


Abstract
Human hepatic microsomes were used to investigate the carboxylesterase-mediated bioactivation of CPT-11 to the active metabolite, SN-38. SN-38 formation velocity was determined by HPLC over a concentration range of 0.25–200 μM CPT-11. Biphasic Eadie Hofstee plots were observed in seven donors, suggesting that two isoforms catalyzed the reaction. Analysis by nonlinear least squares regression gave KM estimates of 129–164 μM with a Vmax of 5.3–17 pmol/mg/min for the low affinity isoform. The high affinity isoform hadKM estimates of 1.4–3.9 μM withVmax of 1.2–2.6 pmol/mg/min. The lowKM carboxylesterase may be the main contributor to SN-38 formation at clinically relevant hepatic concentrations of CPT-11.
Using standard incubation conditions, the effects of potential inhibitors of carboxylesterase-mediated CPT-11 hydrolysis were evaluated at concentrations ≥ 21 μM. Positive controls bis-nitrophenylphosphate (BNPP) and physostigmine decreased CPT-11 hydrolysis to 1.3–3.3% and 23% of control values, respectively. Caffeine, acetylsalicylic acid, coumarin, cisplatin, ethanol, dexamethasone, 5-fluorouracil, loperamide, and prochlorperazine had no statistically significant effect on CPT-11 hydrolysis. Small decreases were observed with metoclopramide (91% of control), acetaminophen (93% of control), probenecid (87% of control), and fluoride (91% of control). Of the compounds tested above, based on these in vitro data, only the potent inhibitors of carboxylesterase (BNPP, physostigmine) have the potential to inhibit CPT-11 bioactivation if administered concurrently.
The carboxylesterase-mediated hydrolysis of α-naphthyl acetate (α-NA) was used to determine whether CPT-11 was an inhibitor of hydrolysis of high turnover substrates of carboxylesterases. Inhibition of α-NA hydrolysis by CPT-11 was determined relative to positive controls BNPP and NaF. Incubation with microsomes pretreated with CPT-11 (80–440 μM) decreased α-naphthol formation to approximately 80% of control at α-NA concentrations of 50–800 μM. The inhibitors BNPP (360 μM) and NaF (500 μM) inhibited α-naphthol formation to 9–10% of control and to 14–20% of control, respectively. Therefore, CPT-11-sensitive carboxylesterase isoforms may account for only 20% of total α-NA hydrolases. Thus, CPT-11 is unlikely to significantly inhibit high turnover, nonselective substrates of carboxylesterases.
Irinotecan (CPT-11,2Camptosar, Pharmacia & Upjohn, Kalamazoo, MI) is a carboxylesterase-labile, carbamate prodrug of the antineoplastic topoisomerase I inhibitor SN-38 (1-3). CPT-11 and SN-38 exist in a pH- and protein-dependent equilibrium between active lactone and inactive hydroxy acid anion forms (4). Bioactivation of CPT-11 by microsomal carboxylesterases (E.C. 3.1.1.1) occurs primarily in the liver (5, 6) and has been studied previously using purified human carboxylesterase (7, 8). In this study, an in vitro model for the assessment of CPT-11 bioactivation by carboxylesterases was developed using human liver microsomes. Michaelis-Menten kinetic constants were determined and a new high affinity isoform was discovered.
Since CPT-11 will be used clinically with a variety of other drugs, anin vitro screen for potential drug interactions involving CPT-11 bioactivation was developed.
Some chemicals were chosen based on possible clinical use and have no known carboxylesterase-inhibiting ability (e.g. 5-FU, ethanol (EtOH), caffeine, acetaminophen, coumarin, dexamethasone, cisplatin, and probenecid). Others were chosen based on previously described effects on carboxylesterases (e.g. ASA (9), fluoride (10), HMB, CMB (6), physostigmine (6, 11), bis-nitrophenylphosphate (BNPP) (7), metoclopramide (12), loperamide (8), and prochlorperazine (13)). To avoid misleading conclusions about the possible clinical significance of in vitro experiments conducted at high inhibitor concentrations, statistically significant results were discussed in the context of clinically relevant concentrations of inhibitor.
As a carbamate, CPT-11 is a relatively poor substrate for carboxylesterases. This was proposed to be a result of slow decarbamylation of the serine esteratic site, inferring that CPT-11 is a slowly reversible competitive inhibitor, possibly also with an allosteric inhibitory effect on other substrates at a modulator site of the enzyme (8). Therefore, CPT-11 could act as an inhibitor of endogenous or xenobiotic high turnover substrates of carboxylesterases. To investigate this, CPT-11 was tested as an inhibitor of microsomal carboxylesterase-mediated hydrolysis of the model substrate, α-naphthyl acetate (α-NA) (14,15).
Methods
Reagents and Materials.
Irinotecan hydrochloride trihydrate (CPT-11) and SN-38, were supplied by Pharmacia and Upjohn, Inc. Camptothecin and inhibitors were obtained from Sigma or Aldrich (St. Louis, MO). Transplant quality human liver tissue was obtained through the International Institute for the Advancement of Medicine (Exton, PA). Microsomes were prepared by standard methods (16) and stored in 0.25 M sucrose at −70°C. Protein was determined by the Bicinchoninic Acid (BCA) assay (Pierce Chemical, Rockford, IL) and standardized relative to bovine serum albumin.
Kinetics and Inhibitors of CPT-11 Bioactivation.Incubations.
