


Abstract
Cytochrome P450 4F12 is a drug-metabolizing enzyme that is primarily expressed in the liver, kidney, colon, small intestine, and heart. The properties of CYP4F12 that may impart an increased catalytic selectivity (decreased promiscuity) were explored through in vitro metabolite elucidation, kinetic isotope effect experiments, and computational modeling of the CYP4F12 active site. By using astemizole as a probe substrate for CYP4F12 and CYP3A4, it was observed that although CYP4F12 favored astemizole O-demethylation as the primary route of metabolism, CYP3A4 was capable of metabolizing astemizole at multiple sites on the molecule. Deuteration of astemizole at the site of O-demethylation resulted in an isotope effect of 7.1 as well as an 8.3-fold decrease in the rate of clearance for astemizole by CYP4F12. Conversely, although an isotope effect of 3.8 was observed for the formation of the O-desmethyl metabolite when deuterated astemizole was metabolized by CYP3A4, there was no decrease in the clearance of astemizole. Development of a homology model of CYP4F12 based on the crystal structure of cytochrome P450 BM3 predicted an active site volume for CYP4F12 that was approximately 76% of the active site volume of CYP3A4. As predicted, multiple favorable binding orientations were available for astemizole docked into the active site of CYP3A4, but only a single binding orientation with the site of O-demethylation oriented toward the heme was identified for CYP4F12. Overall, it appears that although CYP4F12 may be capable of binding similar ligands to other cytochrome P450 enzymes such as CYP3A4, the ability to achieve catalytically favorable orientations may be inherently more difficult because of the increased steric constraints of the CYP4F12 active site.
Introduction
CYP4F12 is a member of the cytochrome P450 (P450) family of drug-metabolizing enzymes that is primarily expressed in the liver, small intestine, colon, kidney, and heart (Bylund et al., 2001; Hashizume et al., 2001; Guengerich, 2005). Currently, there is limited information available on the relative protein expression levels of CYP4F12; however, initial analyses would suggest that it is a widely expressed but relatively minor P450 isoform (Stark et al., 2004). Sequence alignment of the amino acid sequence of CYP4F12 indicates it is approximately 78–83% homologous with the other members of the CYP4F family. To date, seven allelic variants of the enzyme have been identified, with two of the seven exhibiting altered kinetics (Bylund et al., 2001; Cauffiez et al., 2004). CYP4F12 expression has been shown to be inducible through activation of the p53 pathway, a sequence-specific transcription factor known to regulate the expression of multiple P450 isoforms, and also exhibits an increased binding to the pregnane-X receptor in the presence of rifampicin (Hariparsad et al., 2009; Goldstein et al., 2013).
The P450 family of drug metabolizing enzymes is capable of catalyzing the metabolism of substrates with a wide array of structural and physical chemical attributes (Lewis et al., 1998, 1999; Johnson and Stout, 2005). CYP4F12 exhibits similar pharmacophore attributes to other CYP4 isoforms as well as P450 BM3, with known endogenous substrates including arachidonic acid, leukotriene B4, and prostaglandins (Bylund et al., 2001; Stark et al., 2004; Kikuta et al., 2007). CYP4F12 also catalyzes the metabolism of a number of xenobiotics including astemizole, terfenadine, and ebastine and is known to metabolize a number of the same substrates as CYP3A4 and CYP2J2 (Hashizume et al., 2002; Parikh et al., 2003; Lee et al., 2010). The comparison of the relative catalytic and inhibitory promiscuities of P450 isoforms identified a unique ligand profile for CYP4F12, with its susceptibility to inhibition being similar to P450 isoforms such as CYP3A4 but having a catalytic selectivity similar to that of substrate-specific homeostatic isoforms such as CYP26A1 (Foti et al., 2011).
The objective of the work presented here was to characterize the active site properties of CYP4F12 that result in a decreased catalytic promiscuity through detailed kinetic analyses and molecular modeling. A thorough structural assessment was conducted to determine which physicochemical properties result in an increased inhibitory potency for CYP4F12. Metabolite identification assays were carried out to identify the sites of metabolism for astemizole with CYP4F12 compared with CYP3A4, a P450 isoform with a well-documented ability to catalyze a wide range of substrates. By using kinetic isotope experiments, the ability of CYP4F12 to allow for metabolic switching for a given substrate in its active site was probed. Finally, a homology model of CYP4F12 was designed and compared with crystal structure data of CYP3A4 in an attempt to rationalize the current in vitro findings and ultimately to characterize the active site properties of CYP4F12.
Materials and Methods
Astemizole and astemizole-d3 were obtained from United States Biologic (Salem, MA). All other drug standards were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant cytochrome P450 3A4 and 4F12 were purchased from BD Biosciences (San Jose, CA). NADPH was ordered from Calbiochem (San Diego, CA). An Amplex Red hydrogen peroxide fluorometric detection kit and all high-performance liquid chromatography (HPLC)-grade solvents (acetonitrile, methanol, and water) were from Thermo Fisher Scientific (Waltham, MA).
In Vitro Clearance and IC50 Determination.
