


Abstract
The organic anion-transporting polypeptide (OATP) 1B3 is a membrane transport protein that mediates hepatic uptake of many drugs and endogenous compounds. Currently, determination of OATP-mediated drug-drug interactions in vitro is focused primarily on direct substrate inhibition. Indirect inhibition of OATP1B3 activity is under-appreciated. OATP1B3 has putative protein kinase C (PKC) phosphorylation sites. Studies were designed to determine the effect of PKC activation on OATP1B3-mediated transport in human hepatocytes using cholecystokinin-8 (CCK-8), a specific OATP1B3 substrate, as the probe. A PKC activator, phorbol-12-myristate-13-acetate (PMA), did not directly inhibit [3H]CCK-8 accumulation in human sandwich-cultured hepatocytes (SCH). However, pretreatment with PMA for as little as 10 minutes rapidly decreased [3H]CCK-8 accumulation. Treatment with a PKC inhibitor bisindolylmaleimide (BIM) I prior to PMA treatment blocked the inhibitory effect of PMA, indicating PKC activation is essential for downregulating OATP1B3 activity. PMA pretreatment did not affect OATP1B3 mRNA or total protein levels. To determine the mechanism(s) underlying the indirect inhibition of OATP1B3 activity upon PKC activation, adenoviral vectors expressing FLAG-Myc-tagged OATP1B3 (Ad-OATP1B3) were transduced into human hepatocytes; surface expression and phosphorylation of OATP1B3 were determined by biotinylation and by an anti–phosphor-Ser/Thr/Tyr antibody, respectively. PMA pretreatment markedly increased OATP1B3 phosphorylation without affecting surface or total OATP1B3 protein levels. In conclusion, PKC activation rapidly decreases OATP1B3 transport activity by post-translational regulation of OATP1B3. These studies elucidate a novel indirect inhibitory mechanism affecting hepatic uptake mediated by OATP1B3, and provide new insights into predicting OATP-mediated drug interactions between OATP substrates and kinase modulator drugs/endogenous compounds.
Introduction
Organic anion-transporting polypeptides (OATPs) are members of the solute carrier organic anion (SLCO) transporter superfamily. OATP1B1 and OATP1B3 are expressed predominantly in the sinusoidal membrane of the liver (König et al., 2000a,b); they mediate hepatic uptake of a diverse array of endogenous compounds, environmental toxins, and many clinically important drugs, including lipid-lowering statins, antibiotics, immunosuppressants, cardiac glycosides, and antidiabetic and anticancer agents (König, 2011). Recently, OATP1B1 and OATP1B3 have been recognized as important determinants of transport-mediated drug-drug interactions (DDIs) (Tweedie et al., 2013). Many drugs that are potent OATP inhibitors in vitro [e.g., cyclosporine (Letschert et al., 2006) and ritonavir (Annaert et al., 2010)] may cause clinically significant DDIs in vivo; for example, significant increases in the systemic exposure of statins have been reported when these OATP inhibitors are coadministered with statins, which are OATP substrates (Shitara et al., 2003; Kiser et al., 2008). OATP1B3 shares a variety of common substrates with OATP1B1, such as statins (Hirano et al., 2004; Kitamura et al., 2008), rifampicin (Vavricka et al., 2002), and bromosulfophthalein (BSP) (Cui et al., 2001). However, some OATP1B3-specific substrates have been identified, including the octapeptide cholecystokinin 8 (CCK-8) (Ismair et al., 2001; Kullak-Ublick et al., 2001; Hirano et al., 2004).
Transport protein function may be regulated at the transcriptional, translational, or post-translational level. Currently, data regarding regulation of hepatic OATP1B3 transport function are scarce, and most studies have been focused on transcriptional regulation of OATP1B3. At the transcriptional level, the promoter of hepatic OATP1B3 can be repressed by hepatic nuclear factor (HNF) 3β (Vavricka et al., 2004) and transactivated by HNF1α (Jung et al., 2001), farnesoid X receptor (FXR) (Jung et al., 2002), and growth hormone– and prolactin-activated transcription factor Stat5 (Wood et al., 2005). To date, regulation of OATP1B3 function at the post-translational level has not been reported.
Protein kinase C (PKC) is a family of highly related protein kinases. Increased activity of select PKC isozymes has been observed in many disease states—for example, nonalcoholic fatty liver disease (Birkenfeld and Shulman, 2014), diabetes, cancers, ischemic heart disease, Parkinson’s disease, and Alzheimer’s disease [reviewed by Mochly-Rosen et al. (2012)]. Modulation of PKC activity presents an attractive target for drug development (Mochly-Rosen et al., 2012; Sun and Alkon, 2012; Sanchez-Bautista and Nicolas, 2013). Consequently, PKC activators and inhibitors have been developed for clinical trials for many diseases, either alone or in combination with other drugs (Mochly-Rosen et al., 2012). The function of several human transport proteins is regulated by PKC activation. For example, PKC activation downregulates transport function of organic anion transporter (OAT) 1 (Zhang et al., 2008), OAT3 (Duan et al., 2010), and OAT4 (Zhou et al., 2007; Zhang et al., 2010); OATP2B1 (Köck et al., 2010) and OATP1A2 (Zhou et al., 2011); efflux transport protein P-glycoprotein (P-gp) (Miller et al., 1998); and multidrug resistance protein (MRP)2 (Kubitz et al., 2001). OATP1B3 has putative PKC phosphorylation sites as predicted by Scansite 3 (Obenauer et al., 2003). We hypothesized that transport function of OATP1B3 is regulated by PKC. The current study was designed to investigate regulation of transport function of OATP1B3 by phorbol 12-myristate 13-acetate (PMA)–induced PKC activation as well as the underlying mechanism(s) in human hepatocytes.
Materials and Methods
[3H]Cholecystokinin (CCK)-8 (specific activity 96 Ci/mmol) was purchased from PerkinElmer (Boston, MA). Unlabeled CCK-8, Hanks’ balanced salt solution (HBSS), dexamethasone, dimethyl sulfoxide (DMSO), BSP, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). PMA, 4 α-phorbol-12, 13-didecanoate (4αPDD), and bisindolylmaleimide I (BIM I) were purchased from EMD Millipore (Billerica, MA). PhosSTOP Phosphatase Inhibitor and cOmplete Protease Inhibitor Cocktail Tablets were purchased from Roche Diagnostics (Indianapolis, IN). Bovine serum albumin (BSA)–based membrane-blocking solution was purchased from Life Technologies (Carlsbad, CA). Bio-Safe II liquid scintillation mixture was obtained from Research Products International (Mount Prospect, IL). BioCoat culture plates, insulin/transferrin/selenium (ITS+), and Matrigel were purchased from BD Biosciences (Bedford, MA). Fetal bovine serum (FBS) was obtained from Hyclone Laboratories (Logan, UT). Phenol red-free Dulbecco’s modified Eagle’s Medium (DMEM), minimum essential medium (MEM) nonessential amino acids (NEAA), l-glutamine, insulin, and penicillin-streptomycin were purchased from Gibco/Life Technologies (Grand Island, NY).