Kinetic parameters were determined using CPT-11 that was at equilibrium in pH 7.4 phosphate buffer. Under these conditions the lactone/hydroxy acid anion ratio for CPT-11 was fixed at 87/13 (4). Kinetic studies were done in 2 ml Eppendorf Safe-lock polypropylene tubes (Brinkman, Westbury, NY) tubes using human hepatic microsomes and CPT-11, incubated in a final volume of 0.25 mL sodium phosphate (0.1 M, pH 7.4) at 37°C, with 80 rpm mixing. CPT-11 solutions were pre-incubated for 5 min and incubations were initiated by the addition of pre-warmed microsomes. Reactions were quenched and analyzed for SN-38 content by HPLC as described under sample preparation. All concentrations refer to total (lactone plus hydroxy acid anion) forms of CPT-11 and SN-38. Incubation times (5–60 min) and protein concentrations (0.125–1.8 mg/ml) were tested to identify conditions that resulted in quantifiable and linear formation of SN-38.
Determination of Michaelis-Menten kinetic parameters.
Incubations were done in duplicate using microsomal protein (1 mg/ml) and CPT-11 (0.25–200 μM, 17 concentrations), incubated for 15 min. A 50 μM CPT-11 control in phosphate buffer (without microsomes) and a boiled microsome control were run with each experiment. The hydrolysis of CPT-11 in phosphate buffer, in the absence of microsomal protein, and in the presence of boiled microsomal protein, was negligible over the incubation time course; however, the CPT-11 stock solution contained a small amount of SN-38 (approximately 0.04%). Levels of the SN-38 impurity at other substrate concentrations were calculated by linear interpolation of a calibration line passing through the experimentally-determined 50 μM blank data point and the origin. Total SN-38 formation in the presence of microsomes was corrected for the SN-38 impurity at each substrate concentration. Substrate depletion in kinetic studies was maximally 10% at 0.25 μM CPT-11.
Determination of the effect of potential inhibitors on SN-38 formation.
Incubations were done in triplicate using pooled human liver microsomes, as described in Incubations, at a CPT-11 concentration of 10 μM. Microsomes (25 μl) were mixed with potential inhibitors (0–25 μl) and phosphate buffer (100–125 μl) and pre-incubated for 5 min. Inhibitor concentrations were ≥ 21 μM and were chosen to represent clinically attainable concentrations, with the exception of loperamide, where the 25 μM test concentration was approximately 1000-fold higher than clinically relevant plasma levels. Incubations were initiated by the addition of prewarmed CPT-11 solution (100 μl). In the final incubation, DMSO content was 0.0016% and MeOH content was 0% or 1%. Reactions were quenched and analyzed as described under sample preparation. SN-38 formation was measured as ng/ml/15 min. Inhibition was expressed as per cent of control.
HPLC and analytical standard preparation.
A Perkin-Elmer ISS-200 autoinjector and PE 410 pump were used (Perkin Elmer, Norwalk, CT). Chromatography was done on a Zorbax SB-C8 column (4.6 × 250 mm, 5 μm particle size, Mac Mod Analytical Inc., Chadds Ford, PA) with a Brownlee Newguard RP8 guard column using a mobile phase of 74:26, v:v triethylamine buffer:acetonitrile at a flow rate of 1 mL/min. Detection was done using a Waters Model 474 fluorescence detector (Millipore Corp., Milford, MA). Calibration standards (9–10 concentrations) were prepared in duplicate in phosphate buffer to cover a concentration range of approximately 1 to 4000 ng/ml SN-38. Quality control (QC) standards were prepared in duplicate from separately prepared stock solutions at nominal SN-38 concentrations of 23, 76, and 1900 ng/ml. The stability of stock and control solutions in MeOH and in phosphate buffer at 4°C (storage conditions) were proven by HPLC analysis after storage periods of 0, 5, 22, 28, and 41 days. SN-38 recovery from microsomes relative to phosphate buffer was determined to be complete by comparing triplicate incubations of microsome suspensions spiked with 4, 90, 300, and 1900 ng/ml SN-38 with triplicate QC samples prepared identically in phosphate buffer.
Sample preparation.
A 250 μl aliquot of incubation sample, calibration standard or QC sample was mixed with 500 μl quench solution (acetonitrile/acetic acid/methanol (95.6/4.0/0.4, w/w/w) containing 24 ng/ml camptothecin (CPT) internal standard). The samples were heated in a water bath at 37°C for 20 min to convert SN-38 to the lactone form. Samples were mixed with 1.0 ml of 50 mM, pH 4.2 triethylamine/acetate buffer. Precipitated protein was separated by centrifugation at 14,000 rpm for 2 min. A 50 μl aliquot of supernatant was analyzed by HPLC. Retention times were sensitive to small changes in mobile phase composition. The detector was programmed to change wavelengths and detector gain. From 0–10 min the excitation and emission wavelengths were 372 and 425, respectively (gain 10, measures CPT-11). From 10–14 min the excitation and emission wavelengths were 372 and 535, respectively (gain 100, measures SN-38). From 14 min onward the excitation and emission wavelengths were 372 and 425, respectively (gain 100, measures CPT).
Data collection and analysis.
Data were collected and processed using the UPACS (Upjohn) chromatography system, Version 5.2. Quantitation was done using peak height ratios of the analyte to internal standard. Calibration curves were determined by using linear regression through-the-origin best fit with a 1/concentration weighting factor. Unknown SN-38 concentrations in incubation solutions were determined by comparison of the peak height ratios with the calibration curve. Correction factors for the SN-38 impurity at each concentration/time datapoint were calculated by subtracting background SN-38 from the total SN-38 formed in the presence of microsomes. Unknown concentrations were determined as corrected (carboxylesterase-mediated) ng/ml SN-38 produced in 15 min and were converted to units of velocity (pmol/mg/min) using a formula weight of 410.4 g/Mol for SN-38 monohydrate.
Analytical validation.
Standards of CPT-11 in MeOH or in phosphate buffer, prepared in polystyrene centrifuge tubes and stored at 4°C, were stable over a period of 41 days. Because of a limited supply of human microsomes, standards and QC samples prepared in phosphate buffer were used to quantify microsomal concentrations of SN-38. Recoveries of SN-38 from microsomes spiked in triplicate with SN-38 at concentrations of 4, 90, 300, and 1900 ng/ml were approximately 100% in both microsomes and phosphate buffer. Correlation coefficients (r2) for linear standard curves in kinetic assays were ≥ 0.998. QC data were typically within ± 15%. The assay lower limit of quantitation (LLOQ) was 1.1–2.4 ng/ml SN-38.
