


Abstract
The P450 2A6 catalyzed 7-hydroxylation of coumarin proceeded with a mean Km of 0.40 (±0.13) μM andVmax of 6.34 nmol/nmol P450/min (36-fold variation) in microsomal preparations from a panel of 12 human livers. Substrate depletion was avoided during the kinetic determinations. 8-Methoxypsoralen (8-MOP) is a potent mechanism-based inactivator of human liver P450 2A6 and reconstituted purified recombinant P450 2A6 based on the following evidence: 1) 8-MOP causes time, concentration, and NADPH-dependent loss of P450 2A6 activity that is not reversed by potassium ferricyanide or extensive dialysis, 2) loss of P450 2A6 activity is associated with a loss of spectrally observable P450, 3) addition of nucleophiles or reactive oxygen scavengers to the incubations does not prevent inactivation of P450 2A6, and 4) 8-MOP-dependent P450 2A6 inactivation is inhibited (concentration dependent) by the addition of a competitive inhibitor (pilocarpine). Inactivation is selective for P450 2A6 at low concentrations of 8-MOP (2.5 μM) after short incubation time periods (3 min) and was characterized by a KI of 0.8 and 1.9 μM in a reconstituted and microsomal system, respectively, and akinact of 1 min−1 and 2 min−1 in a reconstituted and microsomal system, respectively. A substrate depletion partition ratio of 21 was calculated for the inactivation of recombinant P450 2A6. Potency and selectivity suggest that 8-MOP could be a useful tool in vitro for evaluating P450 2A6 activity in various enzyme preparations.
Cytochrome P450 (P450)12A6, the major coumarin 7-hydroxylase (fig.1) present in human liver (1-6), is known to metabolize a variety of other compounds including quinoline (7), nicotine (8), cotinine (9), aflatoxin B1(10), 2,6-dichlorobenzonitrile (11), butadiene (12), and variousN-nitroso compounds present in cigarette smoke (13). Since the P450 2A6-mediated metabolism of several of these compounds leads to the formation of toxic metabolites, an individual’s adverse response to exposure to one of these compounds may depend on the level of P450 2A6 present in that individual. Although P450 2A6 is thought to comprise only a small percentage of the total liver P450 present in humans (2, 6), a marked variability has been observed in both the degree of coumarin 7-hydroxylase activity [in vivo (13) andin vitro (1, 2)] and in P450 2A6 protein levels (2, 5, 14). This suggests that the expression of P450 2A6 is controlled by environmental and/or genetic factors and that its variability has the potential of compromising quantitative assessment of its catalytic activity toward any particular compound. Coumarin 7-hydroxylation is believed to be an isoform-specific activity for P450 2A6 (1-6), suggesting that it should be an excellent marker activity for establishing the presence of this enzyme. However, available literature values indicate a 50-fold variation (0.2–10 μM) in theKm value for this activity in human liver microsomes (1, 15). Because uncertainty inKm compromises the usefulness of coumarin 7-hydroxylation as a marker P450 2A6 activity, a major goal of this investigation was to establish a firm value forKm for this substrate.

Chemical structures of coumarin and 8-MOP.
The arrow indicates the site of coumarin hydroxylation mediated by P450 2A6.
8-Methoxypsoralen (8-MOP, fig. 1) is a furanocoumarin used in combination with long wavelength ultraviolet light for the treatment of psoriasis, vitiligo, and cutaneous T-cell lymphoma (16-18). The usefulness of 8-MOP in treating these particular disease states resides in its ability to be photoactivated to a species that can covalently adduct to nucleic acids and lymphocytes, thereby inhibiting cell synthesis and proliferation. 8-MOP is also known to be a potent mechanism-based inactivator of several rat and mouse P450-mediated activities (19-23) and a time-dependent inhibitor of various human P450-mediated activities (24). Other studies indicated that 8-MOP is a potent inhibitor of human P450 2A6 (25-29) but did not explore the possible time dependency of its inhibition. Although both the in vivo and in vitro metabolic profiles of 8-MOP have been reported for several mammalian species, including man (30-33), the structure of the reactive species remains unknown, and the specific P450 isoform(s) responsible for its formation has not been identified. Thus, the second goal of this investigation was to test the ability of 8-MOP to specifically inhibit human P450 2A6 and to determine if the inhibition is time dependent and mechanism-based. The results provide strong evidence that 8-MOP, at concentrations observed in vivo (34, 35), is a selective and potent mechanism-based inactivator of human P450 2A6.