In vitro clearance values were determined for a set of 53 compounds using a substrate depletion approach. Incubations consisted of 10 pmol recombinant CYP4F12, 3 mM MgCl2, and 1 µM test compound in 100 mM potassium phosphate buffer (pH 7.4). After a 3-minute preincubation period at 37°C, incubations were initiated with 1 mM NADPH (final concentration). Aliquots were removed at 0, 2.5, 5, 10, 20, and 30 minutes and quenched with 2 volumes of ice-cold acetonitrile containing 500 nM tolbutamide as an internal standard. All in vitro clearance incubations were performed in duplicate. Samples were subsequently centrifuged for 10 minutes at 1130g in a Beckman Allegra 6R table top centrifuge (Beckman Coulter, Fullerton, CA). The resulting supernatants were transferred to 96-well polypropylene plates for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
Substrate depletion rates were determined by first normalizing the analyte concentrations to a percent remaining based on the T0 value and subsequently using linear regression to determine the slope (−k) of the ln(percent remaining) values as a function of incubation time (Obach et al., 1997). The rates (k) were converted to half-life values (t1/2 = 0.693/k) and ultimately used to calculate an in vitro clearance with units of milliliter per minute per nanomole P450 value using eq. 1.
 (1)
(1)Inhibition potency (IC50) was also determined for each of the 53 test compounds in a recombinant CYP4F12 incubation. In brief, incubations consisted of 1 pmol recombinant CYP4F12, 3 mM MgCl2, 0.25 µM terfenadine as the probe substrate, and 0–25 µM test compound. For incubations intended to determine the inhibition potency of terfenadine in recombinant CYP4F12, astemizole (0.15 µM, final concentration) was used as the probe substrate. Final organic solvent content was limited to less than 1% (v/v), and product formation was linear with respect to time and protein concentration. Reactions were initiated with 1 mM NADPH and were allowed to proceed for 5 minutes (terfenadine as probe substrate) or 1 minute (astemizole as probe substrate) before being quenched with 2 volumes of ice-cold acetonitrile containing 500 nM tolbutamide as internal standard. IC50 determinations were performed in duplicate. Samples were subsequently centrifuged for 10 minutes at 1130g in a Beckman Allegra 6R table top centrifuge (Beckman Coulter), and the resulting supernatants were transferred to 96-well polypropylene plates for LC-MS/MS analysis.
Inhibition parameters were estimated using Graphpad Prism (version 6.03; GraphPad Software Inc., San Diego, CA). Data were expressed as a percent of control (no inhibitor) and fit to a fixed-slope three parameter inhibition model to determine the IC50 (eq. 2), where max and min refer to the metabolite formation rates of the probe substrate at the lowest and highest inhibitor concentrations, respectively, and [I] is the concentration of inhibitor in the incubation.
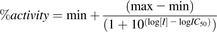 (2)
(2)Metabolite Identification in Recombinant CYP4F12 and CYP3A4.
Incubations were performed in recombinant CYP3A4 and CYP4F12 to identify the metabolites of astemizole formed by each enzyme. Reactions consisted of 20 pmol recombinant protein, 3 mM MgCl2, and 10 µM astemizole or astemizole-d3 in 100 mM potassium phosphate buffer (pH 7.4). After a 5-minute preincubation period at 37°C, the incubations were initiated with 1 mM NADPH (final concentration) and allowed to proceed for 20 minutes. Metabolite identification incubations were extracted into 3 volumes of ethyl acetate and vortexed for 5 minutes. The organic layer was collected and evaporated to dryness under a gentle stream of N2. Samples were reconstituted in water:acetonitrile (95:5) and placed into vials for LC-MS/MS analysis.
Kinetic Isotope Effects on Intrinsic Clearance and Metabolite Formation.
O-Demethylation of the methoxyphenyl moiety of astemizole was previously reported to be a major metabolic pathway for astemizole by CYP4F12 (Matsumoto and Yamazoe, 2001). Substrate depletion (intrinsic clearance) and enzyme kinetic experiments were conducted in recombinant CYP3A4 or CYP4F12 to determine the effects of converting the methoxy group (−OCH3) of astemizole to a d3-methoxy group (−OCD3) on the in vitro metabolism characteristics of astemizole. Briefly, intrinsic clearance incubations consisted of 1 pmol CYP4F12 or CYP3A4, 3 mM MgCl2, and 0.15 µM astemizole or astemizole-d3 in 100 mM potassium phosphate buffer (pH 7.4). After a 3-minute preincubation at 37°C, incubations were initiated with 1 mM NADPH (final concentration). Aliquots were removed at 0, 2.5, 5, 10, 20, and 30 minutes and quenched with 2 volumes of ice-cold acetonitrile containing 500 nM tolbutamide as an internal standard. In vitro half-lives were obtained using linear regression from 0 to 5 minutes for astemizole and from 0 to 20 minutes for astemizole-d3, and resulting in vitro clearance values calculated using eq. 1. Enzyme kinetic experiments contained 1 pmol recombinant CYP4F12 or CYP3A4, 3 mM MgCl2, and varying concentrations of astemizole or astemizole-d3 from 0 to 100 μM depending on the metabolic pathway. Enzyme kinetics in recombinant CYP4F12 and CYP3A4 incubations were terminated after 1 and 2.5 minutes, respectively, to ensure Michaelis-Menten reaction conditions and product formation linearity with respect to time and protein concentration. Reactions were quenched with 500 nM tolbutamide in ice-cold acetonitrile, centrifuged (1160g for 10 minutes), and transferred to 96-well polypropylene plates for LC-MS/MS analysis. Enzyme kinetic parameters were determined using nonlinear regression analysis fit to either a Michaelis-Menten model (eq. 3) or a substrate inhibition model (eq. 4). For the enzyme kinetic equations, Km refers to half the substrate concentration ([S]) at maximal reaction velocity (Vmax). Ki is the dissociation constant for a single substrate molecule binding to the enzyme in such a way as to decrease the rate of product formation. Kinetic isotope effects are expressed as the ratio of [(Vmax/Km),H/(Vmax/Km),D].
