


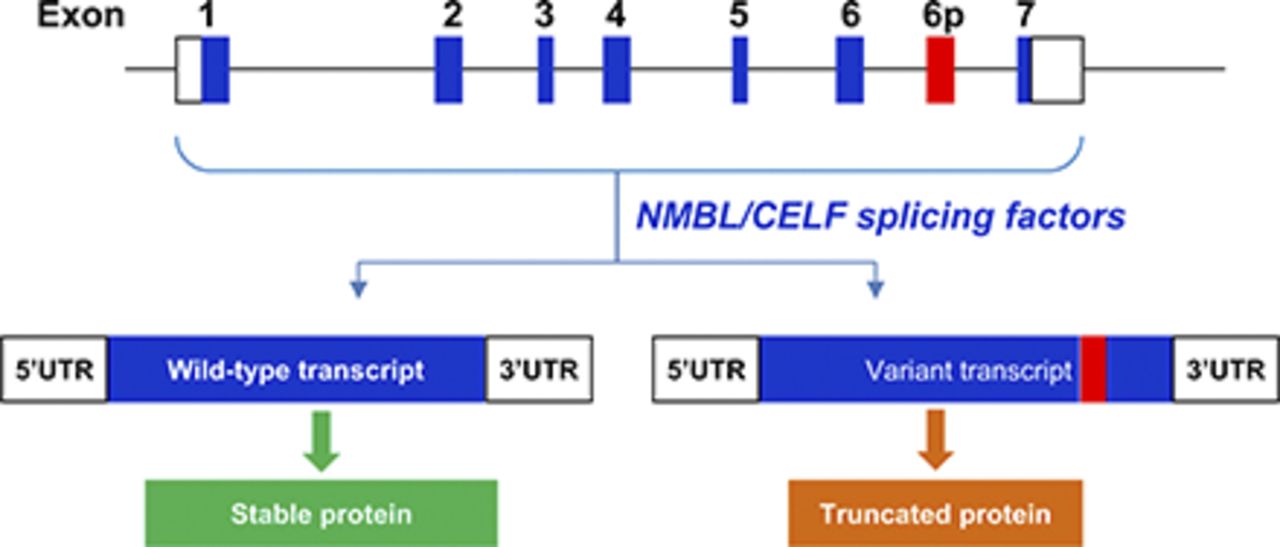
Visual Overview

Abstract
Sulfotransferase 4A1 (SULT4A1) is a sulfotransferase-like protein that is highly conserved between species. In human tissues, there are two transcripts, one that produces a full-length protein and one that produces an unstable truncated protein. The second transcript, which includes a pseudo-exon between exons 6 and 7 (6p), is widely expressed, whereas the first is more restricted. Differentiation of neuronal cells results in the removal of the pseudo-exon and subsequent SULT4A1 protein expression. Recent studies with SULT4A1 knockout mice showed that the protein is essential for normal development and that its absence leads to a severe neurologic phenotype. Here, the regulation of SULT4A1 6p splicing was investigated during neuronal differentiation using SH-SY5Y cells, human induced pluripotent stem cells, and mouse embryonic tissue. In all three models, pseudo-exon 6p was removed during differentiation, resulting in stable SULT4A1 protein expression. Using a minigene splicing assay, a region upstream of pseudo-exon 6p was identified that is essential for correct splicing of SULT4A1 mRNA. Within this region, there were binding motifs for four RNA processing factors (MBNL-1, MBNL-2, CELF-1, and CELF-2). Time-dependent changes in SULT4A1 protein and MBNL/CELF protein during differentiation supported their role in correctly splicing the SULT4A1 mRNA. Furthermore, ectopic expression of each factor produced efficient splicing in the minigene assay as well as correct splicing of the endogenous SULT4A1 mRNA. These results show that SULT4A1 mRNA is a target for MBNL/CELF-dependent splicing, which may be essential in producing stable, functional SULT4A1.
