral administration. An interesting metabolic cyclization and aromatization pathway led to the tricyclic core of M9, i.e., 2-ethyl-7-(2-fluoro-6-methoxyphenyl)pyrimido[5,4-c]cinnolin-4(3H)-one. Further oxidative metabolism generated M10 via O-demethylation and M42 via hydroxylation. An authentic standard of M9 was synthesized to confirm the novel structure of M9 and that of M10 and M42 by liver microsomal incubation of the M9 standard. Metabolites M9, M10, and M42 were either minor or absent in plasma samples after a single dose; however, all became major metabolites in human and preclinical animal plasma after repeated doses and circulated in humans longer than 48 hours after the end of seven repeated doses. The absence of these long circulating metabolites from selected patients’ plasma samples was used to demonstrate patient noncompliance as the cause of unexpected lack of drug exposure in some patients during a Phase IIb outpatient clinical study. The observation of late-occurring and long-circulating metabolites demonstrates the need to collect plasma samples at steady state after repeated doses when conducting metabolite analysis for the safety testing of drug metabolites. All 12 major nonconjugate metabolites of AZD7325 observed in human plasma at steady state were also observed in dog, rat, and mouse plasma samples collected from 3-month safety studies and at higher exposures in the animals than humans. This eliminated concern about human specific or disproportional metabolites.
Introduction
Selective allosteric modulators of GABAAα2,3 were discovered and developed at AstraZeneca, aiming for anxiety disorder treatment with benzodiazepine-like efficacy and fast onset but reduced sedation and abuse liabilities (Alhambra et al., 2011; Jucaite et al., 2017). Among a few candidate drugs discovered, 4-amino-8-(2-fluoro-6-methoxyphenyl)-N-propylcinnoline-3-carboxamide (AZD7325) was advanced to Phase IIb clinical trials. In support of the drug development of AZD7325, drug metabolite profiling and identification were conducted in plasma samples of humans and animal species used for preclinical safety testing.
Since 2008, quantitative assessment of exposure coverage of human metabolites in safety testing animals has been recommended by regulatory agencies (Gao et al., 2013; Timmerman et al., 2016), apparently derived from an earlier initiative by the pharmaceutical industry on drug metabolites in safety testing (MIST; Baillie et al., 2002). Both the revised guidance by the US Food and Drug Administration in 2016 (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079266.pdf) and the ICH guideline M3(R2) that was previously adopted by the European Medicines Agency (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf) have recommend an adequate exposure coverage in preclinical animal species for human metabolites observed at exposures greater than 10% of the total drug-related exposure. Various analytical approaches alternate to quantitative bioanalysis have been practiced for MIST by pharmaceutical industry laboratories. LC-MS-based approaches without the need of metabolite standards have been nicely summarized in the literature (Ma and Chowdhury, 2011; Gao et al., 2013). One approach adjusts the LC-MS signal of a metabolite with its MS signal response factor relative to the parent drug, as determined by correlating the MS and radiochromatographic signal responses in radiolabeled metabolite studies (Yu et al., 2007; Yi and Luffer-Atlas, 2010). Another method more efficient in time and resources is the direct comparison of the LC-MS signal of metabolite samples between different species after adding blank plasma of different species to each other to eliminate potential differences in sample matrix effect (Gao et al., 2010; Ma et al., 2010, Haglund et al., 2014).
This present study does not introduce any new approach in quantitative evaluation on MIST. Instead, some interesting findings arise from the analysis of drug metabolites of AZD7325 after repeated doses, i.e., the MIST analysis at steady state as recommend by the United States regulatory agency (Gao et al., 2013). It has been known in the literature that the level and ratio of metabolites at steady state may be different from that after a single dose (Griffini et al., 2010; Gong et al., 2016). Some metabolites may have a much longer half-life in the systemic circulation than their parent drug. For example, two slowly formed and slowly eliminated active metabolites were reported for the calcium sensitizer levosimendan (Antila et al., 2004). Also reported was a trace but long-circulating metabolite, thiocyanate anion, that was formed by decyanation of the checkpoint kinase 1 inhibitor GDC-0425 (Takahashi et al., 2017). In the present study, three metabolites of AZD7325 were either minor or absent from plasma samples after a single dose; however, they were all major circulating metabolites after repeated doses in humans and preclinical animals. Furthermore, they are unusual metabolites formed via a metabolic cyclization and aromatization pathway. These unusual metabolites would have passed uncharacterized or even unnoticed if we had only conducted metabolite analysis after a single dose.
Materials and Methods
AZD7325 Materials and Metabolite Standards
AZD7325 is a candidate drug of AstraZeneca (Chapdelaine et al., 2007, Example Number 64 in the patent; Alhambra et al., 2011, Compound 40 in the article). The preparation of [14C]-AZD7325 and the chemical synthesis of metabolite M9’s authentic standard are reported elsewhere (Artelsmair et al., 2018). However, the synthesis route for the M9 standard and a portion of two-dimensional NMR 1H–15N heteronuclear multiple bond correlation spectrum of the M9 synthetic standard are available in the supplemental material (Supplemental Fig. 1).
The M4 standard was prepared by the following procedures.
4-Amino-8-bromocinnoline-3-carboxylic acid.
A solution of 5.00 g (16.2 mmol) of 4-amino-8-bromo-N-propylcinnoline-3-carboxamide in 20 ml of 80% sulfuric acid was heated at 100°C for 18 hours. The reaction was cooled to room temperature and poured into 200 ml of ice water. The yellow solid was collected, washed with cold water, and dried to afford 5.25 g (14.3 mmol, 89%) of 4-amino-8-bromocinnoline-3-carboxylic acid as the sulfate salt. The compound was characterized by 1H NMR in DMSO-d6.
4-Amino-8-bromo-N-(2-(tert-butyldimethylsilyloxy)propyl)cinnoline-3-carboxamide.
A solution of 740 mg (2.02 mmol) of 4-amino-8-bromocinnoline-3-carboxylic acid hydrogen sulfate and 459 mg (2.83 mmol) of 1,1′-carbonyldiimidazole in 5 ml of dry dimethylformamide at 0°C was warmed to room temperature and stirred for 4 hours. The resulting suspension was cooled to 0°C, and 574 mg (3.03 mmol) of 2-(tert-butyldimethylsilyloxy)propan-1-amine was added. The reaction mixture was warmed to room temperature and stirred for 2 hours. The reaction was diluted with water and extracted with ethyl acetate. The combined organic extracts were dried with sodium sulfate, filtered, and concentrated. The residue was purified by flash chromatography on silica gel (ethyl acetate-hexane) to afford 749 mg (1.70 mmol, 84%) of 4-amino-8-bromo-N-(2-(tert-butyldimethylsilyloxy)propyl)cinnoline-3-carboxamide as a white powder. The compound was characterized by 1H NMR in DMSO-d6.
4-Amino-N-(2-(tert-butyldimethylsilyloxy)propyl)-8-(2-fluoro-6-methoxyphenyl)cinnoline-3-carboxamide.
A solution of 200 mg (0.46 mmol) of 4-amino-8-bromo-N-(2-(tert-butyldimethylsilyloxy)propyl)cinnoline-3-carboxamide, 741 mg (2.28 mmol) of cesium carbonate, 193 mg (1.14 mmol) of 2-fluoro-5-methoxy-phenylboronic acid), and 32.3 mg (45.5 µmol) of bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II) in 6 ml of a 7:2:3 mixture (v/v) of tetrahydrofuran (THF), isopropanol, and water was heated at 60°C for 1 hour. The reaction was then cooled to room temperature, diluted with water, and extracted with ethyl acetate. The organic residue was purified by flash chromatography on silica gel to afford 200 mg (0.41 mmol, 91%) of 4-amino-N-(2-(tert-butyldimethylsilyloxy)propyl)-8-(2-fluoro-6-methoxyphenyl)cinnoline-3-carboxamide as a yellow oil. The compound was characterized by 1H NMR in CDCl3.