Curve fitting methods.
KM and Vmaxwere determined by nonlinear least squares regression using Systat Version 5.2.1 for Macintosh (Version 7.0). SN-38 formation V versus [S] curves were fit for a two enzyme system with 1/V2 weighting. α-NA hydrolysis V versus [S] curves were best fit by a single enzyme Michaelis-Menten equation.
Determination of inhibitor potency.
Data were analyzed using Microsoft Excel Version 4.0a (Microsoft Corp., Redmond, WA). Means and standard deviations of uncorrected SN-38 production in the presence and absence of inhibitors were compared with a Student’s t test. Mean SN-38 production was corrected for mean SN-38 impurity measured in the presence of heat denatured (boiled) microsomes and SN-38 production was expressed as per cent of enzyme-catalyzed control. When MeOH was necessary as an aid to solution for inhibitors, an additional control containing an equal amount of MeOH was run. The carboxylesterase inhibitor BNPP and a phosphate buffer control (no protein) were used as positive control and blank, respectively.
Kinetics of α-naphthyl acetate hydrolysis and inhibition by CPT-11
An assay for the carboxylesterase-mediated formation of α-naphthol from α-NA was developed based on the formation of an azo dye from the reaction of fast blue RR (a diazonium salt, Sigma, St. Louis, MO) with α-naphthol (17). Enzyme kinetics were determined using modified literature methods (14, 15) on a THERMOmax Microplate Reader (Molecular Devices, Sunnyvale, CA) (18). Optical Density (O.D.) was measured at 450 nm. Initial velocities were recorded in units of mO.D./min and were corrected for background hydrolysis in buffer, determined simultaneously. CPT-11 (80–440 μM) was tested as an inhibitor of α-NA hydrolysis. Control carboxylesterase-mediated hydrolysis was determined on the same plate, simultaneous with each inhibitor concentration, since preincubation effected the control kinetics. Results were expressed as per cent of control sample initial velocity (mO.D./min).
Reagent Preparation.
BNPP and NaF were prepared in 0.1 M, pH 7.4 phosphate buffer. Stock solutions of α-naphthol for standard curves were prepared in acetone. Final acetone concentration in the standard curve wells was 3.3%. Stock CPT-11 solutions were prepared at 360 μM in phosphate/MeOH or at 80, 140, and 440 μM in phosphate/dimethylsulfoxide (DMSO) at 90/10 w/w. The co-solvent concentration in each incubation well was 0.33%. Controls containing an equal amount of co-solvent were prepared. Stock α-NA solutions were prepared in acetone/phosphate at 50/50 (w/w). The final acetone concentration of 1.7% in all wells does not effect enzyme activity (14). Fast Blue RR was prepared fresh in phosphate buffer at 0.25% v/v for each experiment and was filtered prior to use.
Preliminary Experiments.
Low protein concentrations and short reaction times at 23°C and substrate concentrations of ≥ 40 μM were needed to avoid substrate depletion in excess of 15–20%.
At α-NA concentrations ≥ 40 μM product formation was linear for 3–4 min. Estimates of initial velocity (mO.D./min) were determined using 13 data points acquired over 2 min. Initial velocity measured over 2 min was linear from 1–10 μg/ml microsomal protein. Subsequent experiments were conducted at 5 μg/ml protein. Calibration curves of O.D. versus concentration (μM) for the α-naphthol (product)/dye complex were linear (r2 ≥ 0.98) over the concentration range 0–150 μM. Kinetic experiments were conducted in quadruplicate in two male (donors 10M and 24M) and three female (donors 14F, 17F, and 20F) donor microsomes over an α-NA concentration range of 5–800 μM. Means of quadruplicate experiments had percent coefficient of variation values of typically less than 15% for each concentration ≥ 40 μM. Thereafter, in inhibitor studies, incubations were conducted in duplicate in sodium phosphate buffer (pH 7.4, 0.1M). When MeOH or DMSO were used an aid to solution for CPT-11, control experiments were also conducted in parallel with the appropriate concentration of co-solvent. In all experiments, duplicate controls were run simultaneously for each substrate concentration on the same plate.
Incubation Conditions.
In a 96 well microplate, each well contained 260 μl buffer, 20 μl microsomes (0.075 mg/mL) or buffer, 10 μl fast blue RR (0.25 mg/ml), and 10 μl substrate or buffer. The total volume was 300 μl. Substrate was added last using a multi-channel pipette. Immediate automatic mixing was employed and incubations were conducted with continuous assessment of optical density within the thermostatted compartment of the plate reader. Preincubations with inhibitors were done inside the plate reader.
Kinetics of α-Naphthyl Acetate Hydrolysis.
Substrate concentrations of 0, 5, 10, 20, 40, 60, 80, 100, 200, 400, 600, and 800 μM (12 concentrations/lane) were incubated in quadruplicate on the same plate with a matching quadruplicate set of enzyme-free controls. Substrate depletion, calculated from the α-naphthol standard curve, was maximally 17% at 2 min at 60 μM α-NA for donor 20F, and was 13% or less in all other donors at α-NA ≥ 40 μM. Data were fit to a single enzyme Michaelis Menten equation using substrate concentrations of 40–60 μM to 600–800 μM. Vmax data were compared toVmax data for low and highKM isoforms of the CPT-11 hydrolyzing carboxylesterase by linear regression comparing donors 10M, 24M, 17F, and 20F.
Inhibition by CPT-11.