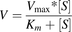 (3)
(3)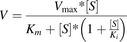 (4)
(4)Estimation of NADPH Consumption/H2O2 Formation.
Incubations consisted of 1 pmol recombinant CYP3A4 or CYP4F12 and 2 μM astemizole or astemizole-d3 in 100 mM potassium phosphate buffer (pH 7.4). Reactions were initiated with 1 mM NADPH (final concentration). A Cary 4000 UV-visible spectrophotometer (Agilent, Santa Clara, CA) was used to measure NADPH consumption using previously reported methods by monitoring absorbance at 340 nm (Xiao et al., 2011). A value of 6.22 mM−1⋅cm−1 for Δε340 was used in calculating the NADPH consumption rate. Hydrogen peroxide formation was monitored using an Amplex Red hydrogen peroxide assay following guidelines provided by the manufacturer (Mishin et al., 2010). A hydrogen peroxide detection cocktail of 100 μM Amplex Red reagent, 0.2 U/ml horseradish peroxidase, and 1× reaction buffer (final concentrations) was added to the incubation after 2 minutes at 37°C, and the formation of resorufin was monitored at an excitation wavelength of 545 nm and an emission wavelength of 590 nm.
In Vitro Sample Analysis.
Sample analysis for in vitro clearance and IC50 samples was conducted using LC-MS/MS technology. Briefly, the LC-MS/MS platform consisted of an Applied Biosystems 4000 QTrap fitted with an electrospray ionization source (Applied Biosystems, Foster City, CA). Generic instrument parameters included curtain gas (40 arbitrary units), CAD gas (medium), ion spray voltage (4700 V), source temperature (500°C), and ion source gas 1 and gas 2 (35 arbitrary units each). Dwell time was 200 ms and interface heaters were kept on for all analytes. Positive ionization multiple reaction monitoring transitions for astemizole, terfenadine, and tolbutamide were 459.2/135.1, 471.9/436.3, and 271.2/91.1, respectively. Specific instrument parameters for the 53 test compounds are listed in Supplemental Table 1.
The QTrap mass spectrometer was coupled to a liquid chromatography system consisting of two LC-20AD pumps with an in-line CBM-20A controller and DGU-20A5 solvent degasser (Shimadzu, Columbia, MD) and to a LEAP CTC HTS PAL autosampler equipped with a dual-solvent self-washing system (CTC Analytics, Carrboro, NC). HPLC separation was achieved using a Gemini C18 2.0 × 30 mm 5-µm column (Phenomenex, Torrance, CA). Rapid gradient elution (flow rate = 400 µl/min) was carried out using a mobile phase system consisting of (A) 5 mM ammonium formate with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. HPLC flow was diverted to waste for the first minute to remove any nonvolatile salts. An injection volume of 10 μl was used for all analytes.
For metabolite identification analyses, chromatographic separation was achieved using a Kinetex C18 2.1 × 100 mm 2.7-μm column (Phenomenex). Gradient elution (flow rate = 200 µl/min) was carried out using a mobile phase system consisting of (A) water with 0.1% formic acid and (B) acetonitrile:methanol (1:1) with 0.1% formic acid. HPLC flow was diverted to waste for the first 2 minutes to remove any nonvolatile salts. An injection volume of 15 μl was used for all analyses. Mobile phase B was held at 2.5% for the first 2 minutes, increased linearly from 2.5 to 33% from 2 to 6.5 minutes, from 33 to 45% from 6.5 to 16 minutes, increased to 95% at 17 minutes, and held at 95% for 1 minute before re-equilibration at 2.5% B for 2 minutes. Metabolites were initially identified using a Q1 scan (100–800 amu). Additional analysis was conducting using multiple reaction monitoring transitions for astemizole and its metabolites. The analytes monitored were astemizole (459.2/135.1), O-desmethylastemizole (445.3/121.4), N-desalkylastemizole (325.3/109.5), and hydroxylation at the 6-OH position of the benzimidazole moiety (475.3/135.2), fluorophenyl moiety (475.3/135.2), or methoxyphenyl moiety (475.2/151.4).
CYP4F12 Homology Modeling.
A three-dimensional homology model of CYP4F12 was developed using Prime (Schrodinger, Portland, OR). The CYP4F12 amino acid sequence was obtained from GenBank (AAQ89336.1) with the crystal structure of P450 BM3 (PDB 1JPZ) serving as the template for the homology model (30% sequence identity). While BLAST searches identified CYP3A4 as another plausible template based on sequence identity and total sequence alignment score, BM3 was chosen as the template as to not bias the subsequent comparison with the CYP3A4 crystal structure. The structural plausibility of the CYP4F12 homology model was determined through evaluation of the Ramachandran plot and through visualization of bond angles and lengths. Local flexibility/rigidity was assessed by visual inspection of helices compared with loop regions and through secondary structure predictions performed in PSIPRED (University College London, UK) and SSPro (Schrodinger). The active site of the CYP4F12 homology model was defined using SiteMap (Schrodinger) and a docking grid generated using a 14 × 14 × 14 Å3 box that was used to constrain the center of mass of the docked ligand. The astemizole structure was prepared before docking using LigPrep 2.0 (Schrodinger), which converts two-dimensional .mol files to three-dimensional molecular structures with defined stereocenters. After conversion, the astemizole structure underwent a minimization step using a distance dependent dielectric model and the OPLS_2005 force field. Astemizole was then docked into the homology model of CYP4F12 or the crystal structure of CYP3A4 (PDB 1W0G) using Glide (Schrodinger) and evaluated by GlideScore and eModel. Induced-fit docking algorithms (Sherman et al., 2006a,b), which allow for reduced van der Waals radii, removal, and reorientation of nonrigid protein side chains and a second energy minimization step upon ligand docking, were used to dock astemizole into the homology model of CYP4F12 and for two of the three orientations within the active site of the CYP3A4 crystal structure model (O-demethylation and N-dealkylation orientations).