4-Amino-8-(2-fluoro-6-methoxyphenyl)-N-(2-hydroxypropyl)cinnoline-3-carboxamide.
A solution of 200 mg (0.41 mmol) of 4-amino-N-(2-(tert-butyldimethylsilyloxy)propyl)-8-(2-fluoro-6-methoxyphenyl)cinnoline-3-carboxamide in THF (3 ml) was stirred at room temperature as 4.1 ml (4.1 mmol) of a 1 M solution of NH4F in THF was added, and the resulting mixture was stirred for 2 hours. The reaction was diluted with water and extracted with ethyl acetate. The organic extracts were combined, dried with sodium sulfate, filtered and concentrated. The residue was purified by flash chromatography on silica gel (dichloromethane-methanol). Product containing fractions were combined and concentrated, and the residue was repurified by flash chromatography on silica gel (acetonitrile-chloroform). Product containing fractions were combined and concentrated to give 105 mg (0.28 mmol, 67%) of 4-amino-8-(2-fluoro-6-methoxyphenyl)-N-(2-hydroxypropyl)cinnoline-3-carboxamide as a white powder. High-resolution mass spectrometry measured for C19H19FN4O3, [M + H]+ found at m/z 371.1509 (calculated m/z 371.1514). 1H NMR (600 MHz, DMSO-d6) δ ppm 1.09 (d, J = 6.2 Hz, 3H), 3.20–3.26 (m, 1H), 3.35–3.40 (m, 1H), 3.648* and 3.652* (s, total 3H), 3.80–3.84 (br m, 1H), 4.85 and 4.86 (d, J = 4.5 Hz total 1H), 6.91 (t, J = 8.6 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 7.43–7.47 (m, 1H), 7.75 (d, J = 6.8 Hz, 1H), 7.77–7.80 (m, 1H), 8.21 (br s, 1H), 8.46 (dd, J = 8.3,1.1 Hz, 1H), 8.94 (t, J = 5.4 Hz 1H), 9.09 (br s, 1H). 13C NMR (151 MHz, DMSO-d6) δ ppm 21.13* and 21.14*, 46.0, 56.0, 65.05* and 65.07*, 107.09* and 107.11* (d, J = 2.4 Hz), 107.31 and 107.33 (d, J = 22.7 Hz), 115.50* and 115.52* (d, J = 19.6 Hz), 116.2, 122.5, 127.6, 128.2, 129.7 (d, J = 10.5 Hz), 131.3, 133.61 and 133.63, 143.8, 146.7, 158.21* and 158.23* (d, J = 7.4 Hz), 160.04* and 160.06* (d, J = 241.6 Hz), 167.15* and 167.16*.
Clinical Plasma Samples
Human plasma samples were obtained as part of a multiple-ascending dose study in 48 healthy male volunteers (n = 12 on placebo and n = 36 on active drug, age 21–54) dosed orally with placebo, 5, 10, 20, or 50 mg AZD7325 once followed by a 2-day washout and then daily for 7 consecutive days. Metabolite identification was only carried out in pooled plasma samples from eight subjects each in the 10 and 50 mg cohorts.
Human plasma samples were also obtained from a Phase II, multicenter, double-blind, randomized, placebo-controlled, parallel group study examining safety, efficacy, and pharmacokinetics of AZD7325 in adult patients with generalized anxiety disorder aged 18–65 years. The study was conducted at 48 study centers in the United States. The study consisted of a 28-day, double-blind treatment period with one of four treatment regimens: 1) AZD7325 5 mg twice daily, 2) AZD7325 15 mg twice daily, 3) lorazepam 2 mg twice daily, or 4) placebo twice daily. AZD7325 plasma exposure following multiple-doses in subjects with generalized anxiety disorder was assessed in this study. A single plasma sample was taken at the scheduled office visit on days 7, 14, 21, and 28 after starting treatment on day 1. The exact blood sampling time and date were recorded, and the exact time and date of the last AZD7325 dose were collected from subjects. Metabolite identification was only carried out in plasma samples from selected patients of the 15 mg twice daily treatment.
Both clinical studies were performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with ICH/Good Clinical Practice and applicable regulatory requirements and the AstraZeneca policy on Bioethics.
Analysis of Radiolabeled Metabolites in Rat Plasma after a Single Oral Dose of [14C]-AZD7325
Male and female Wistar Hanover rats were orally administered with 2 mg/kg [14C]-AZD7325 at the specific activity of 130 μCi/kg using a solution formulation in 20% hydroxypropyl-β-cyclodextrin. Rat plasma samples were pooled by sex to obtain representative samples for male and female rats. The Hamilton pooling method (Hamilton et al., 1981) was adopted to result in a pooled sample representing the average concentration of area under the curve. Aliquots of 1.5 ml pooled rat plasma samples were mixed with 1.5 ml acetonitrile to precipitate proteins. After centrifugation, the resulting clear supernatant was transferred to a new vial and was evaporated under vacuum at 30°C for 4 hours using a centrifugal vacuum evaporator. The dried residues were reconstituted in 300 μl mixed solvent of water and acetonitrile containing 0.05% formic acid.
An aliquot of 50 μl reconstituted samples was injected for the radiochromatographic and LC-MS analysis. The LC was conducted using a Luna C8, 5 µm, 4.6 × 150 mm column (Phenomenex, Torrance, CA) with an Agilent 1100 LC system (Agilent Technologies, Santa Clara, CA). Mobile phase consisted of water (A) and acetonitrile (B), both containing 0.05% formic acid, at flow rate of 1.0 ml/min with linear gradients that started from and held at 5%B during 0−2 minutes, followed by 5−25%B at 2−40 minutes, 25−85%B at 40−45 minutes, 85−95%B at 45−50 minutes, and held at 95%B until 55 minutes, then returned to the initial composition for equilibration prior to the next sample injection. LC eluate post UV cell was split at an approximately 1:9 ratio between a LTQ-Orbitrap XL mass spectrometer equipped with an electrospray ionization source (Thermo Scientific, San Jose, CA) and a fraction collector. The LC fractions were collected by the time interval of 0.13 minute into a total of four LumaPlates—96-well yttrium silicate scintillator-coated microplates (PerkinElmer, Waltham, MA) after each injection. The solvent in collected LumaPlates was evaporated in a fume hood at room temperature overnight. The radioactivity of collected fractions was read using a TopCount NXT microplate scintillation counter (PerkinElmer) at count time of 5 minutes. The mass spectrometer was operated in data-dependent scan mode. Each data acquisition cycle was led by a high-resolution MS scan. The acquisition of LC-MS/MS product ion spectra, either collision-induced dissociation (CID) or the higher energy collisional dissociation, was triggered by matching the detected likely 12C:14C isotope pattern with the dosing ratio of AZD7325: [14C]-AZD7325, i.e., M: (M + 2.003) = 1:0.52 ± 0.15 in this particular case.
Analysis of Plasma Metabolites after Multiple Oral Doses of AZD7325 in Human, Dog, Rat, and Mouse
The Hamilton pooling method was used (Hamilton et al., 1981). After bioanalysis for pharmacokinetics, residual human plasma samples collected from eight healthy men over 24 hours at the end of a multiple ascending dose clinical study were pooled to obtain representative samples for 10 and 50 mg once daily dose groups, respectively. After bioanalysis for toxicokinetics, residual animal plasma samples collected over 24 hours on day 91 of 3-month oral safety studies in dog, rat, and mouse were pooled to obtain a respective pooled sample for beagle dog (45 mg/kg per day dose group of three males and three females), Han Wistar rat (100 mg/kg per day dose, the toxicokinetics satellite group of three males and three females), and CD-1 mouse (400 mg/kg per day dose, the toxicokinetics satellite group of 18 males and 18 females, with the sampling from three males and three females at each time point). Water was added to pooled animal plasma samples to give a fivefold dilution. Aliquots of pooled human and preclinical animal plasma samples were combined with two volumes of acetonitrile, respectively. After centrifugation to pellet proteins, the supernatants were transferred to new vials and evaporated under vacuum at 30°C for 4 hours to dryness using a centrifugal vacuum evaporator. The dried residues were reconstituted in a mixed solvent of water and acetonitrile containing 0.05% formic acid back to the same sample volume as that prior to the protein precipitation.