Microsomes from donor 17 were preincubated with CPT-11 (360 μM), or BNPP (360 μM), or NaF (500 μM) for 10 min before the addition of α-NA (0–120 μM, 8 concentrations/plate lane) to start the reaction. The experiment was repeated with CPT-11 (80, 140, and 440 μM) preincubated for 1 h at 23°C over α-NA concentrations of 0–800 μM.
Results
Hydrolysis Kinetics of CPT-11 in the Presence of Human Liver Microsomal Carboxylesterases
Incubation Conditions.
In pilot studies, heat denaturation (boiled microsomes) and the organophosphate esterase inhibitor, BNPP reduced SN-38 production by human hepatic microsomes to zero, providing evidence for the involvement of esterase enzymes. An optimal protein concentration of 1 mg/ml and incubation time of 15 min were chosen from the linear regions of SN-38 production. These conditions were necessary to surpass the assay limit of quantitation at low substrate concentrations and to adequately differentiate enzyme-catalyzed SN-38 formation from the SN-38 impurity.
Kinetic Results for SN-38 Formation.
HPLC chromatograms showing the effect of CPT-11 concentration on the formation of SN-38 by microsomes are shown in fig.1. In microsomes from seven different liver donors, Eadie Hofstee plots were clearly biphasic over 0.25–200 μM CPT-11 (data not shown). In accord with the linear plots, V versus [S] data, modeled using nonlinear least squares regression analysis, were best fit by a two enzyme equation.KM and Vmaxestimates are presented in table 1. A representative Eadie Hofstee plot for the pooled microsomes, comparing fitted with actual data is shown in fig.2. The actual and theoretical curves were superimposed. The calculated KM andVmax for each carboxylesterase isoform are individually represented in the same figure by the calculated linear plots for each isoform.

Superimposed HPLC chromatograms showing SN-38 formed by donor 10M microsomes (1 mg/mL protein, 15 min) at CPT-11 concentrations of 0.5 (lower trace), 5 (second trace), 25 (third trace), and 100 μM (upper trace).
Summary of Michaelis-Menten kinetic parameters for SN-38 formation from CPT-11 by human liver microsomes. V vs. [S] data for each donor were fit by nonlinear regression to a 2 enzyme Michaelis-Menten equation to obtain KM and Vmax estimates

Eadie Hofstee plot generated following nonlinear curve fitting of V versus [S] data for pooled microsomes to a 2 enzyme Michaelis-Menten equation.
Linear interpolations for each isoform, based on nonlinearKM and Vmaxestimates, illustrate graphically the correct intercept and slope for the individual isoforms. KM andVmax estimates are in table 1.
Data for all donors (N = 7) gave a well defined apparent KM of 1.4–3.9 μM for the low KM isoform and 129–164 μM for the high KM isoform. TheVmax/KM of the low KM isoform was higher than the high KM enzyme.
A histogram comparing the relative contribution of the low and highKM CPT-11 carboxylesterases to the total production of SN-38 is shown versus CPT-11 concentration in fig. 3. The lowKM enzyme is the dominant source of SN-38 at CPT-11 concentrations up to 20 μM. Thus, at physiologically relevant hepatic concentrations of CPT-11, the lowKM isoform will be the predominant enzyme responsible for CPT-11 bioactivation.
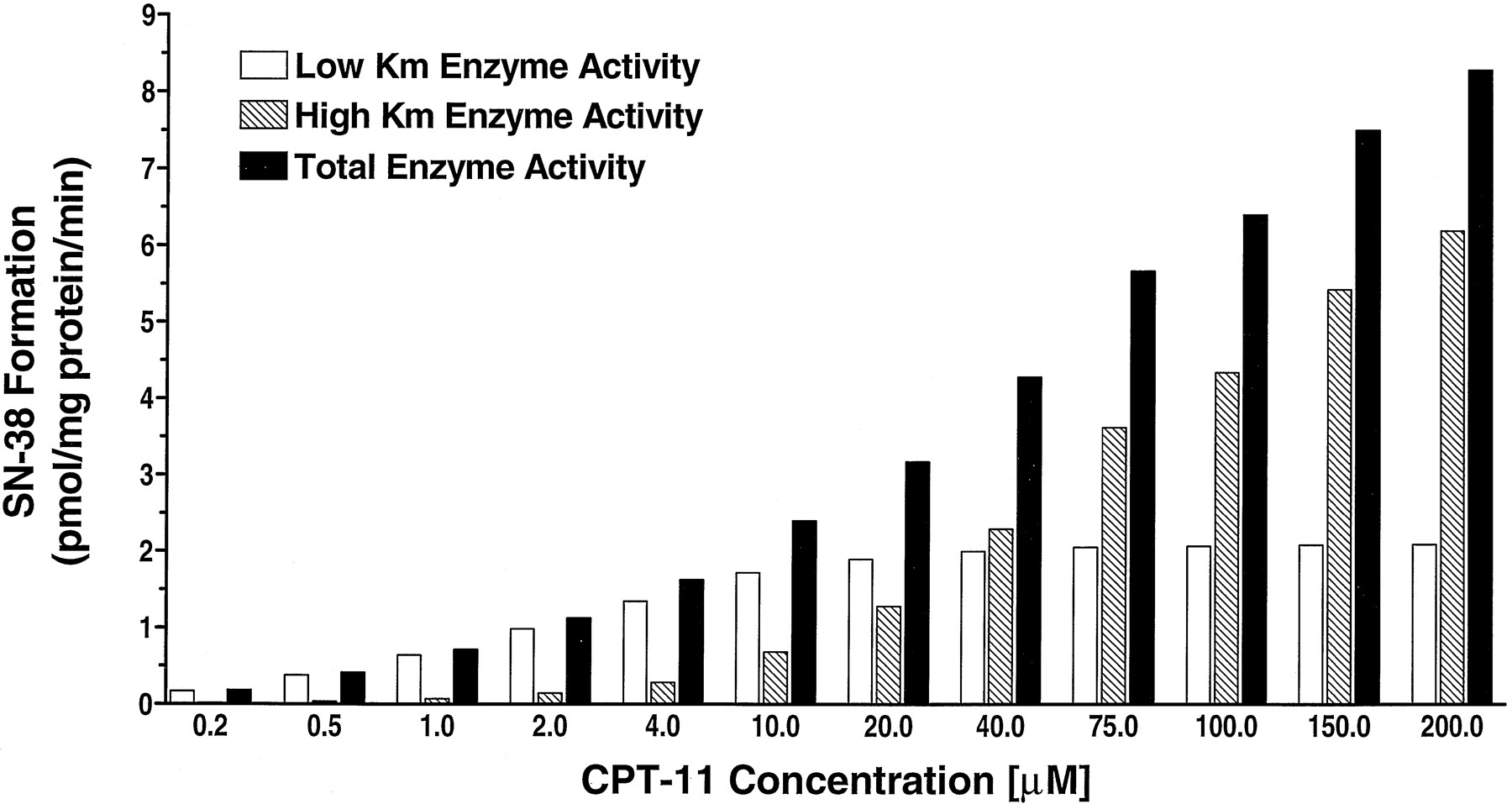
Histogram showing the calculated relative contributions of each esterase isoform to the total production of SN-38 at increasing concentrations of CPT-11.