Introduction
Sulfotransferase 4A1 (SULT4A1) is a cytosolic sulfotransferase expressed primarily in neuronal cells. It is highly conserved between species but its biologic function remains elusive. SULT4A1 polymorphisms have been associated with neurologic diseases such as schizophrenia (Brennan and Condra, 2005; Meltzer et al., 2008), and there has been conflicting evidence suggesting that SULT4A1 may be a biomarker for response to antipsychotic drugs such as olanzapine (Ramsey et al., 2014; Wang et al., 2014). Two recent studies highlighted the physiologic importance of SULT4A1. First, Falany’s group successfully deleted the gene in a murine model and observed a severe phenotype that included tremors, ataxia, abnormal gait, and absence seizures (Garcia et al., 2018). This important work suggests that SULT4A1 is fundamental to normal neuronal development. Second, two reports independently showed that SULT4A1 protein expression is dependent on alternative splicing of its transcript (Sidharthan et al., 2014; Hashiguchi et al., 2018). Whereas the SULT4A1 protein is mostly confined to neuronal tissue, its mRNA is more widely expressed (Falany et al., 2000; Sidharthan et al., 2014). Two different transcripts have been reported, one that includes a pseudo-exon between exons 6 and 7 (6p), termed the variant transcript, and one where 6p has been removed, termed the wild-type (WT) transcript. The variant transcript introduces a premature stop codon resulting in an unstable, truncated protein (Sidharthan et al., 2014). In human tissues, SULT4A1 protein expression correlates well with the WT transcript, consistent with the notion that the variant transcript does not produce measurable protein levels (Sidharthan et al., 2014). Thus, alternative splicing appears to be an important mechanism for regulating SULT4A1 expression.
In both neuroblastoma SH-SY5Y and SK-N-MC cells, the switch from the variant to the WT transcript is seen during cellular differentiation (Sidharthan et al., 2014). This alternative splicing is also observed over time in cultured neuron-glia mixed cells (Hashiguchi et al., 2018). Alternative splicing of RNA results in multiple protein isoforms from a single gene. Of the approximately 20,000 protein-coding genes in human cells, more than 90% generate multiple transcripts, providing greater diversity to the human genome (Scotti and Swanson, 2016). Splicing is regulated by RNA processing factors that recognize explicit RNA binding motifs and direct the splicing machinery to specific splice sites. These factors are expressed in a cell-specific and a temporal-dependent manner to regulate protein function. During embryonic development, alternative splicing is central to cell reprogramming. Thus, it is not surprising that errors in splicing are associated with many human diseases such as Duchenne muscular dystrophy, amyotrophic lateral sclerosis, dilated cardiomyopathy, and frontotemporal dementia (Scotti and Swanson, 2016).
In this study, the regulation of SULT4A1 splicing was investigated to identify potential splicing factors responsible for the maturation of SULT4A1 mRNA. Several models of cell differentiation were used, including retinoic acid (RA)–treated neuroblastoma cells, differentiated human induced pluripotent stem cells (hiPSCs), and mouse embryonic tissue.
Materials and Methods
Cell Culture and Differentiation.
SH-SY5Y cells were grown in advanced Dulbecco’s modified Eagle’s medium/F12 medium containing 10% fetal calf serum, 4 mM glutamine, and 1% penicillin/streptomycin in a humidified atmosphere of 95% air, 5% CO2 at 37°C. The cells were differentiated in the same medium with 10 μM trans-RA (Sigma, St. Louis, MO) as previously described, in which differentiation was confirmed morphologically and by the upregulation of NeuN expression (Encinas et al., 2000; Sidharthan et al., 2014). hiPSCs (C11; WiCell, Madison, WI) were cultured in mTeSR/1 (STEMCELL Technologies, Tullamarine, VIC, Australia)/Matrigel (BD Biosciences, North Ryde, NSW, Australia) as previously described (Ovchinnikov et al., 2015) and differentiated with 10 μM trans-RA for up to 28 days. The neuronal marker NeuN increased with time, as determined by Western blot (Supplemental Fig. 1A). Differentiation of the hiPSCs was also evident from the morphologic changes where the cells formed spheroids with prominent dendritic outgrowths (Supplemental Fig. 1B).
Cell Transfections.