An aliquot of 50 μl reconstituted individual samples was injected into the same type of C8 column using the same mobile phases as described above for radiolabeled rat metabolites of [14C]-AZD7325. An Acquity ultraperformance liquid chromatography (UPLC) system (Waters, Milford, MA) was used this time. Linear gradients started from and held at 5%B during 0−2 minutes, followed by 5−25%B at 2−40 minutes, 25−98%B at 40−55 minutes, and held at 98%B until 60 minutes, then returned to initial composition for equilibrium before the next sample injection. Postcolumn LC flow of 1 ml/min was split at an approximate 1:9 ratio between the LTQ-Orbitrap XL mass spectrometer and the waste line. The mass spectrometer was operated in data-dependent scan mode. Automatic acquisitions of LC-MS/MS CID product ion spectra were triggered by detecting the accurate mass of any metabolite known from previous studies and those calculated by possible combinations of metabolic pathways. Otherwise, the CID spectra were obtained for the top three most intensive ions detected in the high resolution MS scan of each data acquisition cycle.
Analysis of Representative Plasma Samples Selected from a Phase IIb Clinical Study of AZD7325
After the bioanalysis and pharmacokinectics data processing, residual plasma samples collected from nine selected patients in the 15 mg twice daily dose group were analyzed by LC-MS to obtain metabolite profiles. An aliquot of individual plasma samples was combined with two volumes of acetonitrile containing an internal standard (IS) compound. The IS was a closely related structural analog of AZD7325, differing only by an isobutylamine amide group in the IS instead of the propylamine amide in AZD7325. The exact mass of the IS ([M + H]+ m/z 369.1721) does not interfere with any of the AZD7325 metabolites. After centrifugation, the resulting clear supernatant was transferred to a new vial and evaporated under vacuum at 30°C to dryness using a centrifugal vacuum evaporator. The dried residues were reconstituted in a mixed solvent of water and acetonitrile containing 0.05% formic acid. An aliquot of 10 μl reconstituted individual samples was injected into an Acquity BEH C8, 1.7 µm, 2.1 × 100 mm UPLC column (Waters) for LC-MS analysis, using an Acquity UPLC system coupled with an LTQ-Orbitrap XL mass spectrometer. Mobile phase consisted of water containing 0.05% formic acid (A) and acetonitrile (B) at the flow rate of 0.2 ml/min with linear gradients of 10−35%B at 0−17 minutes, 35−98%B at 17−22 minutes, and held at 98%B until 24 minutes, then returned to initial composition to equilibrate before the next sample injection. The mass spectrometer was operated in data-dependent scan mode to acquire high-resolution MS spectra and LC-MS/MS CID spectra.
Human Liver Microsomal Metabolites of AZD7325 and the M9 Synthetic Standard
The incubation mixture in a phosphate-buffered saline contained 1 mg protein/ml human liver microsomes (BD UltralPool HLM150, Catalog Number 452117), 10 µM AZD7325 or the M9 synthetic standard spiked from respective 10 mM stock solutions in DMSO, and 1 mM NADPH. Negative control incubations were conducted in the absence of cofactor NADPH. The incubation volume was 250 µl. After 2-hour incubation, the reactions were stopped by mixing with cold acetonitrile of equal volume. After centrifugation, the resulting clear supernatants of 400 µl were transferred into new vials and then diluted with 400 µl water. An aliquot of 5 µl sample was injected into an Acquity BEH C8, 1.7 µm, 2.1 × 100 mm UPLC column for LC-UV-MS analysis, using the same LC-MS instrument and the same LC gradient as that described above for selected plasma samples from a Phase IIb clinical study. High resolution MS and MS/MS spectra were acquired.
Proton NMR of Metabolite M42 Isolated from Dog Plasma
After bioanalysis, residual plasma samples collected on day 28 from the high-dose group of a 1-month oral safety study in beagle dogs were combined and then mixed with acetonitrile to precipitate proteins. After centrifugation, the resulting clear supernatant was collected and concentrated by nitrogen blow-down. LC separation was carried out using a 10 × 100 mm C18 column with the gradient mobile phase consisting of water/acetonitrile containing 0.1% formic acid at the flow rate of 5 ml/min. The LC fraction of metabolite M42 was collected from multiple sample injections, and the solvent was evaporated under vacuum till dryness. The 1H NMR and two-dimensional correlation spectroscopy (COSY) spectra were recorded at 303 K in dimethylsulfoxided-d6 (DMSO-d6) on a Bruker Avance NMR spectrometer, either 500 or 600 MHz (Bruker, Billerica, MA). The COSY spectrum for the low-level sample was obtained over 3 days. After the COSY experiment suggesting two likely isomers in the M42 sample, the metabolite sample was further separated into two relatively pure diastereomers using the same LC column but a modified gradient. Afterward, 1H NMR spectra were acquired again for each of two pure diastereomers. Two-dimensional COSY was obtained for one of the two pure diastereomers.
Results
Extensive metabolism of AZD7325 has been observed in plasma across species of rat, mouse, dog, and human. For example, the radiochromatographic and LC-MS metabolite profiles of [14C]-AZD7325 in Fig. 1 show two dozen metabolites in pooled male rat plasma collected at 0–12 hours after a single oral dose of 2 mg/kg to the animals. LC-MS extracted ion chromatograms of nonconjugate metabolites and conjugated metabolites are displayed separately in Fig. 1, B and C, setting apart some nonconjugate and conjugate metabolites that coeluted. The comparison of LC-MS metabolite profiles versus the radiochromatogram indicates that MS signal response factors of conjugate metabolites were lower than that of the parent drug and nonconjugate metabolites in the positive-ion electrospray mass spectrometry. For example, it is estimated from the data shown in Fig. 1 that sulfate M19 would give a MS signal response only at approximately 15% of the parent drug AZD7325 at the same concentration. On the other hand, the relative MS signal responses between a nonconjugate metabolite and the parent drug AZD7325 were either similar or had a smaller difference than the conjugate metabolites. Hence, the LC-MS metabolite profile of nonconjugate metabolites of AZD7325 may provide qualitative comparison for metabolite abundance relative to unchanged parent drug in individual samples.

Radiochromatographic metabolite profile of [14C]-AZD7325 in male rat plasma acquired with a pooled 0- to 12-hour sample collected after a 2 mg/kg oral dose to animals (A). LC-MS accurate-mass extracted ion chromatograms showing nonconjugate metabolites (B) and conjugate metabolites (C), with y-axis normalized to the same MS intensity scale. The letters S and G in parentheses denote O-sulfate and O-glucuronide respectively.
The complexity of the AZD7325 metabolism originated from ordinary metabolic oxidations in three regions of the AZD7325 molecule (Fig. 2, marked in red), followed by subsequent conjugations and various combinations of different metabolic pathways. The initial metabolic oxidations include O-demethylation (M7), hydroxylation at an aromatic ring (M11 and M12), and oxidations at the N-propyl group such as hydroxylation (M4, M5, and M6) as well as N-depropylation (M2). Subsequently, glucuronidation may follow some or all of these hydroxylations (some in rat and mouse, all in dog and human). Sulfation most likely occurs at the phenolic hydroxyl introduced by the M11 and/or M12 metabolic pathway. There was no direct conjugation metabolism of parent drug AZD7325, thus eliminating the possibility of N-conjugation. Moreover, no conjugate metabolites of O-demethyl-AZD7325 (i.e., M7) were observed, likely due to steric hindrance around the M7 hydroxyl that is ortho to the bulky cinnoline. Unusual metabolite M9 and its subsequent metabolites will be described later. Nevertheless, various combinations of all ordinary metabolic pathways described above would give a biotransformation scheme probably too complex to display here.