Human Sandwich-Cultured Hepatocytes.
Human hepatocytes were purchased from Life Technologies (Carlsbad, CA), Celsis/BioreclamationIVT (Chicago, IL), and Triangle Research Laboratories, LLC (Research Triangle Park, NC); available demographics are listed in Table 1. Sandwich-culture of human hepatocytes was conducted as previously described (Swift et al., 2010). Briefly, the cells were plated at 3.5 × 105 cells per well in 24-well BioCoat culture plates on day 0 in phenol red-free DMEM supplemented with 5% (v/v) FBS, 1% (v/v) MEM NEAA, 2 mM l-glutamine, 100 units of penicillin G sodium/ml, 100 g/ml of streptomycin sulfate, 4 μg/ml insulin, and 1 μM dexamethasone. Cells were cultured at 37°C in a humidified incubator (95% O2, 5% CO2). When cells were attached (5–24 hours after seeding), cells were overlaid with Matrigel (BD Bioscience) at a final concentration of 0.25 mg/ml in phenol red-free DMEM containing 2 mM l-glutamine, 1% (v/v) MEM NEAA, 100 units/ml of penicillin G sodium, 100 g/ml of streptomycin sulfate, 0.1 μM dexamethasone, and 1% (v/v) ITS+ premix. Culture medium was replaced every 24 hours.
Demographics of human liver donors
Packaging and Transduction of Recombinant Adenoviral Vectors.
The open reading frame of OATP1B3 from the OATP1B3-expression vector [pOATP8.31; provided by Dr. Dietrich Keppler (König et al., 2000a)] was subcloned in-frame into the Origene pCMV6-Entry (OriGene, Rockville, MD) vector to add a FLAG-Myc tag at the C-terminus of OATP1B3. The FLAG-Myc tagged-OATP1B3 (FLAG-Myc-OATP1B3) was then subcloned into the pCMV-shuttle vector (Luo et al., 2007), resulting in the plasmid CW101644. Mutagenesis was conducted on CW101644 to remove the Pac I restriction site in the OATP1B3 sequence without changing the OATP1B3 protein sequence as required by the AdEasy adenoviral vector packaging protocol (Luo et al., 2007), and the resulting construct was used for packaging of adenoviral vector (Ad-OATP1B3) at the UNC Vector Core Facility according to the protocol published previously (Luo et al., 2007). Lac operon (LacZ)–expressing control adenoviral vector (Ad-LacZ) was purchased from the UNC Vector Core Facility. The titer of adenoviral vectors was determined by Adeno-X Rapid Titer Kit (Clontech Laboratories, Mountain View, CA). The adenoviral vectors that express OATP1B3 and LacZ control were designated as Ad-OATP1B3 and Ad-LacZ, respectively. After seeding for 5–6 hours, human hepatocytes were transduced overnight with Ad-OATP1B3 or Ad-LacZ at a multiplicity of infection (MOI) of 5 in feeding medium with or without 0.25 mg/ml Matrigel.
Transport Studies.
Human hepatocytes were incubated for designated times at 37°C with medium containing vehicle control 0.1% (v/v) DMSO, PMA, 4αPDD, and/or BIM I. After rinsing with prewarmed HBSS (pH 7.4), cells were incubated with HBSS containing [3H]CCK-8 at designated concentrations for specified times. At the end of each specified time interval, the buffer was aspirated rapidly, cells were rinsed three times with ice-cold HBSS and lysed in ice-cold phosphate buffered saline (PBS) containing 0.5% Triton-X100, and an aliquot was subjected to liquid scintillation counting (Packard Tricarb scintillation counter; Packard Corp., Meriden, CT; and LS6500 scintillation counter; Beckman Coulter, Brea, CA). [3H]CCK-8 accumulation was normalized to protein concentration determined by BCA assay (Pierce Chemical, Rockford, IL) and corrected for nonspecific binding by including a Matrigel-overlaid or nonoverlaid blank plate.
TaqMan Real-Time RT-PCR.
Total RNA from human sandwich-cultured hepatocytes (SCH) was extracted using the ABI RNA isolation system (Applied Biosystems, Foster City, CA). TaqMan real-time reverse transcription–polymerase chain reaction (RT-PCR) was conducted using an ABI Prism 7700 system (Applied Biosystems) to determine OATP1B3 mRNA levels as described previously (Kim et al., 2002). OATP1B3 expression relative to that of control was analyzed with the 2−ΔΔCT method (Livak and Schmittgen, 2001) using GAPDH as an internal control. The TaqMan probe and primer sequences (5′–3′) used for human OATP1B3 were TCCTACAAGCCCCTTGTGCTCCACA (probe), CATGTATGAAGTGGTCCACCA (forward primer), and CAAGTAGACCCTTCCAAAAAATAC (reverse primer). Sequences used for human GAPDH were CAAGCTTCCCGTTCTCAGCC (probe), ACCTCAACTACATGGTTTAC (forward primer), and GAAGATGGTGATGGGATTTC (reverse primer).
Immunoprecipitation.
Cells were lysed in buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 100 mM NaF, 1% (v/v) NP-40, 0.2 mM Na3VO4, and cOmplete protease inhibitor cocktail. Protein concentrations were determined via BCA assay. Immunoprecipitation was performed similarly as published previously (Yue et al., 2004). In brief, whole cell lysates (1.2–1.5 mg) were precleared with normal mouse IgG followed by immunoprecipitation with FLAG monoclonal antibody (clone M2; Sigma-Aldrich). The immunocomplex was then incubated with protein A/G beads (Santa Cruz, Biotechnology, Dallas, TX) overnight in 4°C. After washing the beads, the immunocomplex was eluted by heat at 70°C for 10 minutes. The denatured immune complex was separated by electrophoresis and analyzed by immunoblotting.
Immunoblotting.
Whole cell lysates or immunocomplex were resolved on SDS-PAGE (Bio-Rad, Hercules, CA). The proteins were then transferred to nitrocellulose membranes. Membranes were blocked at 4°C overnight in 5% milk or BSA-based membrane blocking solution (Life Technologies, Carlsbad, CA) in Tris-buffered saline with Tween (TBST), and probed with the following antibodies: rabbit polyclonal OATP1B3 antibody [provided by Dr. Richard B. Kim (Ho et al., 2006) in Fig. 3B, or custom-generated anti-OATP1B3 antibody (Fisher Scientific Company, LLC, Denver, CO) according to a previous publication (König et al., 2000a) in Fig. 6A], mouse FLAG (clone M2; Sigma-Aldrich), phospho-Ser/Thr/Tyr antibody (Anaspec, Fremont, CA), GAPDH (Santa Cruz Biotechnology, Inc.), or β-actin (Sigma-Aldrich). Blots were incubated with horseradish peroxidase (HRP)–conjugated secondary antibody (Santa Cruz Biotechnology, Inc.), and signals were detected by chemiluminescent substrate Supersignal West Duro (Pierce) with a Bio-Rad ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Densitometry analysis was performed using Image Laboratory v4.1 software.