CAVER 3.0.1 (CaverSoft, Brno, Czech Republic) was used to estimate the active site volumes of the CYP4F12 homology model and the CYP3A4 crystal structure as previously described (Petrek et al., 2006; Chovancova et al., 2012; VandenBrink et al., 2012). Protein structures were prepared in Maestro (Schrodinger) before CAVER analysis. Initial optimization steps included correction for missing hydrogen atoms, removal of water molecules, and definition of the formal charge on the heme iron at Fe3+. The Maestro script automatically assigned protonation states for all arginine, glutamine, and histidine residues. After optimization of the hydrogen bonding network, the entire protein structure was subject to a restrained energy minimization after which each protein structure was imported into CAVER. The active site of each enzyme was defined with respect to a water ligand that was positioned axial to the heme prosthetic group. Active site volume estimates were obtained using embedded molecular dynamics trajectory files within CAVER and graphically visualized through PyMOL (Schrodinger).
Results
The in vitro inhibition potency and in vitro clearance of 53 compounds selected to sample a diverse cross section of the chemical space of "drug-like" compounds were investigated using recombinant CYP4F12. The resulting IC50 values of the molecules ranged from 78 nM (amiodarone) to >100 μM (multiple compounds). Only astemizole and terfenadine exhibited measurable turnover in recombinant CYP4F12 incubations, with in vitro clearance values of 70.1 ml/min per nmol P450 and 60.8 ml/min per nmol P450, respectively (Supplemental Table 1). Examination of the structural and physicochemical properties of the 53 test compounds indicated that the lipophilic nature of the compounds (cLogP) had the greatest impact on inhibition potency (Fig. 1). Furthermore, there were no significant trends observed between inhibition potency and molecular weight, polar surface area, pKa, the number of acidic or basic groups, the number of hydrogen bond donors or acceptors, or the number of aromatic rings (Pearson coefficient <0.25; data not shown).

IC50 (μM) and in vitro clearance (ml/min per nmol) values for a set of 53 test compounds incubated in recombinant CYP4F12. Inhibition data are represented on the left axis by open blue circles and clearance data on right axis by open red squares. Astemizole and terfenadine are highlighted with solid symbols.
Experiments carried out to identify the sites of astemizole metabolism in recombinant CYP4F12 confirmed previously published reports that O-demethylation is the primary metabolic clearance pathway for astemizole by CYP4F12 (fraction metabolized [fm] = 0.99). The fraction metabolized was calculated as a ratio of the peak area to internal standard area for each respective metabolite to the sum of the peak area to internal standard areas for all observed metabolites. Trace amounts of 6-hydroxyastemizole, fluorophenyl-OH astemizole, and N-desalkylastemizole were also observed (Fig. 2). Incubations with recombinant CYP3A4 identified multiple oxidative metabolites. In addition to O-demethylation (fm = 0.39), 6-hydroxyastemizole (fm = 0.38), hydroxylation on the methoxyphenyl moiety (fm = 0.16), fluorophenyl hydroxylation (fm = 0.06), and N-desalkylastemizole (fm = 0.01) were also identified in recombinant CYP3A4 incubations.

Sites of metabolism for astemizole by CYP4F12 and CYP3A4. Astemizole is primarily metabolized to O-desmethylastemizole (solid arrow) by CYP4F12 (fm >99%; middle panel) and to multiple metabolites (open arrows) by CYP3A4 (lower panel; peaks highlighted with an asterisk were also observed in control samples).
When deuterium was incorporated at the site of O-demethylation, the in vitro clearance of astemizole in recombinant CYP4F12 incubations decreased by 8.3-fold (Fig. 3). Conversely, the effect of deuteration on the in vitro clearance of astemizole was negligible for incubations conducted with recombinant CYP3A4. An isotope effect [(Vmax/Km),H/(Vmax/Km),D] of 7.1 and 3.8 was observed for the formation of O-desmethyl astemizole by CYP4F12 and CYP3A4, respectively (Figs. 4 and 5; Table 1). For CYP3A4, isotopically sensitive branching resulted in an increased formation of 6-hydroxyastemizole [(Vmax/Km),H/(Vmax/Km),D = 0.80], fluorophenyl-OH astemizole [(Vmax/Km),H/(Vmax/Km),D = 0.47], and methoxyphenyl-OH astemizole [(Vmax/Km),H/(Vmax/Km),D = 0.45]. No effect was observed on the formation of 6-hydroxyastemizole or fluorophenyl-OH astemizole by CYP4F12 or for N-dealkylation by either isoform. Recombinant CYP4F12 incubations with deuterated astemizole also resulted in a NADPH consumption rate that was approximately 1.5- to 2.2-fold higher than the NADPH consumption rate that was observed for astemizole (Fig. 6). No significant increase in the formation of H2O2 was observed for CYP4F12 incubations with deuterated astemizole (data not shown). Similar to the effects on in vitro clearance, deuteration of the methoxy moiety of astemizole had negligible effects on the NADPH consumption rate and formation of H2O2 when incubations were conducted with recombinant CYP3A4.