The initial metabolic sites of AZD7325 (marked in red) and subsequent pathways.
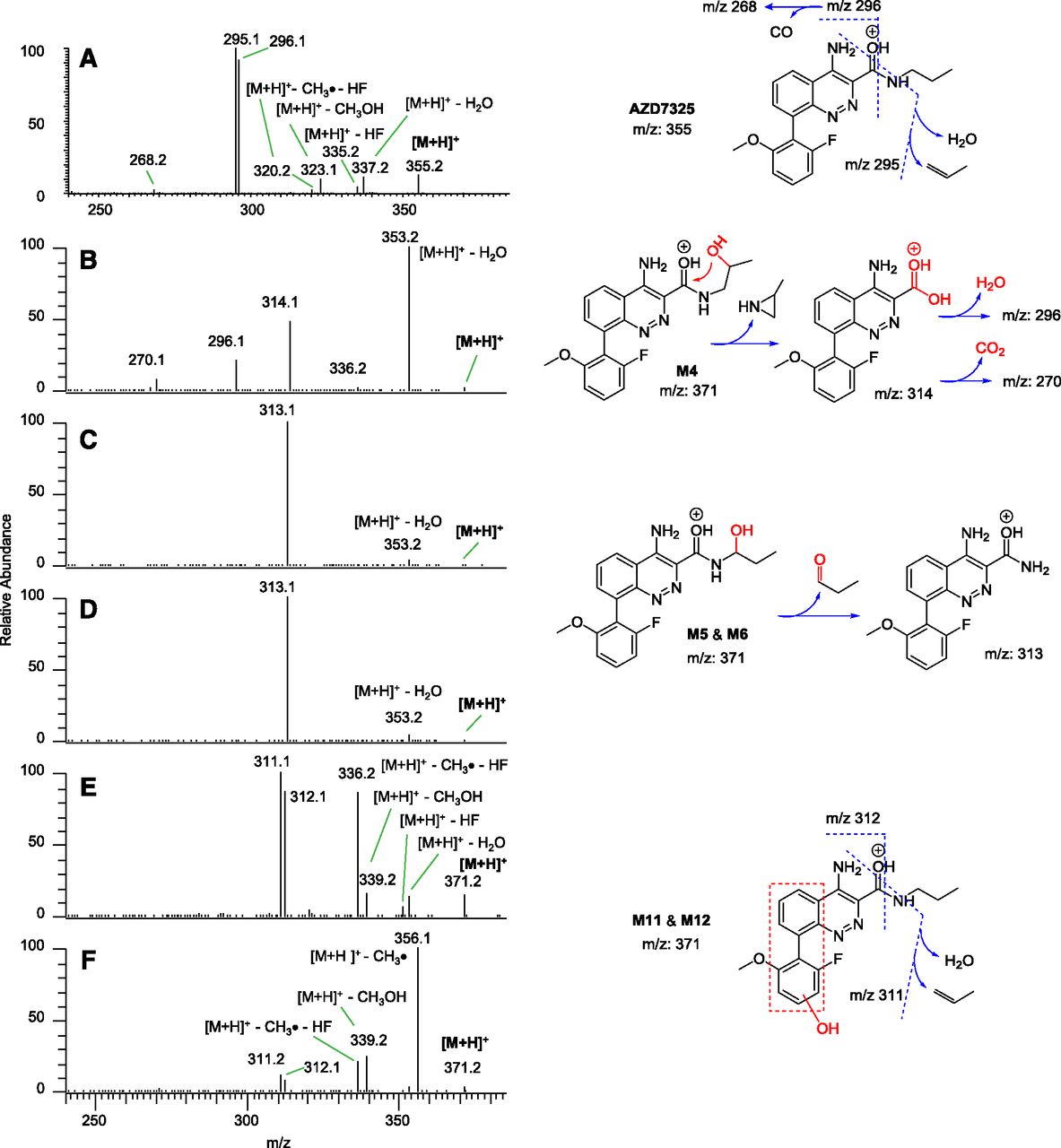
The structural elucidation of isomeric mono-hydroxyl metabolites by LC-MS/MS could be a piece of analytical chemistry useful to metabolite identification. Figure 3 shows LC-MS/MS spectra acquired by CID in a linear ion trap for metabolites M4, M5/6, and M11/12 in a pooled mouse plasma sample in comparison with intact parent drug AZD7325. Distinct fragmentation patterns among the three types of mono-hydroxyl metabolites were recorded. A rearranged fragmentation of protonated M4 led to a carboxylic-acid fragment ion at m/z 314 and further fragmentation typical to a carboxylic acid (Fig. 3B). This kind of rearranged fragmentation reaction could also occur with a terminal hydroxyl metabolite at the propyl group, but the M4 identity was confirmed with the synthetic standard by LC-MS/MS in the present study. Facile cleavage at the carbinolamine (also known as hemiaminal) C-N bond of M5 and M6 was observed in the gas phase fragmentation, comparable with that in solution phase with propyl aldehyde as a leaving group (Fig. 3, C and D). The fragmentation of M11 and M12, producing fragment ions at m/z 311 and m/z 312 (Fig. 3, E and F), resembles that of the parent drug AZD7325 (Fig. 3A), except for an additional oxygen introduced by the metabolism. It is possible that one or both of the M11 and M12 hydroxylations have occurred on the methoxyl fluorophenyl ring, based on intense neutral losses occurring at the moiety as recorded in CID spectra (Fig. 3, E and F) when compared with the parent drug AZD7325 (Fig. 3A). However, this postulation about M11 and M12 has not been confirmed, thus the site of hydroxylation is not specified other than by a dashed box suggesting a larger region.

LC-MS/MS CID spectra of protonated molecular ions of AZD7325 (A), M4 metabolite (B), M5 and M6 metabolites (C and D), M11 and M12 metabolites (E and F) acquired from the same pooled mouse plasma sample. Also provided are spectral interpretations on fragmentations.