At clinically relevant hepatic concentrations of below 20 μM, the high affinity, low KM isoform is the dominant contributor to CPT-11 bioactivation.
Effects of Potential Inhibitors of CPT-11 Bioactivation
Results of four separate inhibition experiments are compared with their respective control values in table2. Inhibition was expressed as per cent of control. Controls containing an equal amount of 1% MeOH were run when MeOH was used to dissolve inhibitors. MeOH decreased activity by 3.5, 6.6, and 18.2% in experiments 2, 3, and 4, respectively. Organic solvent effects on carboxylesterase activity have been documented previously (19).
The effect of various potential inhibitors on the carboxylesterase-mediated production of SN-38 from CPT-11
As expected, BNPP at both 25 and 100 μM reduced CPT-11 hydrolysis to 1.3–1.6% of control values. Physostigmine, a known inhibitor of butyrylcholinesterases (E.C. 3.1.1.8, closely related to carboxylesterases) showed significant (77% decrease, (P < 0.05)) inhibition of CPT-11 hydrolysis, in accord with previous data (6, 11). Probenecid (25 μM) showed a significant (13%, P < 0.01) inhibition of CPT-11 hydrolysis, while 5-FU (25 μM) had no significant inhibitory effect (2%, P < 0.1). Sodium fluoride (NaF), a known carboxylesterase inhibitor (10, 20) inhibited hydrolysis significantly (P < 0.01) by 9% at 25 μM, 14% at 100 μM, and 35% at 481 μM.
Aggressive therapy with loperamide is used to treat CPT-11-induced delayed diarrhea (21). A small decrease (7.5%) in SN-38 formation owing to loperamide was observed; however, the difference was not significantly different from control values. Ethanol at 25 and 100 μM had no significant effect on CPT-11 hydrolysis. Two mercuribenzoate inhibitors of A-esterases (HMB and CMB) also had no significant effect. Caffeine, ASA, and coumarin all showed no significant effect on CPT-11 hydrolysis. Small, but statistically significant, decreases were observed with metoclopramide (9.1% decrease, P < 0.01) and acetaminophen (7% decrease, P < 0.05). Dexamethasone, cisplatin, and prochlorperazine were without significant effect with 3.4%, 5.6%, and 7.0% differences from control values, respectively.
Effect of CPT-11 on α-Naphthyl Acetate, a High Turnover Substrate of Human Liver Carboxylesterase
Fig. 4 shows initial velocityversus substrate concentration plots over 40–800 μM for microsomes from five donors. Data were fit by nonlinear regression to a single enzyme Michaelis Menten equation giving corrected correlation coefficient (R2) values of 0.94–0.98.KM estimates for α-NA hydrolysis were 115 ± 20.8 μM (range 90.6–146 μM, CV = 18%).Vmax was estimated at 1.64 ± 0.45 μMol/mg/min (range 1.1–2.6 μMol/mg/min, CV = 27.3%). Linear regression (forced through the origin) ofVmax for α-NA hydrolysisversus Vmax for low and highKM isoforms of the CPT-11 hydrolase afforded correlation coefficients of 0.94 with the CPT-11 highKM isoform and a lower correlation coefficient of 0.82 for the CPT-11 low KM isoform in five donors. The narrow range ofVmax for the CPT-11 lowKM isoform and the limited number of donors (N = 4, range 2.41–2.61), may be responsible for the lower correlation coefficient. A larger sample size would be needed to conclude that CPT-11 and α-NA carboxylesterase activities are correlated.

Effect of α-naphthyl acetate concentration (40–800 μM) on the initial velocity of human liver microsome catalyzed, carboxylesterase-mediated formation of α-naphthol.
Data shown are mean ± SD for simultaneous quadruplicate determinations on a 96 well microplate. Results for each well are corrected for any measurable background hydrolysis, determined simultaneously in enzyme-free phosphate buffer. Mean data in mO. D./min were converted using a standard curve to units of pmol/mg/min for calculation of KM andVmax.