Materials and Methods
Materials.
7-Hydroxycoumarin, 8-MOP, L-d-phosphatidylcholine, dilauryl (DLPC), catalase, glutathione, deferoxamine mesylate,N-acetylcysteine, superoxide dismutase, NADP+, chlorzoxazone, dextromethorphan, and NADPH were purchased from Sigma (St. Louis, MO). Potassium ferricyanide, sodium cyanide, methoxylamine hydrochloride, and semicarbazide hydrochloride were purchased from Aldrich (Milwaukee, WI). Glucose 6-phosphate and glucose 6-phosphate dehydrogenase (yeast, grade II) were purchased from Boehringer Mannheim (Indianapolis, IN). Coumarin was from Merck (Rahway, NJ), and pilocarpine hydrochloride was from Mallinckrodt (St. Louis, MO). 6-Hydroxychlorzoxazone, (S)-mephenytoin, (R)- and (S)-warfarin, and the hydroxylated mephenytoin and warfarin deuterated internal standards were from laboratory stocks. Dextrorphan was kindly provided by Dr. U. A. Meyer (Biocenter, University of Basel, Switzerland). Slide-A-Lyzer dialysis kits were from Pierce (Rockford, IL). HPLC solvents were of the highest grade commercially available and were used as received. Human liver samples were from the NIH-supported human liver bank at the University of Washington, and microsomes from these liver samples were prepared as previously described (36). A full-length cDNA of P450 2A6 was kindly provided by Dr. Frank Gonzalez (National Institutes of Health, Bethesda, MD). The baculovirus-mediated expression and characterization of P450 2A6 used in this study have been reported previously (10). The purification of P450 2A6 from the crude insect cell paste was accomplished according to published procedures for the purification of P450 2C9 (37), with minor modifications. Escherichia coli bacterial stocks containing the plasmid OR263 for expression of rat NADPH-cytochrome P450 oxidoreductase were kindly provided by Dr. Charles B. Kaspar (University of Wisconsin, Madison). Recombinant rat NADPH cytochrome P450 oxidoreductase was purified according to published procedures (38) with minor modifications. A full-length cDNA of microsomal human cytochrome b5 was kindly provided by Dr. R. Kato (Keio University, Tokyo). Human cytochromeb5 was expressed and purified from bacterial cultures according to previously published procedures (39). Experimental data are presented as the average of duplicate determinations that did not vary by more than 10%.
Km andVmax Determinations.
Potassium phosphate buffer (25 mM, pH 7.4) was used for the experiments involving microsomal systems, and a buffered coumarin stock solution (5 mM) was prepared before each experiment. Initially, theKm and Vmaxconstants for this metabolic event were determined in 12 different human liver microsomal preparations. Coumarin (0.1–25 μM) was incubated with various amounts of microsomal P450 (0.5–5 pmol), depending on the amount of P450 2A6 activity present in each liver. Reaction was initiated after a 3-min preincubation period at 37°C by addition of an NADPH-generating system (NADPH-GS) (final incubation volume, 1 ml). The NADPH-GS consisted of (final concentrations) 10 mM glucose 6-phosphate, 0.5 mM NADP+, and 1 unit of yeast glucose 6-phosphate dehydrogenase ml−1. After 7.5 min, the reaction was quenched with 50 μl of 6 N HClO4 and set on ice. After centrifugation for 10 min at 2500 rpm (HNS II Centrifuge, International Equipment Co.), 100 μl of the supernatant was injected onto an HPLC (Hewlett Packard Series 1050) equipped with a reversed phase C8 column (Econosphere 5 μm, 150 mm × 4.6 mm). Fluorescence detection (Hewlett Packard 1046A programmable fluorescence detector) coupled to the HPLC was used to measure 7-hydroxycoumarin with an excitation wavelength of 323 nm and an emission wavelength of 463 nm. Isocratic elution with 10 mM H3PO4 (pH 2.5):ACN (72:28) yielded a retention time of 4.0 min for 7-hydroxycoumarin. A standard curve composed of five concentrations of 7-hydroxycoumarin that bracketed both the minimum and maximum amounts of metabolite in each particular experiment indicated the method had a detection limit of 1 pmol. Eadie-Hofstee plots (V vs. V/S) were used to ensure single enzyme kinetics and to estimate Km and Vmax parameters. TheKm and Vmaxparameters reported were determined by nonlinear regression analysis of the rate data using the statistical package, SYSTAT 5.0 (40), and the Michaelis-Menten equation.