For minigene splicing assays, SH-SY5Y cells were seeded in six-well plates at 0.5 × 106 cells/well and allowed to adhere overnight. Cells were then transiently transfected with 2 μg minigene plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, Scoresby, VIC, Australia). After 4 hours of transfection, cells were treated with 10 μM RA for 5 days before processing for downstream assays. For experiments with coexpression of splicing factors and minigene constructs, cells were cotransfected with 1 μg minigene plasmid and 3 μg FLAG-tagged MBNL- or CELF-expressing plasmids (kind gifts from Thomas Cooper, Baylor College of Medicine, Houston, TX). To create nonclonal SH-SY5Y cell lines that stably express either MBNL or CELF proteins, FLAG-tagged MBNL or CELF plasmids were first linearized by digestion with the endonuclease enzyme PciI, gel purified, and transfected into SH-SY5Y cells using Lipofectamine 2000. The transfected cells were then subcultured four times under antibiotic selection before the stable expression of the splicing factor proteins was confirmed by Western blot.
Whole Mouse Fetal Samples.
Female CD1 mice were selected on gestational days 8.5, 10.5, 12.5, 14.5, and 18.5 of pregnancy to be euthanized for analysis of fetal tissues. All procedures were reviewed by and received approval from the University of Queensland Animal Ethics Office (approval numbers AE03917 and AE02544). Total RNA was isolated using Trizol (Thermo Fisher Scientific) and converted to cDNA. Polymerase chain reaction (PCR) for SULT4A1 was performed as described below. Protein samples for E8.5, E12.5, and E14.5 fetal tissues were prepared in radioimmunoprecipitation assay buffer (150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, and 50 mM Tris, pH 8.0) by brief (3 × 5 seconds) sonication on ice. Protein samples (50 µg) were electrophoresed and Western blotted for SULT4A1 as described below.
Reverse Transcription PCR.
Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Chadstone, VIC, Australia) and 2 μg was reverse transcribed to cDNA using SuperScript II (Thermo Fisher Scientific). PCRs were performed to study the pattern of expression of transcripts of SULT4A1, minigenes, and various neuronal splicing factors, and β-actin was used as the control. All primers used, along with the PCR conditions for amplification, are listed in Supplemental Table 1. The PCR amplicons were then separated on 2% agarose gels containing HydraGreen (1:20,000; ACTGene, Piscataway, NJ) for visualization.
Minigene Cloning and Splicing Assay.
Genomic DNA was isolated from SH-SY5Y cells using the Wizard Genomic DNA Purification Kit (Promega, Alexandria, NSW, Australia) and the alternative pseudo-exon 6p with its 623 bp 5′-upstream and 821 bp 3′-downstream flanking intronic sequences was amplified using AccuPrime Pfx DNA polymerase (Thermo Fisher Scientific) and forward and reverse primers with engineered KpnI and HindIII restriction sites, respectively (Supplemental Table 2). The PCR conditions used for amplification were as follows: initial denaturation at 95°C for 5 minutes, followed by 35 cycles of melting at 95°C for 15 seconds, annealing at 58°C for 30 seconds, and elongation at 68°C for 1.5 minutes. The PCR product was electrophoresed on a 0.8% agarose gel, purified using the PureLink Quick Gel Extraction Kit (Thermo Fisher Scientific), and then double digested with KpnI and HindIII high-fidelity enzymes in CutSmart buffer (New England Biolabs, Ipswich, MA) for 2 hours. The PCR product was first cloned into p-Bluescript (Agilent Technologies, Santa Clara, CA) and the NdeI restriction site in the intronic region of the insert upstream of pseudo-exon 6p was removed by site-directed mutagenesis using the GeneArt Site-Directed Mutagenesis System (Thermo Fisher Scientific) per the manufacturer’s instructions. The insert was then reamplified from p-Bluescript using forward and reverse primers with engineered NdeI restriction sites and the amplified product was digested with NdeI (New England Biolabs) and recloned into the same site in the pTBNde minigene plasmid, a gift from Franco Pagani (plasmid 15125; Addgene, Watertown, MA) (Pagani et al., 2003). A series of deletion minigene constructs were generated by amplifying the different segments of the parent insert with AccuPrime Pfx DNA polymerase using primers listed in Supplemental Table 2. The sequence of each minigene insert was verified by DNA sequencing prior to use in the minigene splicing assay. Minigene constructs were transiently transfected into SH-SY5Y cells using Lipofectamine 2000 as described above and the splicing patterns examined by reverse transcription (RT) PCR.