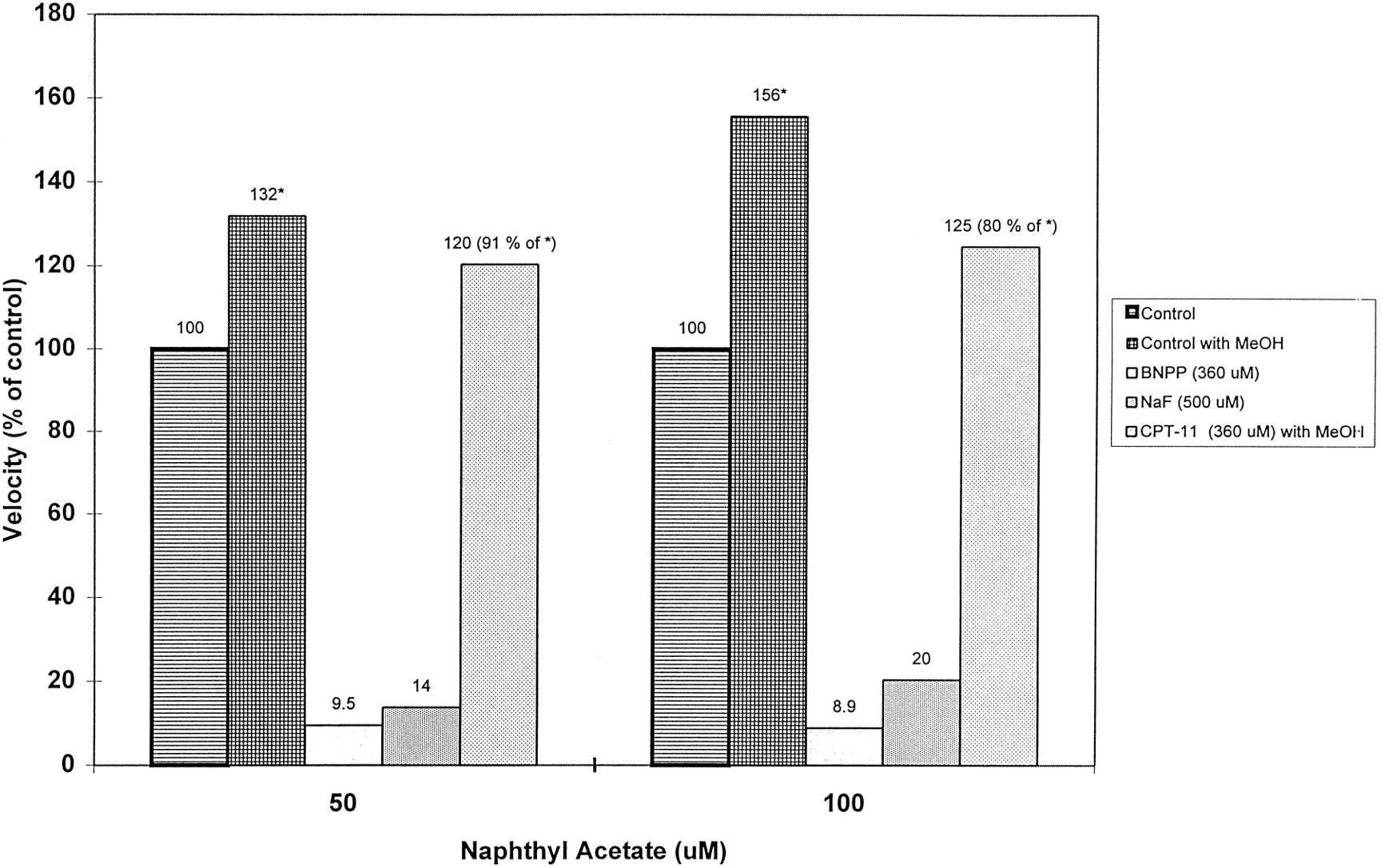
At α-NA concentrations of 0 to 100 μM, hydrolysis was almost completely inhibited by positive control carboxylesterase inhibitors, BNPP (360 μM) and NaF (500 μM), whereas CPT-11 preincubated for 10 min at 360 μM inhibited α-NA hydrolysis by only 9 and 20% relative to control values, at concentrations of α-NA of 50 and 100 μM (fig. 5). In a second experiment, CPT-11 preincubated for 1 h at 80, 140, or 440 μM inhibited α-NA hydrolysis to 69–89% (mean 80 ± 6.6%, N= 9) of control values, at concentrations of α-NA of 200, 400, and 800 μM (data not shown). It is clear from these data that inhibition of α-NA hydrolysis by CPT-11 was maximal at approximately 80% of control values at the lowest CPT-11 concentration tested (80 μM), and therefore this concentration was well above theKi for CPT-11-sensitive carboxylesterases. Thus, CPT-11-sensitive carboxylesterases may only account for 20% of the turnover of the model substrate, α-NA in vitro.

Effect of CPT-11 on the carboxylesterase-mediated hydrolysis of α-naphthyl acetate.
Control experiments with a MeOH cosolvent were done for direct comparison with CPT-11 incubations that required methanol as an aid to solution. The control incubation for NaF and BNPP treatments was free of cosolvent. In a separate experiment (not shown), CPT-11 inhibited α-naphthyl acetate hydrolysis by only 20% (80 ± 6.6% of control, N = 9). The remaining 80% of control activity may be attributed to CPT-11-insensitive carboxylesterase isoforms.
Discussion
CPT-11 Bioactivation
The Enzymology of Carboxylesterases.
To develop a clinically-relevant perspective on the bioactivation of CPT-11, and to understand the implications of carboxylesterase inhibition on antineoplastic activity, it is necessary to understand the basic chemistry of the enzyme class. The mechanism of hydrolysis by serine hydrolases such as the carboxylesterases is well understood from kinetic studies on substrates and inhibitors (19). However, the molecular biology of carboxylesterase enzymes is only now being elucidated, in part because of technical difficulties inherent in subtle differences in sequence, structure, and substrate and inhibitor specificity (22, 23). Two highly similar human liver microsomal carboxylesterases were recently sequenced and expressed by Kroetzet al. (22), and these authors conclude, based on substrate diversity, that additional carboxylesterases must exist. Kettermanet al. have purified and characterized mid and low Pi human hepatic carboxylesterases and have proposed, based on kinetic comparison of different donors, that polymorphism occurs within each purified carboxylesterase (24, 25). Therefore, it is likely that more than one carboxylesterase isoform may contribute to CPT-11 bioactivation.