Microsomal P450 2A6 Inactivation Assays.
Various concentrations (0–2.5 μM) of 8-MOP in 0.5% MeOH (v/v) were preincubated with microsomes (50 pmol of P450) prepared from HL109 for 3 min at 30°C in potassium phosphate buffer (25 mM, pH 7.4). HL109 was chosen because it possessed moderate levels of P450 1A2, 2A6, 2C9, and 3A4 activity. Reaction was initiated by the addition of a freshly prepared NADPH-GS (final incubation volume, 1 ml). At selected time points (0–3 min), 50 μl from each incubate was transferred to a vial containing coumarin (25 μM) and the NADPH-GS (final incubation volume, 1 ml) to assay for residual activity. After 7.5 min, the activity assays were quenched and then analyzed for 7-hydroxycoumarin formation by HPLC as described above. The slopes obtained from the natural log per cent remaining activity vs. time plots were replotted as 1/slope (i.e. 1/rate) vs. 1/(8-MOP concentration). Nonlinear regression analysis of the data was used to determine KI andkinact values using the statistical package, SYSTAT 5.0 (40), and the mechanism-based inactivation equation (41). Experiments involving nucleophilic trapping agents and free iron and reactive oxygen species scavengers were carried out in exactly the same manner. Substrate protection experiments were also carried out in the same manner as described for an inactivation assay, except various concentrations (0–10 μM) of pilocarpine were included. In addition, experiments comparing the effects of 8-MOP on P450 2A6 and other P450s were conducted as described except at 37°C. Nine other human liver microsomal samples were used to assess P450 2A6 inactivation by 8-MOP in the same manner as above, except that these experiments were performed at 37°C, one concentration of 8-MOP (2.5 μM), and two time points (0 and 5 min).
Purified Recombinant P450 2A6 Optimization and Inactivation Assays.
Incubations contained P450 2A6 (1 pmol), rat P450 reductase (0–5 pmol), human cytochrome b5 (0–3 pmol), potassium phosphate buffer (25 or 75 mM, pH 7.4), DLPC (12.5–50 μg), coumarin (25 μM), and the NADPH-GS (final incubation volume, 1 ml). P450 2A6, rat P450 reductase, DLPC, and human cytochromeb5 were added, in that order, at room temperature in 5-min intervals before the addition of substrate and buffer. After a 3-min preincubation at 30°C, reaction was initiated by the addition of the NADPH-GS (final incubation volume, 1 ml). Reactions were quenched after 7.5 min and analyzed for 7-hydroxycoumarin formation by HPLC as described above. Inactivation assays contained various concentrations (0–2.5 μM) of 8-MOP, P450 2A6 (10 pmol), rat P450 reductase (20 pmol), human cytochromeb5 (10 pmol), DLPC (25 μg), potassium phosphate buffer (25 mM, pH 7.4), and the NADPH-GS (final incubation volume, 600 μl). At selected time points (0–3 min), 50 μl was transferred to a vial containing coumarin (25 μM) and the NADPH-GS to assay for residual activity. The activity assay was quenched after 7.5 min and analyzed by HPLC as described above.
P450 1A2, 3A4, and 2C9 Inactivation Assays.
The effect of 8-MOP on the warfarin metabolizing enzymes P450 1A2, 3A4, and 2C9 was also investigated. The 7-hydroxylation of (S)-warfarin, 6-hydroxylation of (R)-warfarin, and 10-hydroxylation of (R)-warfarin were used as selective markers for P450 2C9, 1A2, and 3A4 activities, respectively (42). Microsomal P450 (HL109, 1 nmol) was preincubated for 3 min at 37°C in the presence and absence of 8-MOP (10 μM). The reaction was initiated by the addition of the NADPH-GS (final incubation volume, 500 μl). At 0, 10, and 20 min, 50 μl was transferred to a vial containing the NADPH-GS and either (S)-warfarin (50 μM) to assess for residual P450 2C9 activity or (R)-warfarin (1.5 mM) to assess for residual P450 1A2 and 3A4 activities (final incubation volume, 1 ml). The activity assays were allowed to proceed for 25 min followed by quenching, addition of deuterium-labeled internal standards, pH adjustment, extraction, and GC/MS analysis as previously described (36).