Western Blot Analysis.
Cells were washed twice with cold phosphate-buffered saline and then lysed in radioimmunoprecipitation buffer. The lysates were centrifuged at 14,000g for 10 minutes at 4°C, supernatants were recovered, and protein was quantified by the Bradford assay (Bradford, 1976). Cytosolic and nuclear protein fractions were prepared with a cell fractionation kit (Abcam, Melbourne, VIC, Australia). All protein samples (10 μg) were electrophoresed on 12% SDS-PAGE gels, transferred to nitrocellulose membranes, and blocked in 5% skim milk in phosphate-buffered saline with Tween 20 for 1 hour. Membranes were incubated overnight at 4°C with primary antibody and then with horseradish peroxidase–conjugated secondary antibody for 1 hour. The primary antibodies used in this study were as follows: anti-SULT4A1 (12578-1-AP; ProteinTech Group, Chicago, IL), anti–MBNL-1 (ab108519; Abcam), anti–CELF-1 (ab9549; Abcam), anti–MBNL-2 (sc-136167; Santa Cruz Biotechnology, Dallas, TX), anti–CELF-2 (sc-47731; Santa Cruz Biotechnology), anti–β-actin (3700; Cell Signaling Technology, Danvers, MA), anti–α-tubulin (3873; Cell Signaling Technology), anti-histone H4 (2592; Cell Signaling Technology), and anti–FLAG-M2 (A8592; Sigma).
Data Analysis.
Data were analyzed with either the t test or one-way analysis of variance using GraphPad Prism 7 software (GraphPad Software, San Diego, CA) and are presented as the mean ± S.E.M. Densitometry was performed with ImageJ software (National Institutes of Health, Bethesda, MD) to quantify the level of expression of proteins normalized to loading controls.
Identification of potential binding domains for trans-acting factors within the nucleotide sequences encompassing the alternative pseudo-exon 6p and its surrounding intronic regions was performed using SFmap (Akerman et al., 2009; Paz et al., 2010) in conjunction with SpliceAid 2 (Piva et al., 2012).
Results and Discussion
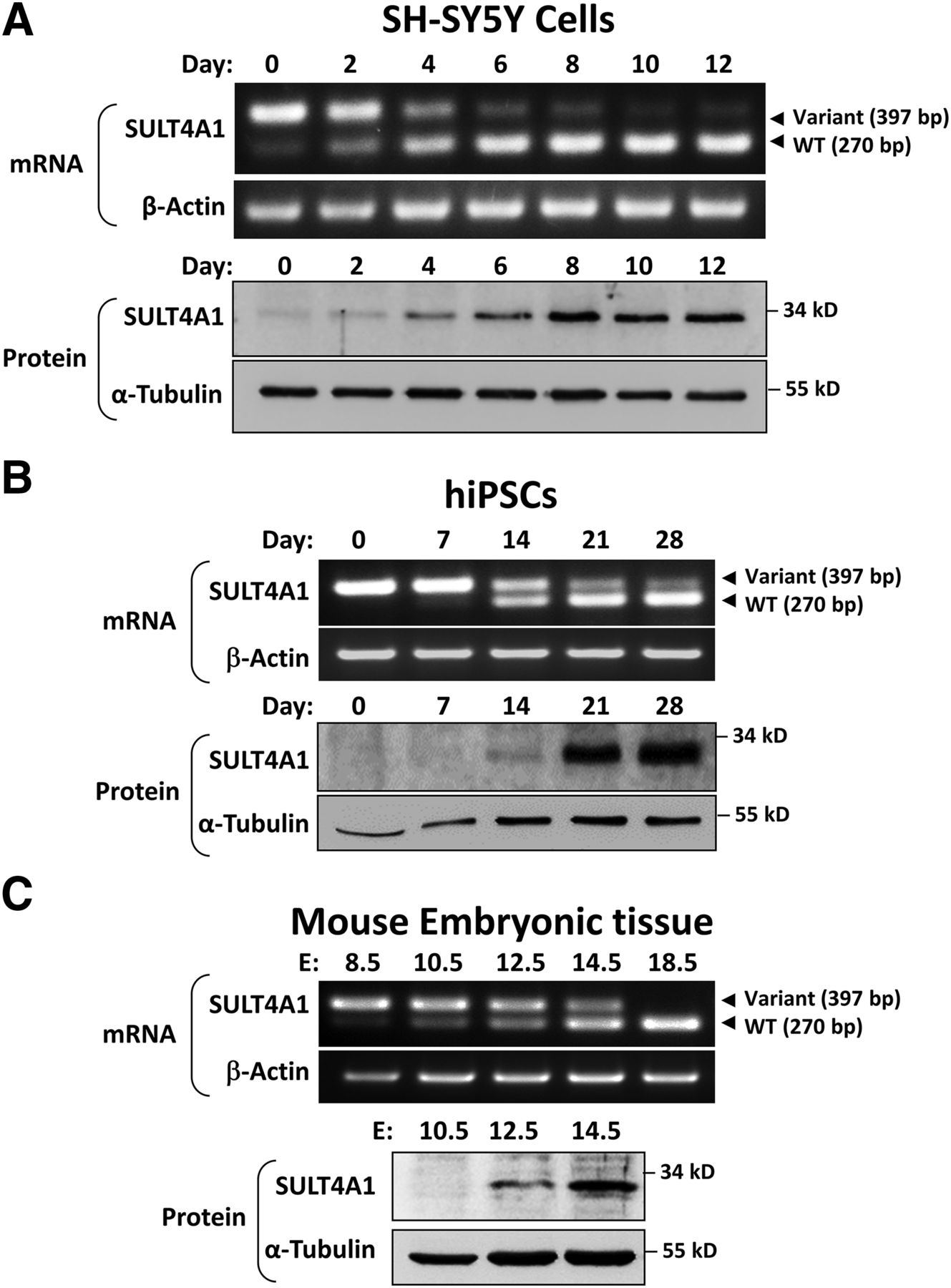