In vitro clearance data for astemizole (AST) and astemizole-d3 (d3-AST). Astemizole clearance by CYP4F12 was reduced 8.3-fold upon deuteration of the α-carbons of the methoxy moiety (top panel). No effect was observed for incubations with CYP3A4 (bottom panel).

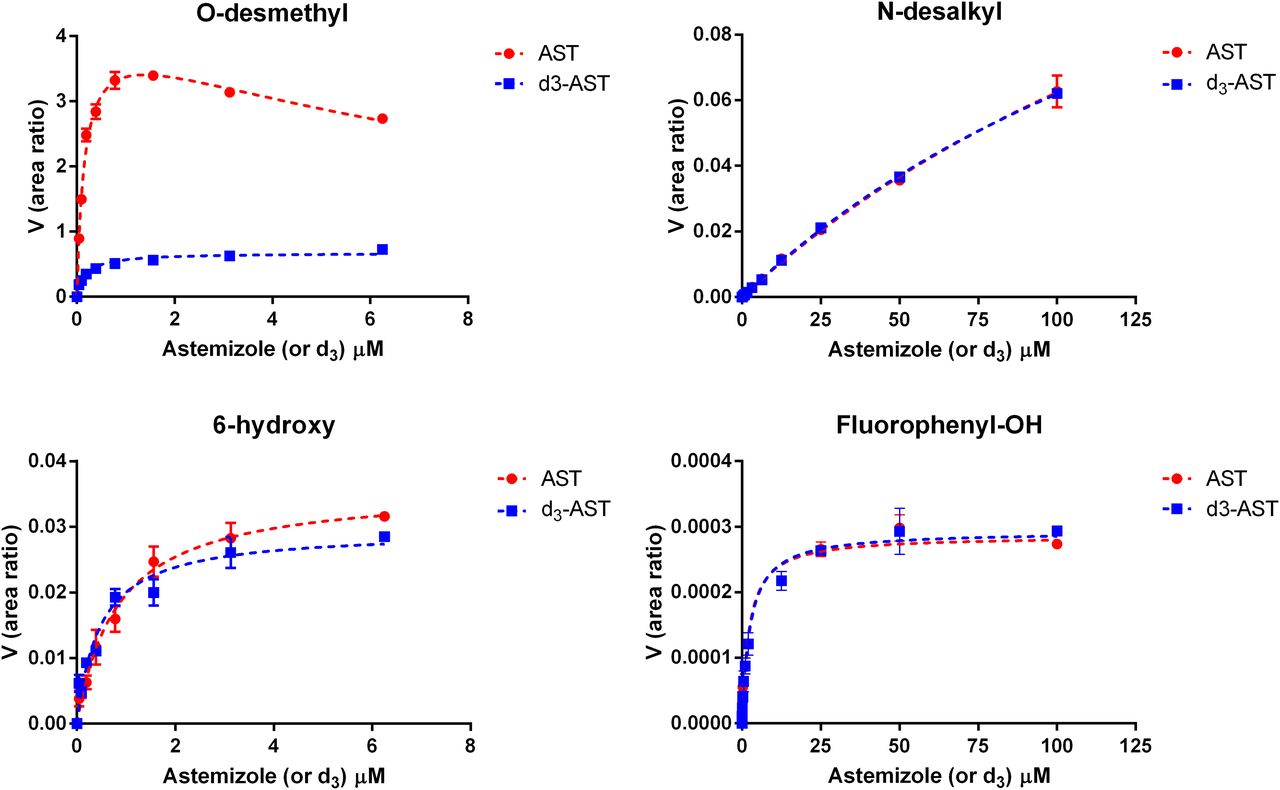
Metabolite formation kinetics for astemizole (AST) and astemizole-d3 (d3-AST) by CYP4F12. An isotope effect of 7.1 was observed for the formation of O-desmethylastemizole with negligible effects noted for the alternative pathways.