Results
PKC Activation by PMA Downregulates OATP1B3 Transport Function.
The time-dependent accumulation of [3H]CCK-8 was characterized in SCH. As shown in Fig. 1A, [3H]CCK-8 (1 μM) accumulation was linear at least up to 7 minutes. The y-intercept of the regression line is slightly greater than zero, which may be attributable to some passive diffusion of CCK-8, as shown in Supplemental Fig. S1 and reported previously (Ismair et al., 2001). When the time-dependent uptake of [3H]CCK-8 in HEK-293 cells overexpressing OATP1B3 (HEK-293-OATP1B3) at 37°C was corrected for 4°C accumulation, the y-intercept of the regression line was approximately zero (Supplemental Fig. S1). Therefore, for CCK-8 uptake, an incubation time of up to 3 minutes was chosen for subsequent experiments. To determine the effect of PKC activation on OATP1B3-mediated transport, [3H]CCK-8 accumulation (1 μM, 3 minutes) was determined in SCH treated with either vehicle control, the PKC activator PMA, or the inactive PMA analog 4αPDD (Peers and Carpenter, 1998), or pretreated with a PKC inhibitor BIM I prior to PMA treatment. [3H]CCK-8 accumulation in SCH pretreated with 0.1 and 1 μM PMA for 30 minutes was significantly decreased to 58.0 ± 4.9% and 43.5 ± 11.6% of vehicle control treatment, respectively, whereas the values in SCH pretreated with the inactive PMA analog 4αPDD (1 μM, 30 min) (Fig. 1B) or with a PKC inhibitor BIM I (1 μM, 20 minutes) prior to PMA treatment were similar to those in the control (Fig. 1C).
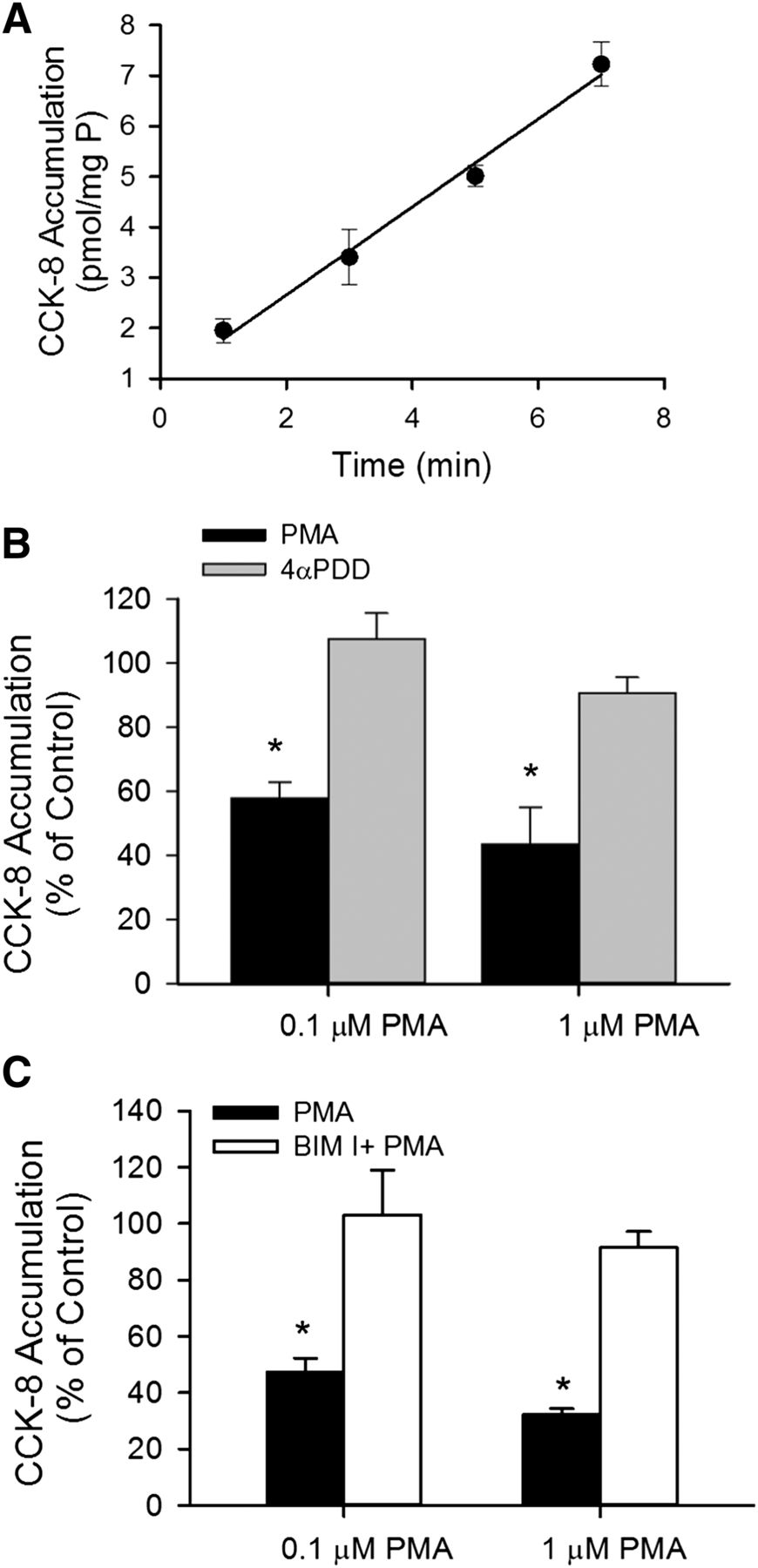
PKC activation by PMA downregulates [3H]CCK-8 accumulation in human SCH. (A) Time-dependent accumulation of [3H]CCK-8 (1 μM) in human SCH. Data represent the mean ± range of duplicate measurements from a single donor. (B) Effect of PKC activator PMA on [3H]CCK-8 accumulation. [3H]CCK-8 accumulation (1 μM for 3 minutes) was determined in human SCH pretreated with PMA (black bars) or a PMA inactive analog, 4αPDD (gray bars), for 30 minutes at 0.1 or 1 μM. (C) Involvement of PKC activation in PMA-induced downregulation of [3H]CCK-8 accumulation. Human SCH were incubated in medium containing 0.1 or 1 μM PMA for 30 minutes in the presence (white bars) or absence (black bars) of pretreatment (20 minutes) and subsequent coincubation (30 minutes) with the PKC inhibitor BIM I (1 μM). [3H]CCK-8 accumulation (1 μM, 3 minutes) in human SCH was expressed as a percentage of the accumulation measured in vehicle-treated cells. Data represent mean ± S.E.M. from n = 3 donors in triplicate. *P < 0.05 versus control by one-way analysis of variance, followed by Dunnett’s t test. Transport experiments were performed on day 5 or 6 of culture.