Initially, the expression of SULT4A1 was determined in SH-SY5Y cells and hiPSCs differentiated into neuronal-like cells. In the SH-SY5Y cells, differentiation was confirmed as described elsewhere (Sidharthan et al., 2014). cDNA from each cell line was amplified using primers located in exons 6 and 7 to give an amplicon of 397 bp when 6p was present or 270 bp when it was absent (Fig. 1A). SULT4A1 mRNA splicing changed over time from the variant to the WT transcript (Fig. 1A, upper panel). At the same time, SULT4A1 protein expression increased (Fig. 1A, lower panel). By day 12, nearly all of the SULT4A1 transcript was WT. In the hiPSCs differentiated over 28 days, neuronal-like cells were evident as shown by their morphologic characteristics and increased expression of the neuron-specific marker NeuN (Supplemental Fig. 1). Similar to that seen in the SH-SY5Y cells, SULT4A1 transcript splicing changed during differentiation (Fig. 1B, upper panel). At day 0, almost all of the transcript included exon 6p, whereas there was a marked shift to the WT transcript by day 14. This was accompanied by an increase in SULT4A1 protein expression. By day 28, the WT transcript was dominant and protein expression was maximal (Fig. 1B, lower panel). To assess SULT4A1 mRNA splicing in vivo, whole embryonic mouse tissue from E8.5 to E18.5 was analyzed (Fig. 1C). At E8.5, only the variant SULT4A1 transcript was present, whereas only the WT transcript was evident by E18.5. SULT4A1 protein expression correlated with the appearance of the WT transcript. These results show that SULT4A1 splicing in both cells and embryonic tissue changes in association with cell differentiation. Because there is little or no protein expression from the variant transcript, differential splicing of pseudo-exon 6p may be an important mechanism for the regulation of SULT4A1 protein expression at the post-transcriptional level. Moreover, the similar results seen in the human and mouse cells, along with the data by Hashiguchi et al. (2018), indicate that the change in SULT4A1 splicing may be consistent across species.