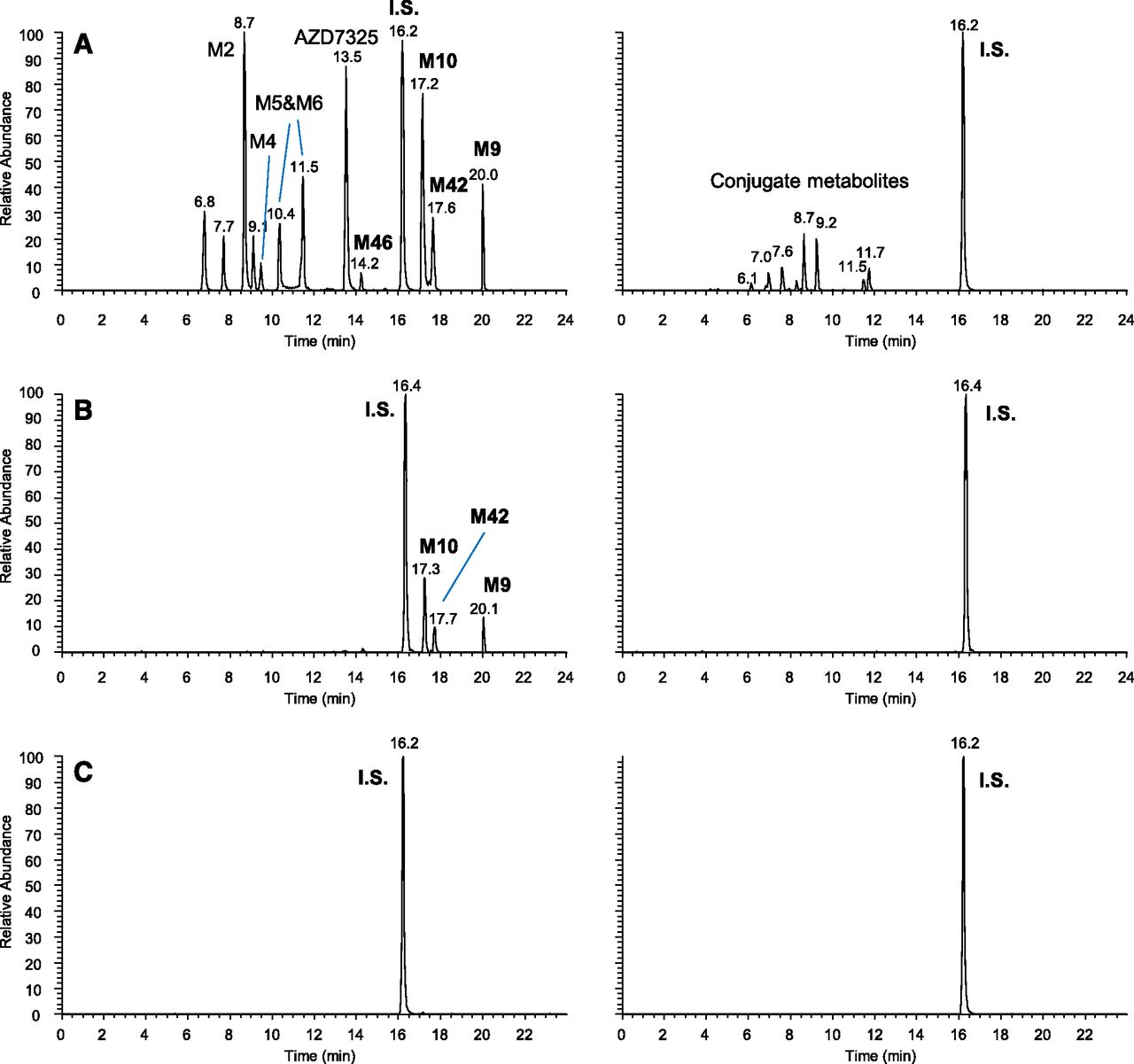
Figure 4 compares the LC-MS abundance of the unusual metabolite M9 and its subsequent metabolites M10, M42, and M46 relative to the parent drug AZD7325 in pooled plasma samples collected at different times from the same dose group during a Phase I clinical multiple ascending dose study. These include the pooled plasma sample collected over the 24-hour dosing interval after the first oral dose of 10 mg/kg (Fig. 4A), the pooled sample collected over 24 hours after multiple daily oral doses (Fig. 4B), and the pooled single time point sample collected at 48 hours after the final dose in the clinical study (Fig. 4C). Note that although M9 was a minor metabolite after the single dose, it became a major metabolite at steady state and circulated at a higher abundance than the parent at 48 hours after the final dose (Fig. 4, A vs. B vs. C). Metabolites M10 and M42 (subsequent metabolites of M9) were absent from pooled plasma samples collected after a single dose but became major metabolites at steady state. Moreover, they and M9 circulated at higher abundance than AZD7325 at 48 hours after the final dose (Fig. 4, A vs. B vs. C). M46, a metabolite subsequent to both M10 and M42, was observed at trace levels when levels of M10 and M42 were high (Fig. 4, C and B). Based on these observations, metabolites M10, M42, and M46 were late-occurring metabolites, and all these metabolites appeared to be circulating longer than the parent drug AZD7325 in plasma. The duration for which these metabolites could be detected in plasma was not determined, but it was certainly longer than 48 hours after the last dose at steady state.

LC-MS metabolite profiles of AZD7325 in human plasma samples collected from a Phase I multiple ascending dose clinical study conducted in clinic: pooled 0- to 24-hour sample after the first dose on day 1 (A), pooled 0- to 24-hour sample after the seven repeated once-a-day doses (B) and at 48 hours after the final of the seven repeated once-a-day doses (C), displayed by accurate-mass extracted ion chromatograms with nonconjugate and conjugate metabolites in left and right columns, respectively, and y-axis normalized to the same MS intensity scale for each sample.
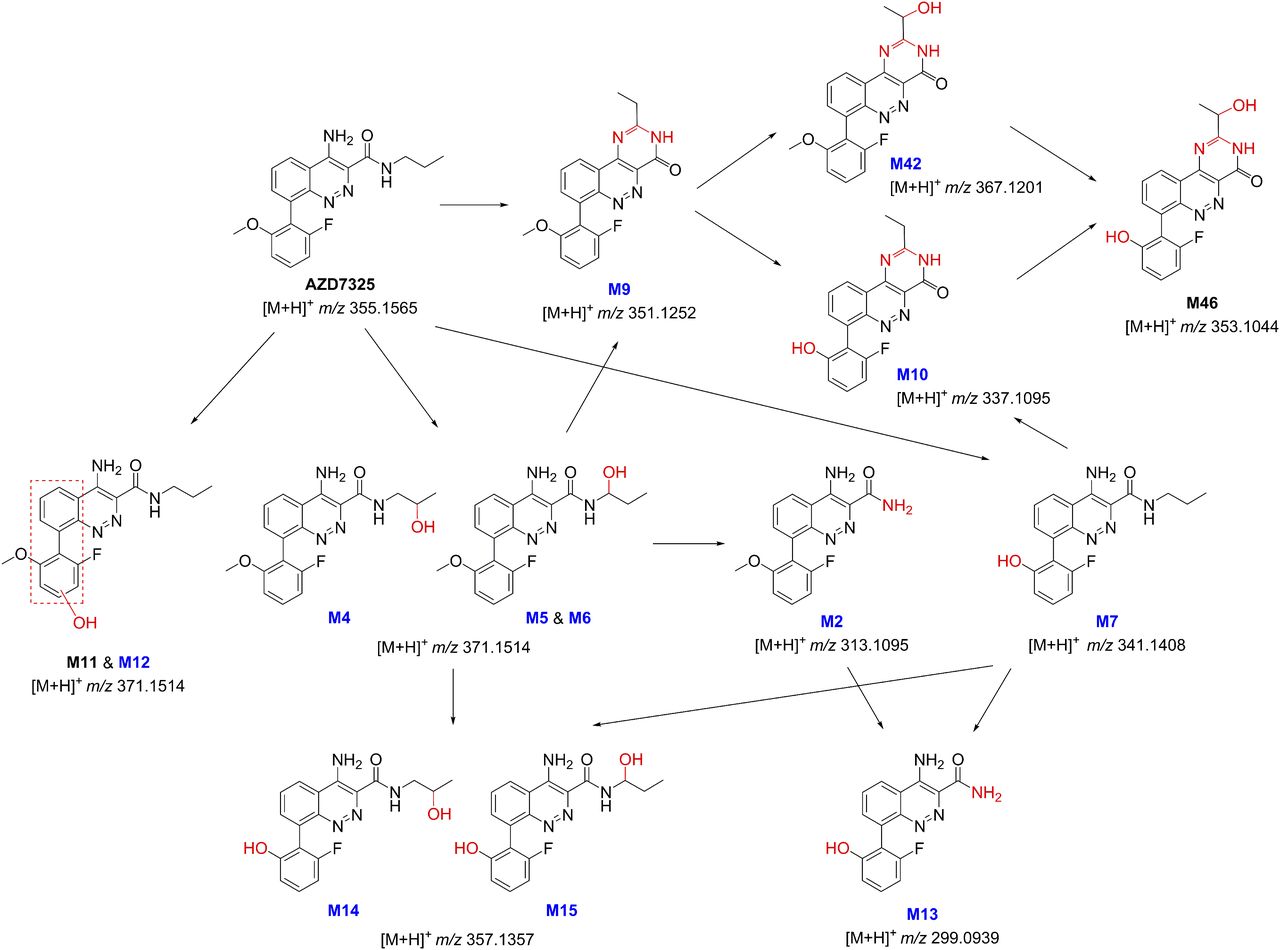
The chemical structures of M9 and its subsequent metabolites M10, M42 and M46 are shown in Scheme 1. A mechanism involving metabolic cyclization and aromatization is proposed for the formation of M9 (Scheme 1). Further oxidative metabolism leads to M10 via O-demethylation and M42 via hydroxylation at the ethyl group of M9 and finally to M46 after both demethylation and hydroxylation (Scheme 1 and Fig. 4). The authentic standard of M9 was prepared chemically to confirm the novel structure (Artelsmair et al., 2018), and a two-dimensional NMR 15N HMBC spectrum of the M9 standard is provided in the supplemental material, showing the evidence for the ring closed structure, i.e., 2-ethylpyrimidin-4-one substructure of the tricyclic core of M9 (Supplemental Fig. 1). The M9 metabolite generated in human liver microsomes has been confirmed to be identical to the synthetic standard by LC-UV-MS/MS, with an excellent match in LC retention time and high-resolution MS/MS spectra showing the accurate mass of fragment ions (Supplemental Fig. 2). In addition, the photodiode array UV spectra of the synthetic M9 and biologically generated M9 are identical and significantly different from that of the parent drug AZD7325 (Supplemental Fig. 3). The CID spectrum of the synthetic M9 is also the same as previously acquired from human and preclinical plasma samples (Supplemental Fig. 2). Furthermore, the incubation of the M9 synthetic standard in human liver microsomes generated M10 and M42 metabolites (Supplemental Figs. 4 and 5), thus verifying the metabolic pathway for M10 and M42 as proposed in Scheme 1. Also demonstrated is the verification of M4 metabolite by the same retention time as the synthetic standard (Supplemental Fig. 2, A vs. B). As mentioned earlier, the identical MS/MS CID spectra between the metabolite and the standard were also acquired (not shown).

Proposed formation mechanism for metabolites M9 and its subsequent metabolites M10, M42, and M46.