Rapid and Indirect Inhibition of OATP1B3 Activity by PMA.
Time-dependent regulation of CCK-8 transport by PMA pretreatment was examined. As early as 10 minutes after PMA pretreatment, [3H]CCK-8 accumulation was significantly reduced to 49.8 ± 5.7% and 37.4 ± 9.4% of control in SCH treated with 0.1 μM and 1 μM PMA, respectively (Fig. 2A). PMA pretreatment for 30 and 60 minutes also significantly decreased [3H]CCK-8 accumulation in SCH compared with control. The direct interaction of PMA and BIM I on OATP1B3-mediated [3H]CCK-8 transport was determined by comparing the uptake of [3H]CCK-8 (1 μM, 3 minutes) in the presence of vehicle control, PMA (0.1 and 1 μM), BIM I (1 μM), or PMA and BIM I in combination. As shown in Fig. 2B, treatment with PMA and BIM I, either alone or in combination, did not affect [3H]CCK-8 uptake compared with vehicle control. The results indicate that PMA and BIM I do not directly affect OATP1B3-mediated transport of [3H]CCK-8 at the concentrations used in current studies.
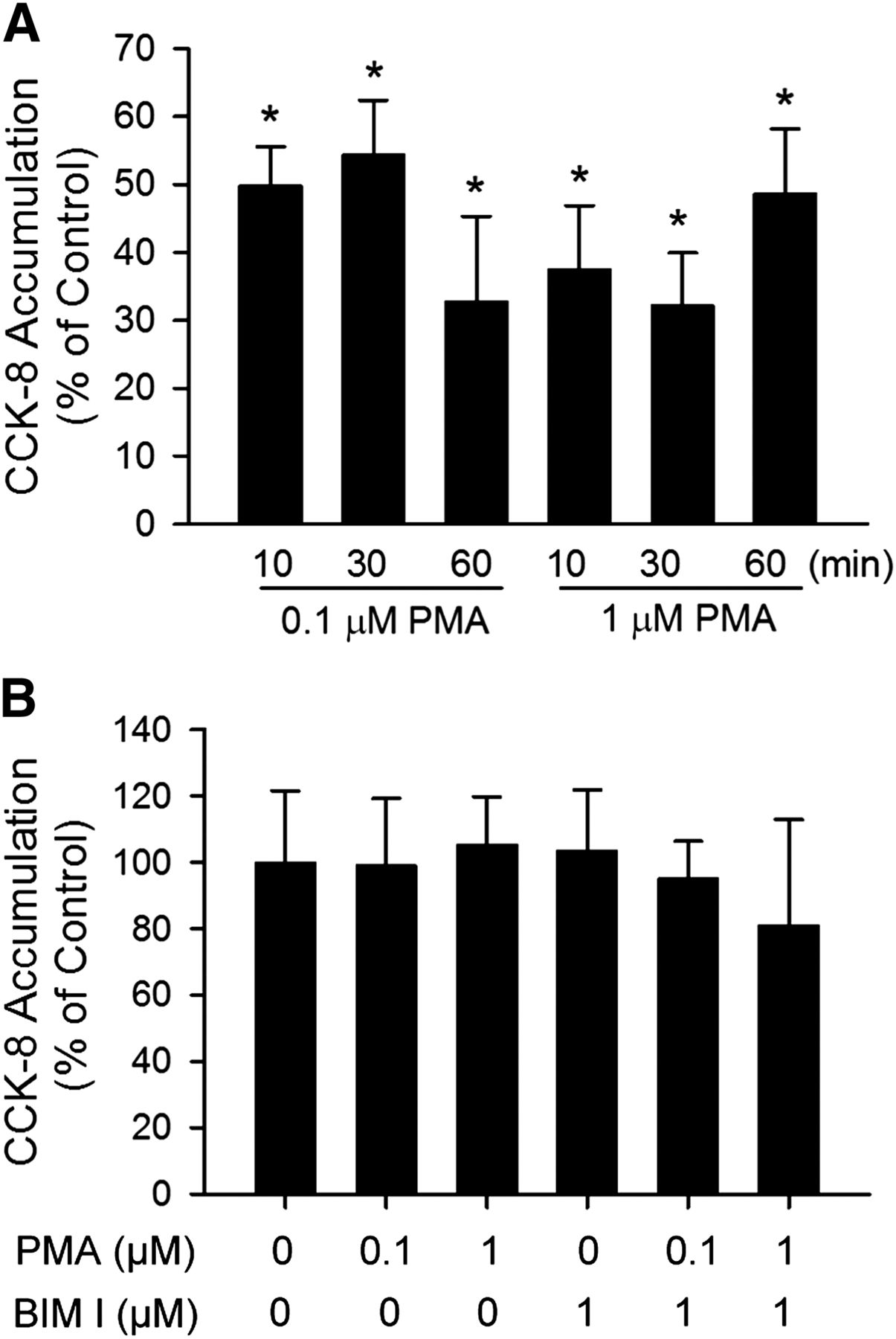
Indirect inhibition of [3H]CCK-8 accumulation by PMA in human SCH. (A) Time-dependent effect of PMA pretreatment on [3H]CCK-8 accumulation in human SCH. Human SCH were pretreated with PMA (0.1 and 1 μM) for various times (10–60 minutes). [3H]CCK-8 accumulation (1 μM, 3 minutes) was expressed as a percentage of the accumulation measured in vehicle control–treated cells. (B) [3H]CCK-8 accumulation in the presence of vehicle control, PMA, or BIM I at indicated concentrations. [3H]CCK-8 accumulation (1 μM, 3 minutes) in human SCH was expressed as a percentage of the accumulation measured in the absence of PMA or BIM I. Data represent mean ± S.E.M. from n = 3 donors in triplicate. *P < 0.05 versus vehicle control treatment by one-way analysis of variance, followed by Dunnett’s t test. Transport experiments were performed on day 5 or 6 of culture.
PMA Treatment Did Not Affect mRNA and Total Protein Levels of OATP1B3 in Human SCH.
The mRNA and protein levels of OATP1B3 in human SCH were compared between 30-minute PMA (0.1 μM) and control treatments by real-time RT-PCR and OATP1B3 immunoblotting. PMA treatment did not significantly affect either OATP1B3 mRNA levels (Fig. 3A) or total protein levels (Fig. 3B).

PMA treatment does not affect OATP1B3 mRNA and total protein levels in human SCH. Human SCH were treated with vehicle control DMSO (CTL) or 0.1 μM PMA for 30 minutes. (A) OATP1B3 mRNA levels were determined by TaqMan real-time RT-PCR with GAPDH as an internal standard; values were expressed as a percentage of CTL. (B) Representative immunoblot (IB) image of OATP1B3 in PMA- and vehicle control–treated SCH (CTL) (n = 3 donors). β-actin was used as a loading control. Densitometry of OATP1B3 was normalized to β-actin expression; results were expressed as fold change versus CTL. Data represent mean ± S.E.M. from n = 3 donors. Statistical analysis was performed versus CTL by Student’s t test. Experiments were performed on day 5 or 6 of culture.