SULT4A1 transcript and protein expression during cell differentiation and mouse embryonic development. (A) SH-SY5Y cells were cultured for up to 12 days in Dulbecco’s modified Eagle’s medium/F12 medium in the presence of 10 µM RA. At each time point, cells were harvested for mRNA and protein determination. RT-PCR was performed using primers located in exon 6 and exon 7. The variant transcript, which contained pseudo-exon 6p, produced a PCR product of 397 bp, whereas the WT transcript produced a PCR product of 270 bp. RT-PCR results are shown in the upper two images, whereas SULT4A1 protein is shown in the lower two images. (B) SULT4A1 mRNA and protein in hiPSCs during RA-induced differentiation. (C) Expression of SULT4A1 mRNA and protein in mouse embryonic tissue from day E8.5 to E18.5. β-actin was used as a housekeeping gene for RT-PCR and α-tubulin as a loading control for Western blots. Molecular weight markers are shown on the right of each Western blot.
To identify possible intronic sequences that regulate SULT4A1 mRNA splicing, a minigene splicing assay was used (Pagani et al., 2003). A genomic fragment containing pseudo-exon 6p with flanking intronic sequences (623 bp 5′-upstream and 821 bp 3′- downstream) was cloned into the minigene construct (Supplemental Fig. 2A). Differentiated SH-SY5Y cells were transiently transfected with the minigene and mRNA was isolated for analysis by RT-PCR using forward and reverse primers located upstream and downstream of the insert. This allowed for identification of mRNA both with and without pseudo-exon 6p (Supplemental Fig. 2B). Next, deletion mutants of the minigene were constructed by sequentially removing sequences in the upstream and downstream regions of pseudo-exon 6p. Each minigene was transiently transfected into SH-SY5Y cells with and without RA-induced differentiation, and mRNA was isolated to evaluate splicing. Endogenous SULT4A1 mRNA was also measured as a positive control (Fig. 2A, right panels). The full-length minigene showed differentiation-dependent splicing of 6p (Fig. 2A). The ratio of the variant to the WT transcript is shown to the right of each gel, and this was 0.69 for the full-length minigene. Deletion of the upstream sequence to 208 bp (5′Δ406) showed splicing of 6p similar to that seen with the full-length construct (ratio = 0.73). However, when the upstream sequence was deleted to only 176 bp, very little splicing of the minigene was observed, although endogenous SULT4A1 mRNA was efficiently spliced in the same cells. Similar deletions in the downstream sequence had little effect on splicing (Fig. 2A, 3′Δ528 and 3′Δ220). These results suggested that a region essential for the splicing of 6p is located between 176 and 208 bp upstream of the pseudo-exon. This 32-bp sequence is shown in Fig. 2B (in red). Analysis of this region using SpliceAid 2 and SFmap databases identified potential cis-elements for two RNA processing factors: MBNL and CELF (Fig. 2B). Both interact with similar sequences comprising CUG/CUU motifs (Lambert et al., 2014).

Minigene splicing assay of SULT4A1 pseudo-exon 6p in SH-SY5Y cells differentiated with RA. (A) Deletion minigene assays: 10 minigene plasmids with sequential deletion of intronic segments of the approximately 1.6 kb full-length minigene were constructed and transfected into SH-SY5Y cells with and without RA treatment. Switching of the endogenous SULT4A1 transcript was used as a positive control (far right panels). The nomenclature of the deletion minigenes is based on the number of base pairs deleted from either the 5′ or 3′ ends of the full-length minigene. RA untreated (−) and treated (+) lanes are labeled and the ratio of the variant to the WT PCR product (V/WT) is shown next to the RT-PCR gels. Results are representative of duplicate experiments. (B) Sequence of the SULT4A1 gene upstream of pseudo-exon 6p (gray arrow) showing the 32-base sequence between 5′Δ406 and 5′Δ438 (red). Putative binding motifs for RNA processing factors are shown in the boxes with blue (MBNL) and green (CELF). Numbers refer to the number of bases upstream of the 6p pseudo-exon. (C) Effect of ectopic expression of the MBNL and CELF genes on splicing of the 5′Δ338 minigene construct in undifferentiated SH-SY5Y cells. Cells were transfected with each RNA splicing factor and the minigene. The upper panel shows the PCR products of the minigene transcript with the variant and WT indicated. Expression of each protein is shown as a FLAG Western blot below. (D) Effect of MBNL and CELF proteins on the splicing of endogenous SULT4A1 in undifferentiated SH-SY-5Y cells. The upper panel shows the variant and WT PCR products. Expression of each protein is shown as a FLAG Western blot below. Results are representative of duplicate experiments. Molecular weight markers are shown on the right of each Western blot. EV, empty vector.