The aryl-aryl single bond of AZD7325 has restricted rotation, thus atropisomers have been previously observed by chiral liquid chromatography. This had a direct consequence for the hydroxylation of the M9 ethyl group to form M42; two diastereomers were partially isolated by typical reversed-phase liquid chromatography from a pooled dog plasma sample. Both pure single diastereomers of M42 were fully converted to a 1:1 mixture after heating at 60°C for 14 hours (Supplemental Fig. 6). Proton NMR and two-dimensional COSY spectra were acquired on both the mixed diastereomers of M42 and partially isolated single diastereomer. NMR data extracted from those spectra of M42 are listed in Table 1.
NMR data extracted from proton and COSY pdf spectra for the M42 metabolite isolated from a pooled dog plasma sample
Two diastereomers of M42 were recorded due to hindered rotation around C4-C11. 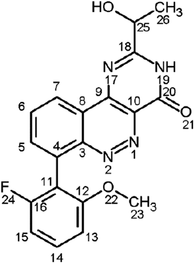
The existence of long-circulating metabolites—M9, M10, and M42 (as well as M46 if detectable)—was used to our advantage when troubleshooting the unforeseen lack of drug exposure in a number of patients during a Phase IIb outpatient clinical study of AZD7325. The concentration of AZD7325 in some plasma samples was unexpectedly below the limit of quantification (LOQ), which complicated data interpretation. To assist in the troubleshooting, plasma samples of nine selected patients were analyzed for metabolite profiles by LC-MS, i.e., to investigate if any extensive metabolizers among patients caused lack of drug exposure. These nine patients from the 15 mg twice daily dose group represent three different categories in terms of bioanalytical data of all four pharmacokinetic plasma samples collected in the treatment period: 1) all samples had measurable levels of AZD7325, 2) some samples were below LOQ, and 3) all samples were below LOQ (Table 2). Figure 5 compares LC-MS metabolite profiles in plasma samples collected on day 21 from three patients, Patients Z, R, and U in Table 2, i.e., one from each of three representative categories. The LC method for Figs. 4 and 5 was specially developed for the analysis of M9 and its subsequent metabolites, giving clear separation. After supposedly 40 or more repeated doses of AZD7325, the day 21 plasma samples collected from these three patients showed striking differences in metabolite profiles (Fig. 5). A full metabolite profile including nonconjugate and conjugate metabolites was observed in the day 21 plasma sample form patient Z (Fig. 5A). Only M9, M10, and M42 were present in the day 21 sample for patient R (Fig. 5B). No metabolite was detected in the sample from patient U (Fig. 5C).
Metabolite profiling results for 36 plasma samples collected from nine representative patients of the 15 mg twice daily dose group in a Phase IIb clinical trial
Patients were designated with arbitrary letters to keep anonymous.
Metabolite profiles of AZD7325 in plasma sample of three different patients collected on Day 21 during a Phase IIb clinical trial (i.e., Patients Z, R and U in Table 2), showing a full metabolite profile in Patient Z (A), only long-circulating metabolites in Patient R (B), and no metabolites in Patient U (C). Data are displayed in LC-MS extracted ion chromatogram, with non-conjugate and conjugate metabolites in left and right columns respectively. The same internal standard (IS) was added to all samples.
As listed in Table 2, all plasma samples with measurable levels of AZD7325 were all similar in that a full metabolite profile was observed in each of the samples. However, not all the plasma samples below LOQ were the same in terms of the metabolite profile. For three patients with partial samples below LOQ, long-circulating metabolites were observed in the samples below LOQ, although other metabolites were not seen. Whereas for three patients where all samples were below LOQ, none of the samples had any detectable metabolites. Since long-circulating metabolites M9, M10, and M42 were present in the plasma at 48 hour after the final dose of seven repeated doses of AZD7325 in a Phase I clinical study conducted in clinic (Fig. 4C), patients with a plasma sample below LOQ in this outpatient Phase IIb clinical study might not have taken the candidate drug within at least 48 hours prior to the blood draw. It is reasonable to conclude that patients with all four samples below LOQ did not take the candidate drug during most of time in the trial period. The absence of long-circulating metabolites helped in revealing the patient noncompliance as the cause of unexpected lack of drug exposure.