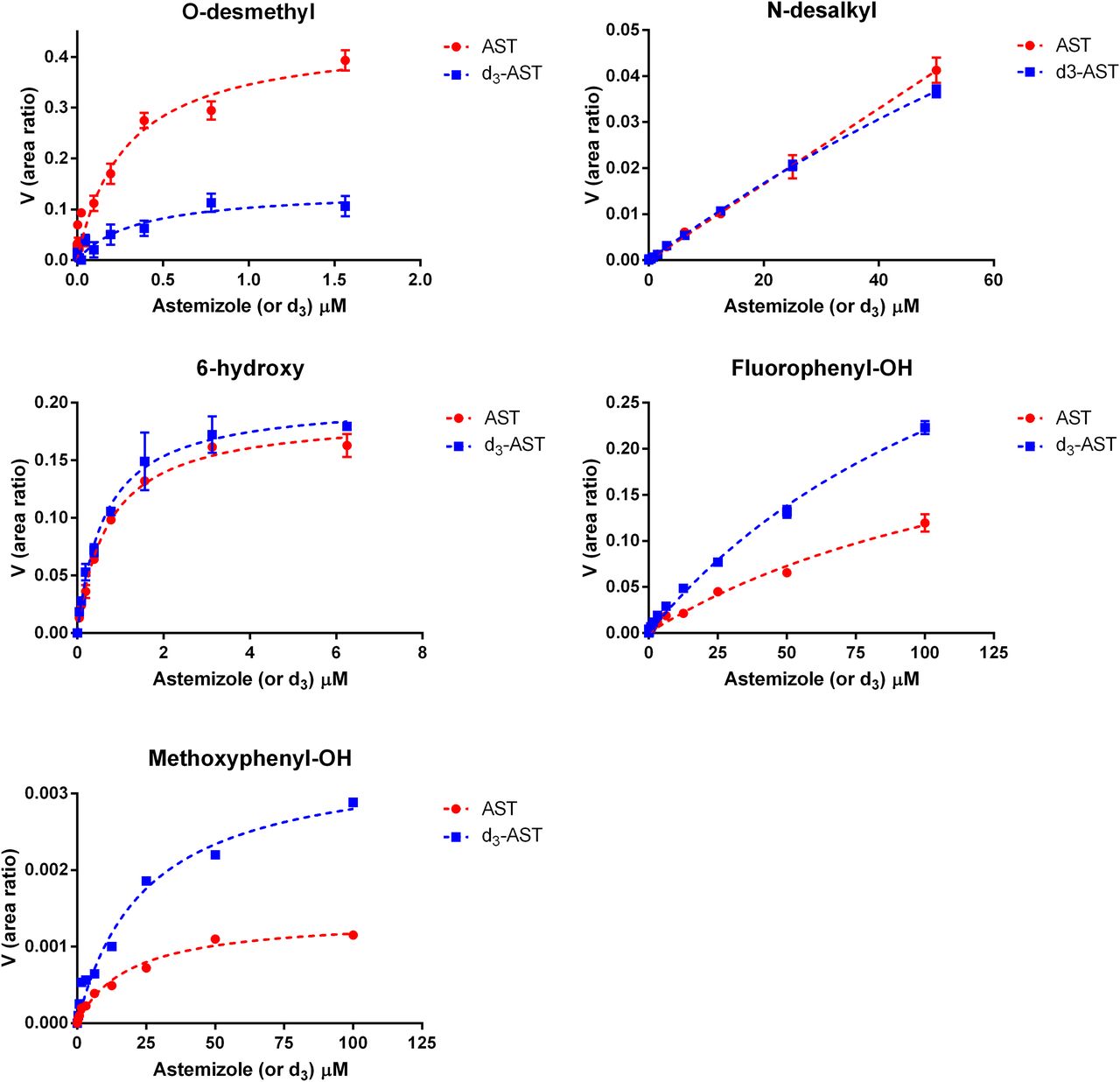
Metabolite formation kinetics for astemizole (AST) and astemizole-d3 (d3-AST) by CYP3A4. An isotope effect of 3.8 was observed for the formation of O-desmethylastemizole with a corresponding inverse isotope effect noted for 6-hydroxyastemizole (0.8), fluorophenyl-OH astemizole (0.47), and methoxyphenyl-OH astemizole (0.45).
Metabolite formation kinetics for astemizole and d3-astemizole
Km and Ki parameters are expressed in μM, Vmax as the ratio of analyte peak area to internal standard peak area, and the kinetic isotope effect (H/D) as the ratio of [(Vmax/Km),H/(Vmax/Km),D].

NADPH consumption rates for astemizole (AST) and astemizole-d3 (d3-AST). An increase in the NADPH consumption rate of approximately 2-fold was observed for astemizole-d3 in incubations with CYP4F12 (top panel). A minimal change was observed for CYP3A4 (bottom panel). DMSO, dimethylsulfoxide.
A homology model was designed for CYP4F12 using P450 BM3 (PDB 1JPZ) as a template and compared with CYP3A4 using crystal structure data (PDB 1W0G). Induced-fit docking was used to orient astemizole in the active site of each model. For CYP4F12, only a single substrate orientation was achieved with the homology model, with the protons of the methoxy carbon located approximately 2.25 Å from the heme iron (Fig. 7A). Key amino acid residues included Phe397 and Leu421, which were located within 2 Å of the fluorophenyl moiety, and Asp121 and Pro396, which were within 2 Å of the benzimidazole and piperidine moieties, respectively. Phe124, Leu137, Phe327, and Thr332 were located within 3 Å of the methoxyphenyl ring. Ile67, Leu74, Ser77, and Ile239 appeared to delineate the boundaries of the CYP4F12 active site beyond the fluorophenyl ring of astemizole. A CAVER analysis predicted the volume of the CYP4F12 active site to be ∼947 Å3. When astemizole was docked in the CYP3A4 in an orientation consistent with O-demethylation (methoxy protons approximately 2.66 Å from heme iron), Phe57 and Phe215 were located within 2 Å of the benzimidazole and fluorophenyl moieties, respectively. Arg105, Ser119, Ala305, Met371, and Arg372 were also located within 2–3 Å of astemizole (Fig. 7B). When the orientation of astemizole was reversed to allow for hydroxylation on either the benzimidazole or fluorophenyl moieties (benzimidazole protons ∼3.23 Å from the heme iron), Arg105 and Arg106 were located within 2 Å of the piperidine backbone of astemizole, with Ser119 located within 2 Å of one of the benzimidazole nitrogens (Fig. 7C). Phe215 was within 3 Å of the methoxyphenyl ring, and Ile369 and Ala370 were within 3 Å of the fluorophenyl moiety. Tyr53, Phe57, and Glu374 were also located within 3–4 Å of astemizole. Finally, when astemizole was positioned in the active site of the CYP3A4 homology model in a pose conducive with N-dealkylation (α-carbon protons ∼5.87 Å from the heme iron), Phe241 and Phe304 were within 3 Å of the benzimidazole ring, with Thr309 located within 3 Å of the methoxyphenyl moiety (Fig. 7D). Arg106, Pro107, Ser119, Arg212, and Ala370 were also located within 3–4 Å of astemizole. The active site volume from the crystal structure of CYP3A4 was estimated to be ∼1239 Å3, similar to previous reports (Ekroos and Sjogren, 2006; Ohkura et al., 2009).