To determine whether the MBNL and CELF RNA processing factors could splice the SULT4A1 minigene, each family member (MBNL-1, MBNL-2, CELF-1, and CELF-2) was ectopically expressed with the 5′Δ338 minigene into undifferentiated SH-SY5Y cells. All four RNA processing factors were able to splice the 5′Δ338 minigene (Fig. 2C, upper panel). Next, SH-SY5Y cells were stably transfected with a construct for each RNA processing factor and the splicing of endogenous SULT4A1 transcript was investigated (Fig. 2D). Again, all four factors were able to remove pseudo-exon 6p from the endogenous SULT4A1 transcript (Fig. 2D, upper panel). Taken together, these results indicate that either of the MBNL and CELF RNA processing factors can promote SULT4A1 6p splicing.
To identify which of the RNA processing factors are expressed during cell differentiation, SH-SY5Y cells were treated with RA over 10 days and MBNL/CELF mRNA was estimated by RT-PCR (Fig. 3A). SULT4A1 splicing with time was evident in these experiments (uppermost panel). Moreover, all four factors were expressed in the untreated cells (day 0) as well as during differentiation. MBNL-2 mRNA is itself subject to exon splicing, which can regulate both its activity as well as its subcellular localization (Sznajder et al., 2016). There are three exons in the C terminus containing 54, 36, and 95 bases, respectively, which are differentially spliced, and these are evident in the RT-PCR products after differentiation of the SH-SY5Y cells (Fig. 3A).

Expression of SULT4A1, MBNL, and CELF transcripts in SH-SY5Y cells during RA-induced differentiation. (A) SH-SY5Y cells were treated with 10 µM RA for 10 days. RNA was isolated at the indicated times and amplified by RT-PCR. For MBNL-2, splice variants were detected. (B) Western blots of protein expression after 10 days of RA treatment. Quantification of protein levels is shown alongside each Western blot. Data are the mean ± S.D. (n = 3). *P < 0.05 (significant differences, t test). CTRL, control.
Since mRNA levels do not always correlate with protein expression, the levels of the CELF and MBNL proteins were quantified in the differentiated SH-SY5Y cells (Fig. 3B). Western blots of whole cell extracts showed increased SULT4A1 expression with RA treatment. By contrast, there was a significant decrease in CELF-1 and MBNL-1 protein expression. There was no change in CELF-2 when normalized to tubulin levels, whereas MBNL-2 protein increased significantly.
To determine changes in MBNL and CELF expression in another model of differentiation, each of the RNA processing factors was measured in the human pluripotent stem cells during differentiation. Similar to that seen in the SH-SY5Y cells, MBNL-2 protein increased with time (Fig. 4). However, changes in the expression of the other RNA processing factors were quite different. Both CELF-2 and MBNL-1 proteins increased until day 14, after which they decreased back toward levels seen in the undifferentiated cells. There was little change in the expression of CELF-1.

Expression of MBNL and CELF proteins in hiPSCs during neuronal differentiation with 10 µM RA. Proteins were identified by Western blots and quantified by densitometry as the ratio of protein to α-tubulin (MBNL proteins) or β-actin (CELF proteins). Molecular weight markers are shown on the right of each Western blot. For MBNL-1 and MBNL-2, closed symbols are the upper band and open symbols are the lower band.