CPT-11 Hydrolysis and Antitumor Activity.
Although CPT-11 has weak antineoplastic activity of its own, a 104-fold increase in cytotoxicity in vitro is realized when SN-38 is released by carboxylesterases in rat serum (26). The conversion of CPT-11 to SN-38 has been studied in a wide variety of tissues, cell lines, and purified enzyme preparationsin vitro (5-8, 11, 27). The sensitivity of proliferating tissues or cell lines to the cytotoxic effects of CPT-11 may be related their carboxylesterase levels (27). Some studies indicate that tumor levels of carboxylesterases are reduced relative to peritumoral normal tissue (28).
A New CPT-11 Hydrolase.
Our observation of biphasic CPT-11 kinetics in human liver microsomes suggests at least two carboxylesterase isoforms are present in our human microsomal preparation. We have observed the lowKM isoform by studying much lower substrate concentrations (250 nM-200 μM) relative to the concentrations used by others (7, 8). Satoh et al. studied a purified enzyme at 25–100 μM CPT-11 givingKM = 169 μM andVmax = 169 pmol/mg/min by Lineweaver-Burk analysis of the linear portion of the plot. Rivory et al.studied the mid Pi purified enzyme at 8–150 μM CPT-11, observing aKM of 52.9 μM by nonlinear regression analysis. Our data fit a two enzyme MM equation extremely well, and we speculate that the characterization of two or more carboxylesterase isoforms by Kroetz et al. (22) and Ketterman et al. (24, 25) may be relevant to the kinetic parameters determined in this study.
The variability in carboxylesterase content of human livers was 33.1-fold in human liver microsomes (N = 12) as shown by Hosokawa et al., using nitrophenyl acetate hydrolysis as a marker (29). Variability was shown by Kroetz et al. to be maximally 3–4 fold for the hCE isoform and 8-fold for the hCEv isoform (N = 8), as determined by mRNA quantitation (22). We observed a 2- to 3-fold difference in individualVmax estimates for CPT-11 hydrolysis in hepatic microsomes from six donors.
Regarding the possibility that carboxylesterase induction could alter CPT-11 bioactivation, little is known about the regulation of carboxylesterases in man. An array of common microsomal inducers, including dexamethasone have no inductive effect or can inhibit expression of rat hydrolases A and B (30). No clinically relevant conclusions about the induction of SN-38 production in man can be drawn from these rat data. This is additionally complicated by the fact that some inducers of carboxylesterases, such as phenobarbital, induce UDPGT isoforms that catalyze the glucuronidation of SN-38 in rats (31) and also induce CYP 3A4 which mediates the metabolism of CPT-11 to the metabolite aminopentanecarboxylic acid (APC) (32).
CPT-11 bioactivation is a relatively slow and inefficient process, withVmax in the low pmol/mg/min range. This observation is supported by low plasma SN-38/CPT-11 AUC ratios in vivo (33). In accord with the carbamate structure shared by CPT-11 with cholinesterase inhibitors such as bambuterol (34), Rivory et al. have proposed that decarbamylation of the serine esteratic site is the rate limiting step in carboxylesterase-mediated CPT-11 hydrolysis (8). Indeed, the carbamate functional group was a molecular design feature (in addition to the solubility enhancement of the outer basic piperidine ring) that allowed the slow and sustained release of SN-38. This may account for relatively superior antineoplastic activity of CPT-11 in vivo, relative to esters of SN-38. (3)
Lactone/hydroxy Acid Anion Equilibria in Relation to CPT-11 Bioactivation.
The lactone form of CPT-11 is stable under acidic, but not neutral, conditions with a half life in pH 7.4 phosphate buffered saline of 25.6 min. At equilibrium, the lactone/hydroxy acid anion ratio is known to be 13/87 (4). Haaz et al. have recently shown that there is an approximately 2-fold greater hydrolysis of the lactone form of CPT-11 relative to the ring-opened hydroxy acid anion form (35). Equilibrium conditions for our kinetic studies were chosen for several reasons: First, the lactone concentration would not be changing rapidly and enzyme activity would be maximal during the incubation at pH 7.4. Second, because the lactone forms of CPT-11 and SN-38 are preferentially bound to and stabilized by serum albumin, the free fraction in plasma may be dominated by the hydroxy acid anion form. Third, the hydroxy acid anion form may hypothetically be more available for extraction by the liver, as shown by the short half life of SN-38 hydroxy acid anion when given intravenously to animals as the soluble sodium salt (36).
It was not experimentally determined whether the biphasic kinetics observed in this experiment were the results of a single carboxylesterase isoform with a differentKM for lactone and hydroxy acid anion forms of CPT-11. If this was the case, however, each form (lactone or hydroxy acid anion) would be a competitive reversible (with a slow off-rate) inhibitor of the turnover of the other. To investigate this possibility, we refit our data to a single enzyme/single inhibitor Michaelis-Menten equation, using a hypothetical constant 13/87 ratio of lactone to hydroxy acid anion to calculate concentrations of lactone and hydroxy acid anion at each substrate concentration. The data could not be fit, in direct contrast to the excellent fit observed for the two enzyme equation. We conclude that any difference in rates of turnover between lactone and hydroxy acid anion forms could not be extracted from our data and that two carboxylesterase isoforms were responsible for the biphasic kinetics.