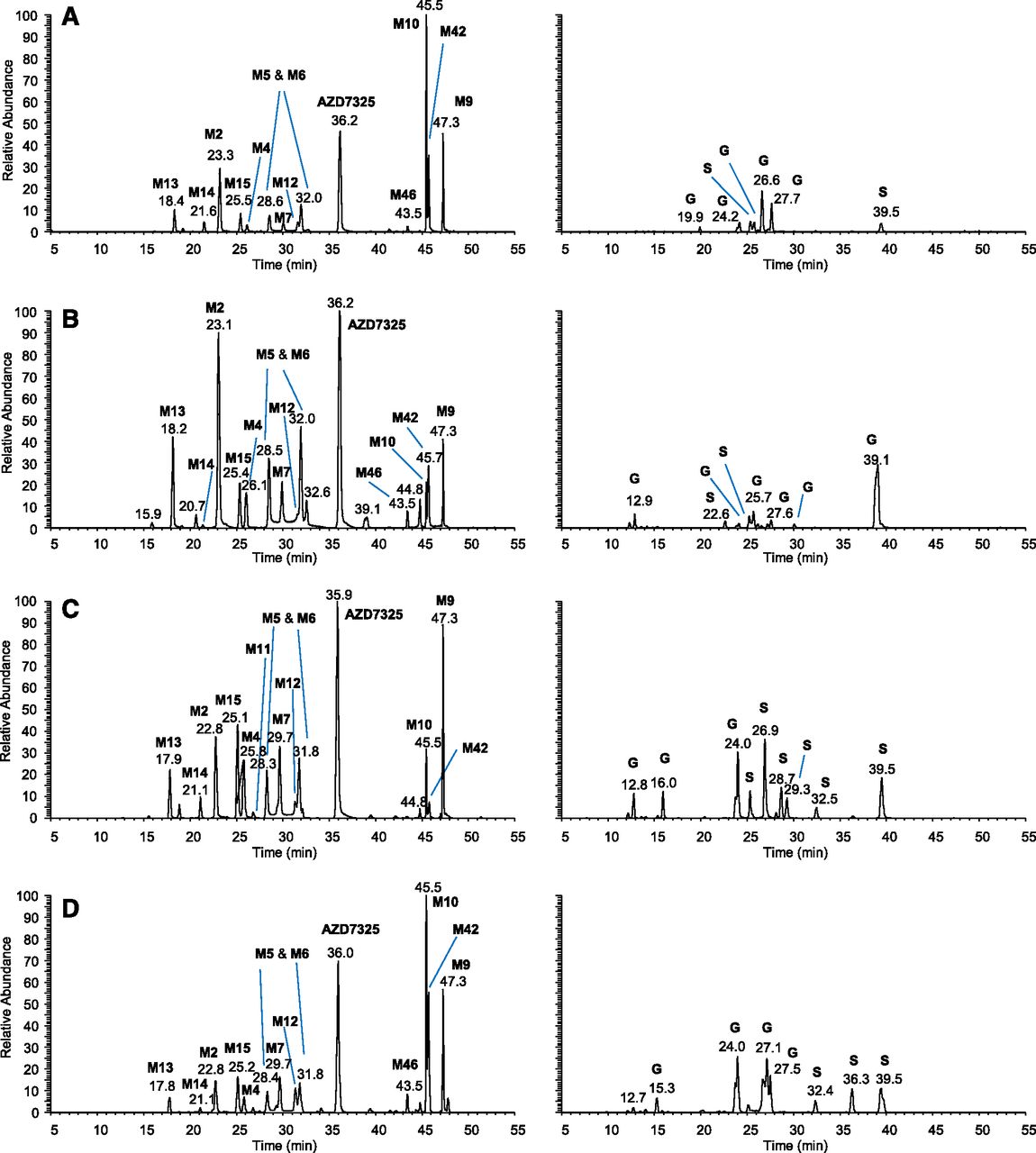
The plasma metabolite profiles of AZD7325 after multiple oral doses in human and preclinical toxicology species, rat, mouse, and dog were compared to evaluate if major human nonconjugate metabolites in the systemic circulation were adequately exposed in preclinical animal species in the safety evaluation of AZD7325. Figure 6 provides the LC-MS extracted accurate-mass ion chromatograms showing AZD7325 metabolites in pooled human plasma collected over 24 hours after the final dose of seven repeated 50 mg once daily doses in a Phase I clinical study (A), in pooled dog plasma of the 45 mg/kg per day dose group (B), rat plasma of the 100 mg/kg per day dose group (C), and mouse plasma of the 400 mg/kg per day dose group (D) collected over 24 hours on day 91 from the respective 3-month safety study. The no observed adverse effect level (NOAEL) in dog and rat was 45 and 100 mg/kg per day, respectively. The NOAEL in mouse was considered to be 200 mg/kg per day. The LC-MS chromatograms for nonconjugate and conjugate metabolites are plotted separately in the left and right columns of Fig. 6. The biotransformation scheme for nonconjugate metabolites is provided in Scheme 2. The conjugate metabolites were either glucuronides (G) or sulfates (S) following a hydroxylation and thus were not further evaluated. Note that M42 was absent from rat plasma after a single dose of [14C]-AZD7325 (Fig. 1) and was not seen in human plasma after a single oral dose of AZD7325 (Fig. 4A). After repeated doses, M42 became a circulating metabolite along with its precursor metabolite M9 and O-demethyl-M9 (i.e., M10) that were both major in all four species (Fig. 6). In summary, all 12 major nonconjugate metabolites observed in human plasma were also found in dog, rat, and mouse plasma samples (Fig. 6, left column).

LC-MS metabolite profiles of AZD7325 in pooled human plasma collected over 24 hours after seven repeated daily doses of 50 mg/kg (A) and pooled dog plasma of 45 mg/kg per day dose group (B), rat plasma of 100 mg/kg per day dose group (C), and mouse plasma of 400 mg/kg per day dose group (D) collected over 24 hours on day 91 from the 3-month safety study in respective animal species. Nonconjugate and conjugate metabolites are displayed in left and right columns, respectively, with y-axis normalized to the same MS intensity scale for each species. The letters S and G denote O-sulfate and O-glucuronide, respectively.
Nonconjugate metabolites of AZD7325 in plasma of humans and preclinical animal species. Calculated exact mass of the protonated molecular ion is provided in m/z for every metabolite. Blue fonts in the metabolite numbering indicate metabolites with an estimated level at approximately 10% or greater than 10% of parent drug AZD7325 in the pooled human plasma.
Discussion
Biotransformation mechanisms considered in the present study were not merely to rationalize the structural analysis outcome for unusual metabolites M9 and M42, rather, the thought process on the formation mechanisms was an integrated part of structural elucidation for M9 and M42. In a previous experience, generating putative structures with envisaged rearrangement mechanisms was an effective way of assisting structural identification on a dramatically rearranged oxidation product in plasma for a candidate drug acting as a human neutrophil elastase inhibitor (Gu et al., 2015). Two intuitive thoughts have guided the proposal for possible mechanisms in the present study. First, M9 might contain a new aromatic structure. The UV spectrum of M9 was significantly changed with a strong new absorption band at 247 nm compared with AZD7325 (Supplemental Fig. 2), suggesting a strong new chromophore in M9. The MS/MS fragmentation of protonated M9 had no water loss from the carboxamide and no C-N cleavages that were observed in the fragmentation of protonated AZD7325 (Supplemental Fig. 2, C and D vs. Fig. 3A), suggesting a stable structure transformed from the N-propylamine amide moiety. The new structure was so stable that fragmentations only occurred elsewhere on the methoxyl fluorophenyl ring (Supplemental Fig. 2). Second, M42, having one more oxygen atom in molecular mass than M9, might be a subsequent metabolite of M9. Both M9 and M42 were unusual metabolites and appeared to be transformed in the same region of the drug molecule; therefore, the chance of them undergoing completely different pathways seemed slim. Additional clues included the CH-CH3 coupling identified in NMR COSY experiment of M42 and measured proton chemical shifts, suggesting a possible bond of an oxygen atom to the CH carbon (Table 1).
The first putative structure considered for M9 and M42 contained a 5-methyloxazole in place of the propylamine amide of AZD7325. We envisioned this as arising from M4 via a five-membered cyclization and aromatization (Supplemental Fig. 7). Afterward, a hydroxylation on the putative oxazole ring, followed by a tautomerization, would give a hypothetical M42 having a CH-CH3 (Supplemental Fig. 7). Interestingly, predicted proton chemical shifts of the CH-CH3 in this putative M42 (δ 5.2 and 1.4, ACD/Laboratories Proton NMR Predictor and DB 2015 in DMSO-d6 solvent) would be almost exactly the same as that of the identified M42 (δ 5.2–5.3 and 1.4–1.5, the same NMR Predictor and DB). However, two experimental results contradicted the putative mechanism: first, the expected oxazole proton was not found in the NMR spectrum of an isolated M10 metabolite sample; second, the incubation of the M4 standard in hepatocytes and liver microsomes did not produce M9 as it would be predicted by the putative mechanism. Consequently, this has directed us to the formation mechanism proposed in Scheme 1 and the M9 structure, which was eventually confirmed with a synthetic standard.