In this study, the MBNL/CELF family of RNA processing factors was shown to splice the SULT4A1 variant transcript. Each of these proteins binds to similar RNA sequences to execute their splicing effects (Kalsotra et al., 2008; Wang et al., 2012; Konieczny et al., 2014). However, they are not always coordinately expressed. For example, in the developing heart, CELF-1 expression is downregulated and MBNL-1 is upregulated during differentiation (Kalsotra et al., 2010). Moreover, there are complex, and often competing, functions for the MBNL and CELF proteins (Kalsotra et al., 2008; Wang et al., 2015). For example, MBNL-1 can behave as a repressor of MBNL-2 activity by inhibiting MBNL-2 splicing. In MBNL-1 null mice, there is an upregulation of MBNL-2 in muscle and heart tissue, with its enrichment in the nuclear compartment (Lee et al., 2013). In this study, differentiation of both the SH-SY5Y cells and the hiPSCs resulted in a decrease in MBNL-1 expression and an increase in MBNL-2 expression. Moreover, the decrease in MBNL-1 was accompanied by alternative splicing of MBNL-2 (Fig. 4). These results support the reciprocal nature of their respective functions. It is noteworthy that variant SULT4A1 mRNA is widespread in human tissues but splicing of exon 6p appears to be controlled in a tissue- and time-specific manner. This may reflect the complexity of the MBNL/CELF splicing machinery, which also shows tissue- and time-specific expression in the body.
There is accumulating evidence suggesting that MBNL proteins have a dominant role in executing a switch from the fetal to adult pattern of mRNA splicing, with their levels increasing during development and differentiation (Han et al., 2013; Konieczny et al., 2014). By contrast, CELF-1 has an opposing role, orchestrating fetal splicing pattern with its level decreasing with development (Lee and Cooper, 2009; Poulos et al., 2011). MBNL proteins show increased expression in differentiated adult tissue, where MBNL-1 is responsible for adult type splicing in heart and skeletal muscle tissues. MBNL-2 is the dominant protein in brain and differentiated neurons (Charizanis et al., 2012; Wang et al., 2012). In this study, SULT4A1 protein was absent early in mouse embryogenesis but was highly expressed by E18.5. This suggests that SULT4A1 is part of the alternative splicing network in the developing nervous system (Raj and Blencowe, 2015), which is supported by the severe neurologic phenotype seen in SULT4A1 null mice (Garcia et al., 2018).
Although an in vivo function for SULT4A1 is currently lacking, the recently reported mouse knockout animals indicate that it is critical for normal development (Garcia et al., 2018). Thus, genetic alterations in the gene may be associated with developmental abnormalities. The human SULT4A1 gene has a very low incidence of mutations (single nucleotide polymorphism frequency <1:700), which supports a vital role in development (Minchin et al., 2008). This study highlights the importance of splicing for correct expression of the SULT4A1 protein. Thus, dysregulation of the RNA processing factors responsible for SULT4A1 splicing may generate a phenotype similar to that described in the mouse knockout model. Although functionally important genetic polymorphisms in the MBNL genes have not been widely reported, sequestration by transcripts with expanded CUG or CCUG repeats has been noted in the onset of myotonic dystrophy (Miller et al., 2000). It will be important in the future to determine whether SULT4A1 splicing is altered in pathologic conditions in which the activities of these splicing factors change.
Acknowledgments
We acknowledge assistance from Dr. James Hudson (University of Queensland) with differentiation of hiPSCs and Dr. Paul Dawson (Mater Research Institute) for mouse embryonic tissue and cDNA. We thank Dr. Thomas Cooper (Baylor College of Medicine) for the kind gift of the MBNL and CELF constructs and Dr. Franco Pagani for the original minigene construct.
Authorship Contributions
Participated in research design: Idris, Butcher, Minchin.
Conducted experiments: Idris, Butcher.
Performed data analysis: Idris, Minchin.
Wrote or contributed to the writing of the manuscript: Idris, Butcher, Minchin.
Footnotes
- Received October 31, 2018.
- Accepted December 19, 2018.
This work was supported by the National Health and Medical Research Council of Australia [Grant 1099135].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- 6p
- pseudo-exon 6
- hiPSC
- human induced pluripotent stem cell
- PCR
- polymerase chain reaction
- RA
- retinoic acid
- RT
- reverse transcription
- WT
- wild type
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}