PKC Activation Rapidly Downregulated [3H]CCK-8 Accumulation in Ad-OATP1B3–Transduced Human Hepatocytes.
OATP1B3 protein expression and the effect of PKC activation on [3H]CCK-8 accumulation was determined in nontransduced, Ad-LacZ–, and Ad-OATP1B3–transduced human hepatocytes. At 48 hours post-transduction, expression of FLAG-Myc-tagged-OATP1B3 was detected by FLAG immunoblot in Ad-OATP1B3-transduced (Ad-OATP1B3) but not in control vector Ad-LacZ or nontransduced (None) human SCH (Fig. 4A). [3H]CCK-8 accumulation (1 μM, 1.5 minutes) was similar in nontransduced and Ad-LacZ–transduced human SCH (data not shown). [3H]CCK-8 accumulation in Ad-OATP1B3-transduced SCH ranged from 3.2- to 8.3-fold higher than in the nontransduced control (n = 3 donors in triplicate), consistent with overexpression of exogenous OATP1B3 in Ad-OATP1B3–transduced SCH. As shown in Fig. 4B, in both nontransduced (None) and Ad-OATP1B3–transduced human SCH (Ad-OATP1B3), PKC activation by PMA treatment (0.1 μM, 30 minutes) significantly inhibited [3H]CCK-8 accumulation to an extent close to the inhibition by 100 μM BSP, a potent inhibitor of OATPs (Annaert et al., 2010).
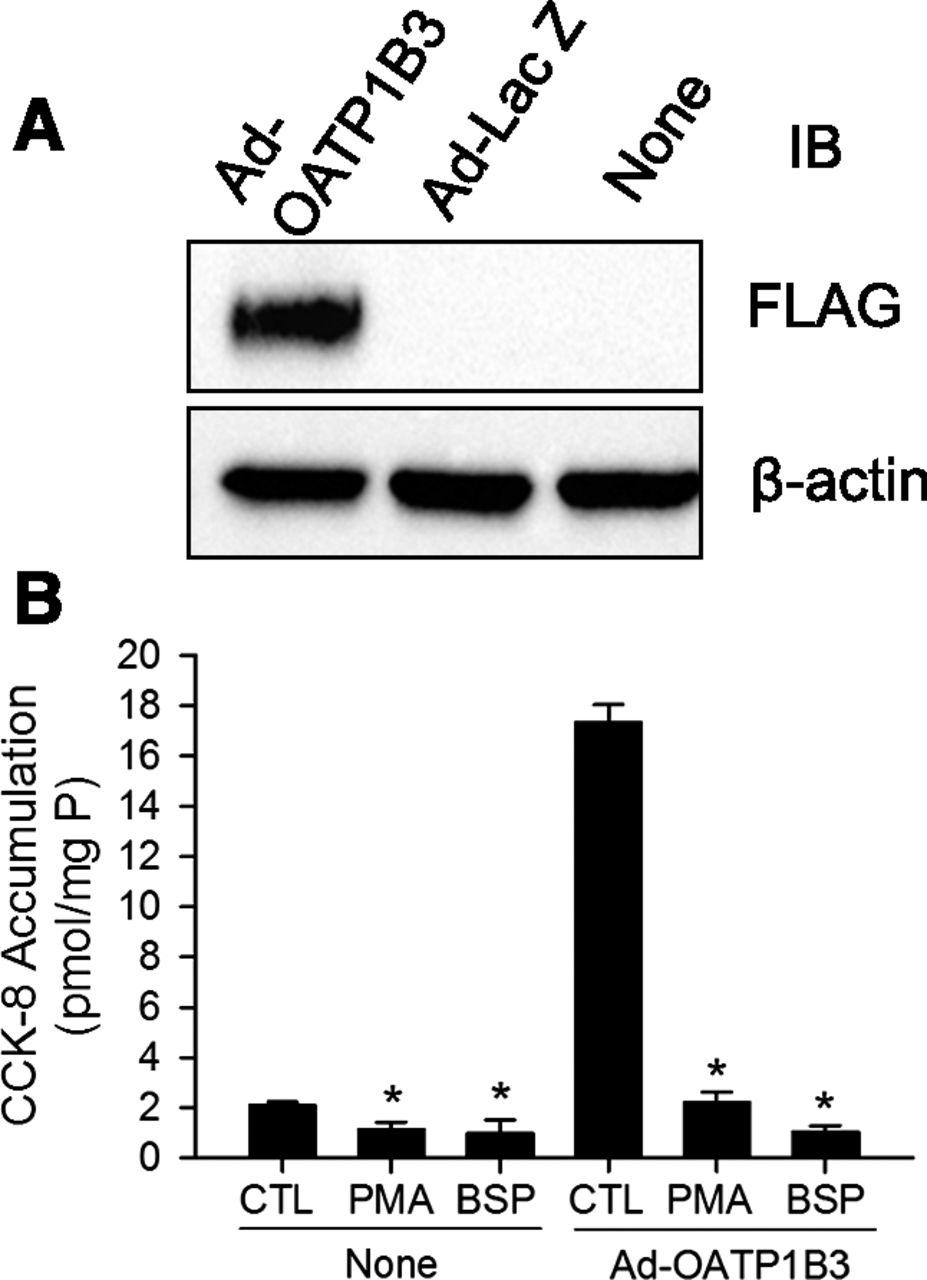
PKC activation potently inhibits [3H]CCK-8 accumulation in nontransduced and Ad-OATP1B3-transduced human SCH. Human hepatocytes were transduced with Ad-LacZ or Ad-OATP1B3 at a MOI of 5 and overlaid with Matrigel. Experiments were conducted 48 hours post-transduction on day 2 of culture. (A) Immunoblot (IB) of FLAG in nontransduced (None), Ad-LacZ–, and Ad-OATP1B3–transduced human SCH. β-Actin was used as a loading control. Representative images from n = 3 donors are shown. (B) Potent inhibition of [3H]CCK-8 accumulation in human SCH upon PKC activation. [3H]CCK-8 accumulation (1 μM, 1.5 minutes) in nontransduced (None) and Ad-OATP1B3-transduced (Ad-OATP1B3) human SCH pretreated with vehicle control (CTL) or PMA (0.1 μM) for 30 minutes), or coincubated with BSP (100 μM) without PMA pretreatment. Data represent mean ± S.D. in triplicate from a representative data set from n = 3 and n = 2 donors for PMA and BSP treatment, respectively. *P < 0.05 versus CTL by one-way analysis of variance in each group of nontransduced and Ad-OATP1B3–transduced cells, followed by Dunnett’s t test.
PKC Activation Did Not Affect Surface or Total Protein Levels of FLAG-Myc-OATP1B3.