Astemizole docked into a homology model of CYP4F12 (single orientation, A) and a crystal structure of CYP3A4 (multiple orientations, B–D). The homology model estimates the active site of CYP4F12 to be approximately 76% of the volume of CYP3A4 active site and more cylindrical in nature, with an increased rigidity in the F and G helix structure compared with CYP3A4.
Discussion
CYP4F12 catalyzes the ω-1 through ω-3 hydroxylations of compounds such as arachidonic acid, 9,11-diazo-prostaglandin H2, and 9,11-methanoepoxy-prostaglandin H2 (Bylund et al., 2001). The enzyme can also metabolize xenobiotics such as astemizole, terfenadine, and ebastine (Parikh et al., 2003; Lee et al., 2010). Although no mechanistic explanation was available at the time, studies to compare the substrate profiles of ten P450 isoforms concluded that, although CYP4F12 is susceptible to inhibition by many of the same ligands as other P450 enzymes, the ability of the enzyme to metabolize the same compounds is relatively restricted (Foti et al., 2011). The objective of this manuscript was to characterize the active site properties of CYP4F12 as a means to rationalize the decreased catalytic promiscuity through kinetic analyses and homology modeling.
Efforts to characterize the inhibitory properties of 53 compounds against CYP4F12 resulted in IC50 values from 78 nM for amiodarone to >100 μM for 13 of the compounds. Lipophilicity is a well-characterized property of P450 ligands that can influence inhibition potency (Lewis and Dickins, 2003; Lewis et al., 2004). This was true for CYP4F12 because the molecular descriptors associated with lipophilicity (cLogP, ALogP, LogD) trended with inhibitor potency, and the most potent inhibitor, amiodarone, was also the most lipophilic (cLogP = 8.95). Of the 53 compounds, 47 had IC50 values for CYP4F12 that were either more potent or similar to the IC50 observed for CYP3A4, indicating that the active site of CYP4F12 is capable of accommodating much of the chemical space that is able to fit in the active site of CYP3A4. Conversely, only two of the compounds (astemizole and terfenadine) exhibited measurable clearance by CYP4F12, suggesting that, although the many of these ligands can act as an inhibitor of CYP4F12, the likelihood of achieving a catalytically favorable orientation in the active site is a less common phenomena.
O-Demethylation has been reported to be a metabolic pathway for astemizole by multiple P450s (Matsumoto and Yamazoe, 2001; Hashizume et al., 2002; Parikh et al., 2003; Lee et al., 2010). Consistent with these results, O-demethylation accounted for over 99% of the metabolites formed by CYP4F12. Trace amounts of 6-hydroxyastemizole, fluorophenyl-OH astemizole, and N-desalkylastemizole were also observed, suggesting that, although it is plausible for astemizole to achieve multiple binding orientations in the active site of CYP4F12, the additional orientations are less catalytically favorable. Although O-demethylation was also catalyzed by CYP3A4, it accounted for only 39% of the metabolites formed, with 6-hydroxyastemizole, fluorophenyl-OH, and hydroxylation on the methoxyphenyl ring all accounting for significant fractions of the overall metabolism. Unlike CYP4F12, the relative similarity of the fm values for CYP3A4 suggests that conditions exist within the active site of CYP3A4 for astemizole to achieve multiple catalytically favorable orientations.
To further compare the active sites of CYP4F12 and CYP3A4, incubations were performed using astemizole-d3. The use of isotope effects to evaluate a ligand’s ability to freely reorient within an active site has been described previously (Iyer et al., 1997; Henne et al., 2001). The basis for these experiments begins with the symmetry of the transition state for the bond-cleaving reaction step (C–H or C–D) and results in an alteration to the rate constant for that step (Westheimer, 1961; Lu et al., 1984; Miwa et al., 1984; Miwa and Lu, 1987; Manchester et al., 1997; Nelson and Trager, 2003; Kim et al., 2006). When astemizole-d3 was incubated with recombinant CYP4F12, an isotope effect of 7.1 [(Vmax/Km),H/(Vmax/Km),D] was observed for the formation of O-desmethylastemizole, with an 8.3-fold decrease in the clearance of the substrate. No alternative metabolites were observed with CYP4F12, suggesting that the active site architecture of CYP4F12 is such that the substrate is unable to rapidly reorient itself. When kinetic isotope effect experiments were conducted with recombinant CYP3A4, an isotope effect was also observed for the formation of O-desmethylastemizole; however, it was reduced to 3.8 [(Vmax/Km),H/(Vmax/Km),D], with no effect on the clearance of astemizole. Inverse isotope effects were observed for the formation of 6-hydroxyastemizole (0.8), fluorophenyl-OH astemizole (0.47), and methoxyphenyl-OH astemizole (0.45) with CYP3A4. One explanation may be that isotopically sensitive branching to the three hydroxylated metabolites is masking the kinetic isotope effect at the site of O-demethylation and that the lack of an effect on the clearance of astemizole is a combination of the metabolic branching and the relatively low fm value for this metabolic pathway (Harada et al., 1984; Jones et al., 1986; Atkins and Sligar, 1987; Miwa and Lu, 1987; Korzekwa et al., 1989; Nelson and Trager, 2003). Furthermore, the P450 catalytic cycle is known to include three nonproductive (uncoupling) pathways, of which only the formation of water from Compound I results in an NADPH to O2 ratio of 2:1. The approximate twofold increase in the NADPH consumption rate that was observed for CYP4F12 and astemizole-d3 with no increase in the formation of H2O2 suggests that the enzymatic reaction may be branching to this uncoupling pathway and resulting in the formation of an additional water molecule from Compound I (Atkins and Sligar, 1988; Grinkova et al., 2013).