Effect of Potential Inhibitors of CPT-11 Bioactivation
Loperamide (24 μM) reduced SN-38 formation from 10 μM CPT-11 by approximately 7%. This difference from control was not statistically significant. Rivory et al. (8), using a purified enzyme, have shown that high concentrations of loperamide (200 μM) inhibits p-nitrophenylacetate hydrolysis, loweringVmax from 60.0 to 41.9 nMol/min and almost doubling the KM (240 to 400 μM). Similarly, 50 μM loperamide reduced SN-38 production by approximately 50% at high CPT-11 concentrations (8). We estimate, based on single dose literature data, that when loperamide is given at 2 mg every 2 hr, that loperamide steady state plasma concentrations are approximately 34 nM in humans (37). We speculate that hepatic concentrations at this dose intensity will be much lower than the 24 μM incubation concentrations used in our study. On the basis of these data, we conclude that, even if administered at high doses at the same time as CPT-11, the likelihood of a clinically significant effect of loperamide on SN-38 production is negligible.
Sodium fluoride (NaF), a known carboxylesterase inhibitor (10, 20), inhibited hydrolysis by 9% at 25 μM. This is a relatively high NaF concentration. Fluoride, which is formed from the breakdown of the 5-FU metabolite, fluoro-β-alanine (FBAL) (38), could potentially decrease SN-38 AUC during co-therapy with 5-FU and CPT-11 (39). In a separate study, at 5-FU doses ranging between 8 and 30 mg/kg, free fluoride was observed in plasma in about one-fourth of patient samples that were examined (median 5 μM, maximum 33 μM) (38). The low fluoride concentrations observed clinically and the extent of in vitro inhibition observed at 25 μM NaF in the present study indicate that 5-FU-derived fluoride is not likely to result in clinically significant decreases in metabolism of CPT-11 to SN-38. A recent clinical report shows that co-administration of 5-FU does not significantly effect the conversion of CPT-11 to SN-38 (40).
Relative to the potent inhibitors, BNPP and physostigmine, which are not expected to be given with CPT-11, inhibition of SN-38 formation by 5–10% in vitro by high concentrations of inhibitor is probably not clinically relevant. Furthermore, any subtle changes would be irrelevant considering the high interpatient variability normally observed in CPT-11 pharmacokinetics (41).
Co-administration of any known potent carboxylesterase inhibitor with CPT-11 is probably rare. However, if a known therapeutic carboxylesterase inhibitor, such as physostigmine or echothiophate, can be safely withheld, staggering co-therapy with CPT-11 would be the prudent and cost effective way to ensure adequate production of SN-38 in patients needing both medications.
α-Naphthyl Acetate Hydrolysis: Effect of CPT-11 on a High Turnover Substrate of Human Liver Carboxylesterases
The kinetics of α-NA hydrolysis were determined. Vmax data were about 50% lower than those determined by Huang et al. (3.89 ± 0.20 μMol/mg/minversus 1.64 μMol/mg/min) (14). In this studyVmax varied from 1.1–2.6 μMol/mg/min. Data from four donors demonstrated a correlation between α-NAVmax and theVmax of the highKM component of CPT-11 hydrolysis. The kinetic difference between ester and carbamate substrates is apparent in the 106-fold higherVmax for α-NA hydrolysis relative to CPT-11 hydrolysis.
In one liver sample, CPT-11 inhibited the carboxylesterase-mediated hydrolysis of α-NA by only 20%. Compared with BNPP and NaF, CPT-11 was therefore a relatively weak inhibitor of α-NA hydrolysis. We did not examine low enough CPT-11 concentrations to establish aKi (presumably this would equal theKM ). Since 80% of the hydrolysis of α-NA continued unabated in the presence of high concentrations of CPT-11, we speculate that nonselective substrates of carboxylesterases will not be appreciably inhibited by CPT-11 in vivo, owing to the contributions of isoforms not affected by CPT-11.
Conclusions
We have characterized a new low KM human carboxylesterase that we propose to be the major isoform responsible for the bioactivation of CPT-11 in vivo. The CPT-11-sensitive microsomal carboxylesterases characterized in this paper are inhibited by potent carboxylesterase inhibitors, but are inhibited to only an insignificant extent by high concentrations of some selected agents that are commonly coadministered with CPT-11. CPT-11 inhibits the carboxylesterase-mediated turnover of the model high turnover ester substrate α-NA, but only by 20% even at the highest CPT-11 concentrations tested. We speculate that, since there is ample remaining activity related to carboxylesterase isoforms that are not sensitive to CPT-11, CPT-11 should not significantly inhibit the hydrolysis of high turnover, nonselective ester substrates of carboxylesterases.
Acknowledgments
We thank Dr. F. Kezdy for advice on curve fitting for Michaelis Menten equations, to Dr. Roger Ulrich and Jim Bacon for help in the early phases of this investigation, and to Dr. Laurent Rivory for a valuable discussion.
Footnotes
-
Send reprint requests to: Dr. J. Greg Slatter, Drug Metabolism Research, Pharmacia and Upjohn Co., 301 Henrietta St., Kalamazoo MI 49007. E-mail: john.g.slatter{at}am.pnu.com.
-
↵1 Present address: Department of Biopharmaceutical Sciences, University of California at San Francisco.
-
A preliminary account of this work was given at the 4th International ISSX Meeting, Abstracts #88, 218 and 219. Seattle WA, Aug 27–31, 1995.
- Abbreviations used are::
- CPT
- camptothecin
- CPT-11
- irinotecan hydrochloride trihydrate
- EtOH
- ethanol
- ASA
- acetylsalicylic acid
- HMB
- hydroxymercuribenzote
- CMB
- chloromercuribenzoate
- HPLC
- high performance liquid chromatography
- KM
- Michaelis constant
- 5-FU
- 5-fluorouracil
- MeOH
- methanol
- BNPP
- bis-nitrophenylphosphate
- α-NA
- alpha naphthyl acetate
- SN-38
- active antineoplastic metabolite of CPT-11
- DMSO
- dimethylsulfoxide
- UPACS
- Upjohn Physical and Analytical Chemistry System
- V
- reaction velocity
- QC
- quality control standard
- [S]
- substrate concentration
- Received March 28, 1997.
- Accepted June 5, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}