As shown in Fig. 5, after PMA treatment (0.1 μM, 30 minutes), the surface and total protein levels of FLAG-OATP1B3 were 0.9 ± 0.1-fold and 1.1 ± 0.1-fold of control, respectively, suggesting that PKC activation did not affect either the surface or the total FLAG-Myc-OATP1B3 protein levels. GAPDH, a marker for intracellular protein (Soundararajan et al., 2009), was only detected in the total protein but not in the surface fraction, suggesting that the surface fraction was not contaminated with intracellular protein.
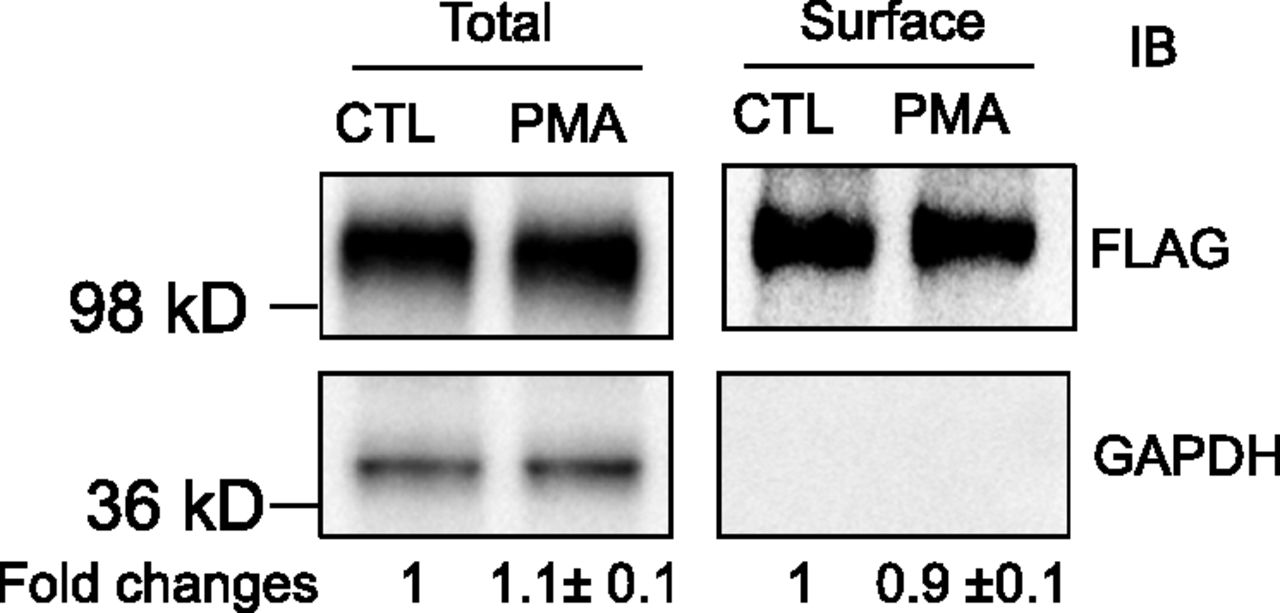
PKC activation does not affect total or surface levels of FLAG-Myc-OATP1B3. Human hepatocytes were transduced with Ad-OATP1B3 at a MOI of 5 and cultured without Matrigel overlay. At 48 hours post-transduction on day 2 of culture, cells were treated with PMA (0.1 μM, 30 minutes) or vehicle control (CTL). Surface levels of FLAG-Myc-OATP1B3 were determined by biotinylation followed by immunoblot (IB) with FLAG antibody. GAPDH was used as a cytoplasmic protein marker. Protein levels were determined by densitometry. Fold change of total and surface levels of FLAG-OATP1B3 (PMA versus CTL) were expressed as mean ± S.E.M (n = 3 donors). Representative images from n = 3 donors are shown.
PKC Activation Increased Phosphorylation of FLAG-Myc-OATP1B3 in Human Hepatocytes.
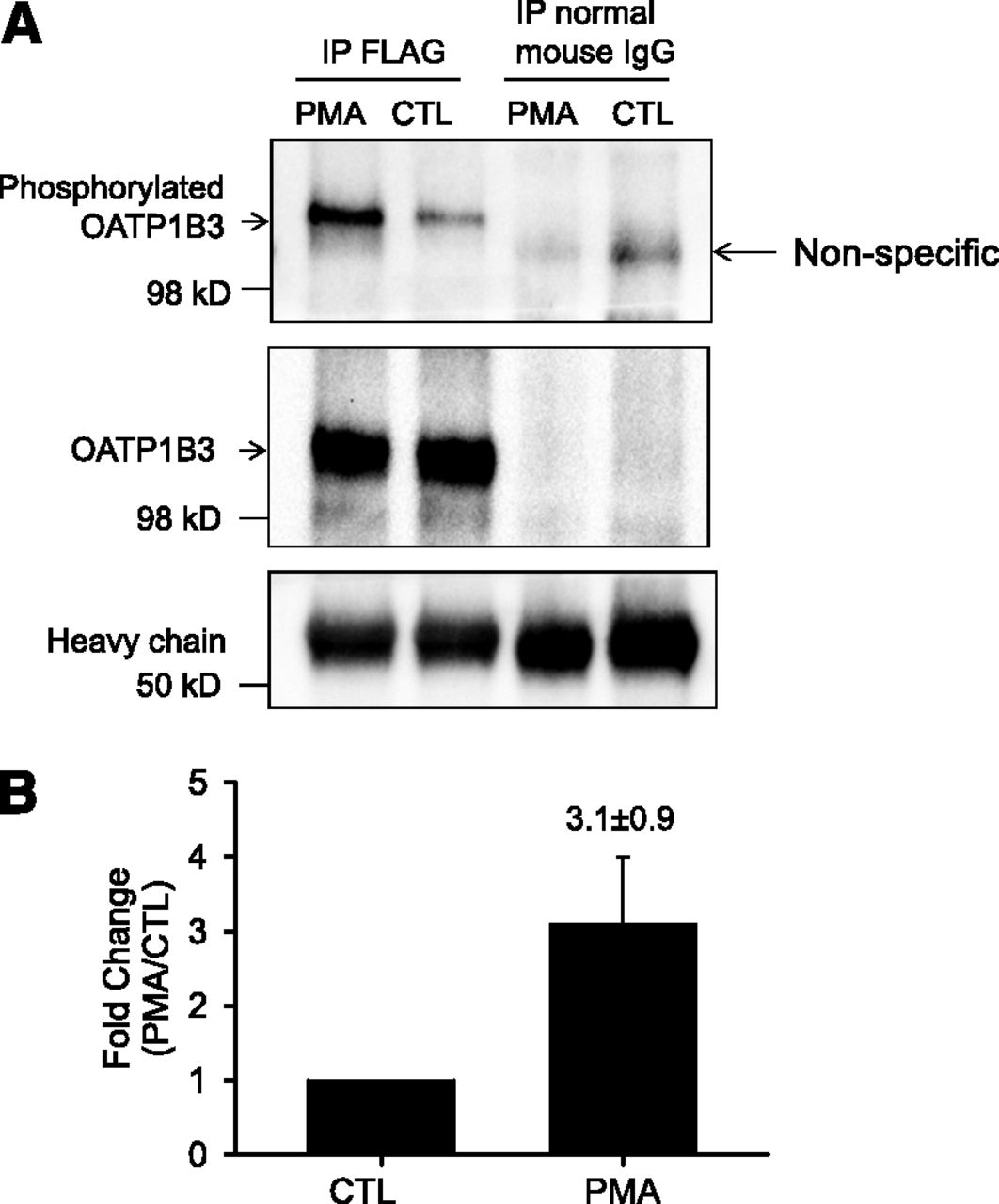
Phosphorylation of OATP1B3 was determined using an anti–phosphor-Ser/Thr/Tyr antibody similar to methods published previously (Köck et al., 2010). In Ad-OATP1B3–transduced nonoverlaid human hepatocytes, following immunoprecipitation with a FLAG antibody, immunoblot with an anti–phosphor-Ser/Thr/Tyr antibody detected a markedly increased phosphorylation signal in PMA-treated (0.1 μM, 30 minutes) cells (Fig. 6A, top panel, and Fig. 6B). After stripping, the blot was probed with OATP1B3 antibody (newly custom-generated by Fisher Scientific Company, LLC, Denver, CO). The signal detected by OATP1B3 antibody (Fig. 6A, middle panel) was superimposable to the phosphorylation signal detected by the anti–phosphor-Ser/Thr/Tyr antibody (Fig. 6A, top panel) in FLAG immunoprecipitation. OATP1B3 was not detected after immunoprecipitation with negative control normal mouse IgG (Fig. 6A middle panel). As indicated by the amount of IgG heavy chain (Fig. 6A, bottom panel), similar amounts of FLAG antibody or normal mouse IgG were used for immunoprecipitation in PMA and DMSO (CTL) treated cells. Pilot experiments confirmed that the newly custom-generated OATP1B3 antibody specifically recognized OATP1B3 in the HEK-293-OATP1B3 stable cell line and in human hepatocytes at the same molecular weight as the previously published OATP1B3 antibody (Ho et al., 2006; data not shown).