Finally, to rationalize the previously discussed results using the active site geometries of CYP4F12 and CYP3A4, a homology model was designed for CYP4F12 and compared with an X-ray crystal structure model of CYP3A4. Upon docking, only a single orientation was achieved with CYP4F12, whereas three orientations were obtained with CYP3A4. Binding of astemizole in the CYP4F12 model required an induced-fit docking approach, indicating that the active site of the enzyme is relatively constrained. The majority of the active site residues that were located within 2–3 Å of astemizole was hydrophobic in nature, consistent with hydrophobic interactions influencing the binding of ligands within the CYP4F12 active site. The active site volume of the CYP4F12 homology model was ∼76% of the total volume of the CYP3A4 active site and was constrained in a long, cylindrical nature. The region of the CYP4F12 active site with the greatest structural divergence from CYP3A4 appeared to be in the region of the F and G helices, where CYP4F12 appears to have a rigid helical motif similar to P450 BM3 as opposed to the more flexible helix-loop-helix motifs of the CYP3A4 F and G helix structures (Joyce et al., 2004; Ekroos and Sjogren, 2006; Ohkura et al., 2009). The overall mobility of the F/G loop in P450 isoforms has been proposed as a determinant of a P450 to bind diverse ligands, and one could argue that rigidity in this region of CYP4F12 contributes to the narrow substrate profile of the enzyme (Poulos, 2003; Scott et al., 2003). The estimated volume of the active site of CYP4F12 suggests it is similar in size to the larger P450 active sites such as CYP3A4 and CYPC8, although the overall flexibility and volume changes upon substrate binding remain to be determined.
Consistent with its metabolic profile, multiple binding orientations were obtained with the CYP3A4 model. Multiple crystal structures and evaluations of the ligand promiscuity of CYP3A4 have been reported, resulting in a prominent role for the enzyme in the biotransformation of xenobiotics and endogenous compounds (Kenworthy et al., 1999; Williams et al., 2004; Yano et al., 2004; Gibbs and Hosea, 2003; Wienkers and Heath, 2005; Ekroos and Sjogren, 2006; Foti et al., 2010, 2011). Binding within the active site of CYP3A4 has also been shown to be related to lipophilicity, and conformational changes to the active site of CYP3A4 upon ligand binding have been reported (Shou et al., 1994; Korzekwa et al., 1998; Ishigami et al., 2001; Kenworthy et al., 2001; Khan et al., 2002; Lu et al., 2001; Riley et al., 2001). Interactions between astemizole and the phenylalanine cluster of CYP3A4 were observed, with Phe215, Phe241, and Phe304 generally involved in π-π interactions with the aromatic ring of astemizole most distal from the site of metabolism. Ser119 and Ala305, previously shown to be involved in the substrate recognition of testosterone, progesterone, and 7-hexoxycoumarin were also located within 3–4 Å of astemizole for all three orientations and may influence binding interactions with the benzimidazole or piperidine ring, depending on the orientation of astemizole (He et al., 1997; Domanski et al., 1998; Khan and Halpert, 2000; Tanaka et al., 2004). The three binding models for astemizole with CYP3A4 also predict significant interactions with a polar region of the active site, which includes Arg106 and Glu374, a region known to be involved in the binding of ketoconazole as well as with Arg212, which is known to undergo significant conformational changes between the ligand-free and ligand-bound structures of CYP3A4 (Yano et al., 2004; Ekroos and Sjogren, 2006; Sevrioukova and Poulos, 2012).
In conclusion, previous studies have identified CYP4F12 as being catalytically restricted compared with other P450 isoforms, and the results presented herein provide a plausible mechanistic explanation for the narrow ligand profile of the enzyme. Metabolite identification studies identified astemizole O-demethylation as the single route of metabolism by CYP4F12, whereas CYP3A4 was capable of catalyzing the formation of multiple metabolic products with relatively low fm values. Kinetic isotope effects demonstrate that deuteration of the primary site of metabolism results in a direct effect on the clearance of astemizole by CYP4F12, whereas CYP3A4 can account for the isotope effect through metabolic branching. Overall, it appears that, although the CYP4F12 active site may be capable of accommodating and binding similar ligands as the other P450 isoforms, the ability to achieve a catalytically favorable orientation may be more difficult, which ultimately results in a decrease in catalytic promiscuity.
Authorship Contributions
Participated in research design: Eksterowicz, D. Rock, B. Rock, Wienkers, and Foti.
Conducted experiments: Eksterowicz, B. Rock, and Foti.
Performed data analysis: Eksterowicz, B. Rock, and Foti.
Wrote or contributed to the writing of the manuscript: Eksterowicz and Foti.
Footnotes
- Received June 18, 2014.
- Accepted July 29, 2014.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- fm
- fraction metabolized
- HPLC
- high-performance liquid chromatography
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- P450
- cytochrome P450
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}