As proposed in Scheme 1, metabolite M9 and its subsequent metabolites would be unusual metabolites generated by plausible metabolic pathways that are known. Guengerich and Munro (2013) previously pointed out that many of the cytochrome P450-mediated unusual reactions are in the context of rearrangements; moreover, many of the rearrangements are initiated by α-hydroxylation to a heteroatom. CYP3A4, 3A5, and 2C19 are the major enzymes that form hydroxypropyl metabolites of AZD7325 with minor contributions of CYP1A, 2C9, and 2D6 (experimental data not shown). Although carbinolamines (also called hemiaminals) are unstable structures in general and tend to break apart, some carbinolamine metabolites have been previously reported to be stable enough for isolation, structural characterization, and even chemical synthesis (Shea et al., 1982; Ross et al., 1983; Perrin et al., 2011). We may have observed the possible on-column interconversion between the carbinolamine-amide M5/M6 and imine-amide intermediate 1 in LC-MS and MS/MS experiments. A wide separation in LC retention time was seen between M5 and M6 using C8 columns (Figs. 4–6). The elevated baseline in LC-MS and MS/MS chromatograms between M5 and M6 suggested on-column interconversion (LC-MS accurate mass m/z 371.151 chromatograms not shown, or LC-MS/MS m/z 371→313 chromatogram shown in Supplemental Fig. 8). Rotation around the aryl-aryl bond to convert diastereomers M5 and M6 is an unlikely explanation due to the wide separation of diastereomers M5 and M6 by LC. The M4 synthetic standard was prepared and diastereomers can be seen in NMR (see Materials and Methods, enantiomers at the 2-hydroxylpropylamine, diastereomers due to atropisomers); however, M4 eluted as a single sharp peak under the LC conditions using C8 columns (Supplemental Fig. 2B). Two diastereomers of metabolite M42 could only be partially separated even with an LC method specifically designed to resolve those diastereomers (Supplemental Fig. 6). Thereby, the wide separation and unequal LC peak heights of M5 and M6 possibly resulted from on-column interconversion between carbinolamine and imine intermediate 1 (Supplemental Fig. 8). The ion corresponding to the intermediate 1 was observed at trace amounts and in synchronization with the occurrence of M4/M5 in LC-MS. However, it was unclear whether the dehydration occurred in the solution during the chromatography or in the gas phase of the MS ionization source. The metabolic cyclization of drug molecules by intramolecular nucleophilic attack of a primary or secondary amino group to an imine or iminium intermediate was previously reported in the formation of six-membered or five-membered cyclic metabolites (Erve, 2008; Nagle et al., 2012). The intermediate 2 contains a cyclic substructure of dihydro pyrimidin-4-one, thus could be prone to metabolic aromatization to form a stable pyrimidin-4-one substructure of the tricyclic core of M9 (Scheme 1).
In the present study, only nonconjugate metabolites were evaluated for the exposure coverage of human metabolites of AZD7325 in the safety testing animals. O-Glucuronidation and O-sulfation reactions make a metabolite more water soluble and pharmacologically inactive, thus these conjugate metabolites do not need to be further evaluated across species (Gao et al., 2013). LC-MS chromatographic profiles of the conjugate metabolites are still presented to provide a little more comprehensive picture of the AZD7325 metabolism (Figs. 1 and 4–6). LC-MS chromatographic peaks of conjugate metabolites under-represent their level in a sample when compared with nonconjugate metabolites, due to lower MS signal response factors. However, adjustments or amplifications were not made on any of the LC-MS chromatograms for conjugate metabolites, so as to remain consistent in displaying data for different samples in this article.
The exposure comparison of drug metabolites should be conducted at the NOAEL of safety testing animals for the assessment of safe exposure of the metabolites (Gao et al., 2013). AZD7325 showed a sufficient NOAEL in the animals, e.g., 100 mg/kg per day in rat and 45 mg/kg per day in dog compared with the targeted therapeutic dose range of 10–50 mg in human. Using the MS signal response factors relative to the parent drug, which were derived from radiolabeled in vitro metabolites and in vivo rat metabolites of [14C]-AZD7325, i.e., an approach that was previously reported for the MIST analysis (Yu et al., 2007; Yi and Luffer-Atlas, 2010), the exposure comparison can be constructed from the LC-MS peak areas of the pooled plasma samples. Based on this comparison (plotted in Supplemental Fig. 9), the exposure multiples for all 12 major nonconjugate metabolites in rat versus human (100 mg/kg per day in rat vs. 50 mg once daily in human) are estimated at ≥6 and up to 200. The exposure multiples for the same metabolites in dog versus human (45 mg/kg per day in dog vs. 50 mg once daily in human) are estimated at ≥3 and up to 40. This basically confirms that if the safety margin is sufficient, the coverage of metabolite exposure in the safety study could also be adequate, given no unique or significantly disproportional human metabolites (Fig. 6)
The estimated sufficient coverages in the exposure of human major metabolites by rat and dog at NOAEL (Fig. 6; Supplemental Fig. 9) have avoided the need for bioanalytical quantification of major metabolites. Furthermore, the observation of late-occurring and long-circulating metabolites for AZD7325 demonstrates the need to assess the circulating metabolite profile at steady state as opposed to after a single dose. Together with a nonintuitive metabolic pathway identified for the AZD7325 drug metabolism, we would hope that this case will serve as a useful cautionary example to others who engage in drug development.
Acknowledgments
This article is dedicated to the memory of Dr. Scott W. Grimm. We thank several DMPK colleagues of the AstraZeneca legacy R&D at Wilmington, DE for contributions to the related in vitro studies, preliminary work on some metabolites, and bioanalysis; Michael Ramaker for selecting representative patient plasma samples for metabolite analysis after a Phase IIb outpatient study; Dr. Jason Shields and Dr. Bin Yang of AstraZeneca, Waltham, MA for helpful discussion; and Dr. Ujjal Sarkar of AstraZeneca, Waltham, MA for helping to retrieve spectral data. The rat mass balance study was carried out by Covance Laboratories at Madison, WI. Three-month safety studies in rat, mouse, and dog were all conducted by Charles River Laboratories at Tranent, Edinburgh, in UK.
Authorship Contributions
Participated in research design: Gu, Elmore, Davis, Christoph, Smith, Chapdelaine, Sunzel.
Conducted experiments: Gu, Artelsmair, Elmore, Lewis, Hall, Dembofsky.
Contributed new reagents or analytic tools: Artelsmair, Elmore, Dembofsky, Chapdelaine.
Performed data analysis: Gu, Elmore, Lewis, Davis, Hall, Christoph, Sunzel.
Wrote or contributed to the writing of the manuscript: Gu, Artelsmair, Elmore, Lewis, Davis, Smith, Sunzel.
Footnotes
- Received October 8, 2017.
- Accepted January 3, 2018.
↵* Separate signals visible from atropisomers. Multiplicity and coupling constants were identical and apply to both peaks. Integrals are the total of both peaks. If not indicated by *, only a single peak was observed (i.e., diastereoisomer peaks were coincident).
This work was partially funded from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 675417.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CID
- collision-induced dissociation
- IS
- internal standard
- LC
- liquid chromatography
- LOQ
- limit of quantification
- MIST
- metabolites in safety testing
- m/z
- mass-to-charge ratio
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- NOAEL
- no observed adverse effect level
- THF
- tetrahydrofuran
- UPLC
- ultraperformance liquid chromatography that is the LC utilizing small particle size and operating at higher pumping pressure
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}