PKC activation increases phosphorylation of FLAG-Myc-OATP1B3 in human hepatocytes. Human hepatocytes were transduced with Ad-OATP1B3 at a MOI of 5 and cultured without Matrigel overlay. At 48 hours post-transduction on day 2 of culture, cells were treated with PMA (0.1 μM, 30 minutes) or vehicle control (CTL). (A) FLAG-Myc-OATP1B3 was immunoprecipitated in hepatocytes treated with PMA or CTL. Immunocomplexes were first probed with anti–phospho-Ser/Thr/Tyr antibody (top panel) and then reprobed with OATP1B3 antibody after stripping (middle panel). Immunoprecipitation (IP) with normal mouse IgG served as negative control. (B) Densitometry of the phosphorylation signal was normalized to OATP1B3 levels in IP and was expressed as fold change (PMA versus CTL). Data represent mean ± S.E.M (n = 3 donors). Representative images from n = 3 donors are shown.
Discussion
Impaired OATP transport function may affect the pharmacokinetics, efficacy, and/or toxicity of drugs that are OATP substrates (e.g., increased risk of muscle toxicity for statins). Consequently, from a drug development and regulatory perspective, there is keen interest in understanding the mechanisms involved in OATP-mediated DDIs. The current studies demonstrate a novel mechanism of indirect inhibition of OATP1B3-mediated hepatic uptake upon PKC activation. This report is the first indication that OATP1B3 is a phosphorylated protein and that PKC activation increased OATP1B3 phosphorylation in human hepatocytes.
Under normal physiologic condition, OATP1B3 is expressed primarily in the human liver. Therefore, in the current studies, regulation of OATP1B3 transport function by PKC activation was studied in primary human hepatocytes using CCK-8 as a probe substrate (Ismair et al., 2001; Kullak-Ublick et al., 2001; Hirano et al., 2004). We confirm in the current studies that [3H]CCK-8 is transported by OATP1B3 but not by OATP1B1 and OATP2B1 in transporter-overexpressing HEK-293 cells (Supplemental Fig. S2). OATP1B3-mediated [3H]CCK-8 transport is significantly decreased by pretreatment with the PKC activator PMA, but not by the PMA-inactive analog 4αPDD (Fig. 1B). Treatment with a PKC inhibitor BIM I prior to PMA treatment blocks the inhibitory effect of PMA on OATP1B3 function, indicating that PKC activation is essential for downregulation of OATP1B3 activity (Fig. 1C). Although PMA (0.1 and 1 μM) did not directly inhibit transport activity of OATP1B3 (Fig. 2B), pretreatment with PMA significantly decreased OATP1B3-mediated [3H]CCK-8 transport (Fig. 1, B and C, Fig. 2A, and Fig. 4B). Taken together, the PMA-induced decrease in OATP1B3 transport function occurs in an indirect and PKC activation–dependent manner.
Activity of membrane transport proteins often is affected by the amount of transport protein localized to the plasma membrane (Köck et al., 2010; Zhou et al., 2011) and post-translational modifications of the transport proteins, which in many cases involves protein phosphorylation (Svoboda et al., 2011). To determine the mechanism(s) involved in rapid downregulation of OATP1B3-mediated transport upon PKC activation, surface levels and phosphorylation status of OATP1B3 were compared between vehicle control– and PMA-treated SCH. Surface expression of endogenous OATP1B3 was not readily detected by biotinylation in nontransduced human hepatocytes with or without Matrigel overlay (data not shown). To the best of our knowledge, determining the surface level of endogenous OATP1B3 by biotinylation in human hepatocytes has not been reported. Previous publications using biotinylation approaches to determine surface expression of transport proteins, including human OATP transport proteins (Tirona et al., 2001; Gui and Hagenbuch, 2008; Köck et al., 2010; Zhou et al., 2011), were conducted primarily in transporter-overexpressing cell lines. In the current studies, FLAG-Myc-tagged OATP1B3 was overexpressed in human hepatocytes by transduction of the human hepatocytes with Ad-OATP1B3 (Fig. 4). Functional studies confirmed that PMA treatment (0.1 μM, 30 minutes) significantly decreased [3H]CCK-8 accumulation in Ad-OATP1B3–transduced human hepatocytes either with (Fig. 4B) or without Matrigel overlay (data not shown). Pilot experiments indicated that the Matrigel overlay interfered with the biotinylation efficiency and might therefore have prevented the detection of surface expression of OATP1B3 in overlaid SCH. This may be because Matrigel is enriched with basement membrane proteins, which may compromise the biotinylation efficiency of the surface proteins of human SCH. Therefore, Ad-OATP1B3–transduced human hepatocytes without Matrigel overlay were used to determine the effect of PKC activation on surface levels and phosphorylation status of the overexpressed OATP1B3. The data reveal that PMA treatment (0.1 μM, 30 minutes) did not significantly affect either surface levels (Fig. 5, right panel) or total protein levels of overexpressed OATP1B3 (Fig. 5, left panel). The latter is consistent with the negligible effect of PMA treatment on total protein levels of endogenous OATP1B3 (Fig. 3B). Taken together, decreased OATP1B3 transport function following PKC activation is not attributable to altered surface levels of OATP1B3.
A rapid downregulation of [3H]CCK-8 accumulation was observed as early as 10 minutes following PMA treatment (Fig. 2A). PKC activation by PMA treatment (0.1 μM, 30 minutes) did not affect either mRNA (Fig. 3A) or total protein levels of endogenous OATP1B3 (Fig. 3B) in human hepatocytes, suggesting a regulatory mechanism at the post-translational level. For the first time, we characterized OATP1B3 as constitutively phosphorylated in Ad-OATP1B3–transduced human hepatocytes. Upon PKC activation, the phosphorylation status of OATP1B3 was markedly increased (Fig. 6A). Notably, PKC activation potently decreased OATP1B3 transport function to approximately the same extent as the potent OATP inhibitor BSP in both nontransduced and Ad-OATP1B3–transduced human hepatocytes (Fig. 4). These results suggest that increased phosphorylation of OATP1B3 may contribute significantly to decreased OATP1B3 transport function upon PKC activation, considering no changes in surface or total protein levels of OATP1B3 were noted.
Decreased surface levels and/or increased phosphorylation of transport proteins has been reported for several OATs and OATPs upon PKC activation. PKC activation decreased surface levels of OAT1, OAT4, OATP2B1, and OATP1A2 without affecting total protein levels of these transport proteins after short-term treatment with a PKC activator for up to 1 hour (Zhou et al., 2007, 2011; Zhang et al., 2008, 2010; Köck et al., 2010). Rapid downregulation of transport function by PKC activation is associated with increased phosphorylation of OATP2B1 (Köck et al., 2010), the brain glutamate/aspartate transporter GLAST-1 (Conradt and Stoffel, 1997), as well as dopamine (Huff et al., 1997) and serotonin transporters (Jayanthi et al., 2005). For serotonin transporter (SERT), pretreatment with PMA for 5 and 30 minutes increased phosphorylated SERT in the total lysates (Jayanthi et al., 2005). Interestingly, surface levels of SERT were unaffected after 5-minute PMA pretreatment but decreased after 30-minute PMA pretreatment, implying a time-dependent regulation of surface levels of the serotonin transport protein upon PKC activation. Our results demonstrating increased phosphorylation and unaffected surface levels of OATP1B3 following 30-minute PMA pretreatment is similar to the trend reported for GLAST-1 (Conradt and Stoffel, 1997) and SERT (Jayanthi et al., 2005) after 30-minute and 5-minute pretreatment with PMA, respectively. Phosphorylation has been reported to affect conformation and function of membrane proteins (Fujitani et al., 2003; Yamada et al., 2013). Although the exact mechanism through which increased phosphorylation of OATP1B3 upon PKC activation decreases transport function of OATP1B3 remains unknown, altered phosphorylation status of OATP1B3 may affect the protein’s conformation and therefore alter its transport function.
PKC may be activated by endogenous compounds including bile acids [e.g., chenodeoxycholic acid (Le et al., 2006) and taurocholic acid (Stravitz et al., 1996)] and in various types of liver disease (Birkenfeld and Shulman, 2014). Modulation of PKC activity is also an attractive therapeutic target (Berman, 2012). Potential changes in PKC activity by endogenous compounds, disease states, or drugs may affect OATP1B3 substrate transport and warrant further characterization in the future.
In conclusion, the present studies report the novel finding of a rapid and potent downregulation of OATP1B3 transport activity via an indirect inhibition mechanism involving modulation of OATP1B3 phosphorylation upon PKC activation. Currently, OATP-mediated DDI determinations in vitro have been focused primarily on direct drug-transporter interactions. However, xeno- and endobiotics modulate transport proteins in a number of ways in addition to direct and competitive interactions at the active site(s) responsible for binding and translocation of substrates. Alterations in transport protein function at the level of transcription, translation, or post-translational modifications do not necessarily require direct interactions of drugs/compounds with the transport protein. Data reported in this study highlight the importance of rational design of in vitro transporter-mediated DDI studies when a perpetrator drug has the potential to modulate transporter function.
Acknowledgments
The authors thank Dr. Dietrich Keppler for providing the OATP1B1- and OATP1B3-expressing construct and the HEK-293-OATP1B3 stable cell line, Dr. Markus Grube for providing OATP2B1-expressing construct, and Dr. Richard B. Kim for providing the OATP1B3 antibody. The SCH studies were conducted under a research agreement between OUHSC and Qualyst Transporter Solutions, LLC (Durham, NC).
Authorship Contributions
Participated in research design: Powell, Farasyn, Köck, Meng, Yue, Brouwer.
Conducted experiments: Powell, Farasyn, Meng, Köck, Pahwa, Yue.
Performed data analysis: Powell, Farasyn, Köck, Meng, Yue.
Wrote or contributed to the writing of the manuscript: Farasyn, Powell, Köck, Meng, Yue, Brouwer.
Footnotes
- Received January 22, 2014.
- Accepted September 8, 2014.
↵1 Current affiliation: Department of Pharmacokinetics and Drug Metabolism, Amgen Inc., Seattle, Washington.
K.K. and X.M. contributed equally to this work.
This research was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants R01-GM094268 and R01-GM41935]; the University of North Carolina University Research Council Grant, the North Carolina Translational and Clinical Sciences [Grant 2KR90907]; College of Pharmacy University of Oklahoma Health Sciences Center start-up support; and Deutsche Forschungsgemeinschaft [Grant Ko4186/1-1]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Part of the work was presented as listed below: Yue W, Köck K, Brouwer KLR (2010) Regulation of organic anion transporting polypeptide (OATP) 1B3 function by protein kinase C, in 2010 FIP Pharmaceutical Sciences World Congress/AAPS Annual Meeting and Exposition; 2010 November 14-18; New Orleans, LA. Poster T3405. Meng X, Köck K, Li J, Brouwer KLR, Yue W (2013) Protein kinase C activation rapidly down-regulates OATP1B3 transport function in primary human hepatocytes, in 2013 AAPS Annual Meeting and Exposition; 2013 November 10-14; San Antonio, TX. Poster W4351.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- αPDD
- 4α-Phorbol-12,13-didecanoate
- BIM I
- bisindolylmaleimide I
- BSP
- bromosulfophthalein
- CCK-8
- cholecystokinin-8
- DDI
- drug-drug interaction
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethyl sulfoxide
- HBSS
- Hanks’ balanced salt solution
- MEM
- minimum essential medium
- MOI
- multiplicity of infection
- NEAA
- nonessential amino acids
- OATP
- organic anion-transporting polypeptide
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate 13-acetate
- RT-PCR
- reverse transcription–polymerase chain reaction
- SCH
- sandwich-cultured hepatocytes
- SERT
- serotonin transporter
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}