P450 2C19 Inactivation Assays.
The effect of 8-MOP on P450 2C19 activity was assessed using the 4′-hydroxylation of (S)-mephenytoin as the marker activity. Microsomal P450 (HL147, 50 pmol) was preincubated for 3 min at 37°C in the presence and absence of 8-MOP (10 μM). The reaction was initiated with the NADPH-GS (final incubation volume, 500 μl). At 0, 10, and 20 min, 50 μl was transferred to a vial containing the NADPH-GS and (S)-mephenytoin (200 μM) to assess P450 2C19 activity. The activity assays were allowed to proceed for 15 min followed by quenching, addition of deuterium-labeled internal standards, pH adjustment, extraction, and GC/MS analysis as previously described (43).
P450 2D6 Inactivation Assays.
An HPLC-fluorescence assay was used to investigate the potential effect of 8-MOP on the O-demethylation of dextromethorphan, a marker for P450 2D6 activity (44). Microsomal P450 (HL119, 1 nmol) was preincubated for 3 min at 37°C in the presence and absence of 8-MOP (10 μM). This human liver was used because it possessed a greater amount of P450 2D6 and 2E1 activity than HL109. Reaction was initiated by the NADPH-GS (final incubation volume, 1 ml). At 0, 10, and 20 min after this addition, 150 μl from each incubate was transferred to a vial containing dextromethorphan (20 μM) and the NADPH-GS (final incubation volume, 500 μl) to assay for residual activity. Reaction was quenched after 30 min with 40 μl of a 70% solution of H3PO4 and the protein precipitated by centrifugation (2500 rpm, 10 min). The supernatant, 200 μl, was injected onto an HPLC equipped with the reversed phase C8 HPLC column described above. Isocratic elution with 20 mM sodium perchlorate (pH 2.5):ACN (62:38) coupled with fluorescence detection using an excitation and emission wavelength of 235 and 312 nm, respectively, was used for analysis. Under these conditions, the retention time for dextrorphan was 5.8 min, whereas that of the parent compound, dextromethorphan, was 12 min.
P450 2E1 Inactivation Assays.
An HPLC assay was used to examine the effect of 8-MOP on the 6-hydroxylation of chlorzoxazone, a marker activity for P450 2E1 (45). The inactivation assay conditions were the same as those described for P450 2D6. At 0, 10, and 20 min, 50 μl from each incubate was transferred to a vial containing chlorzoxazone (500 μM) and the NADPH-GS (final incubation volume, 1 ml). Reaction was quenched after 10 min with 50 μl of a 43% H3PO4 (w/v) solution and assayed by HPLC coupled with UV detection as previously described (45).
Dialysis Experiments.
Microsomal P450 (HL109, 50 pmol) and 8-MOP (2.5 μM) with and without the NADPH-GS (final incubation volume, 1 ml) were incubated at 30°C for 3 min. The mixture was then dialyzed against 1 liter of 25 mM potassium phosphate buffer (pH 7.4) overnight at 4°C using a 0.5–3-ml Slide-A-Lyzer with a molecular mass cutoff of 10 kDa. No significant volume changes were observed, and remaining P450 2A6 activities were reported relative to the control ((−) NADPH-GS) incubation.
Effect of 8-MOP on Cytochrome P450 2A6 Spectral Content.
Because of the small amount of P450 2A6 that is usually present in human liver microsomes, only a minor effect on spectrally detectable microsomal P450 might be expected as a result of P450 2A6 inactivation. Thus, to ensure the observation of a decrease in spectrally detectable P450 accompanying decreased enzyme activity, the effect of 8-MOP on spectral P450 was determined using purified recombinant P450 2A6. P450 2A6 (1 nmol), rat P450 reductase (2 nmol), DLPC (25 μg), and 8-MOP (100 nmol) in 25 mM potassium phosphate buffer (pH 7.4) were incubated at 30°C in the presence and absence of NADPH (1 mM). At 0, 2, 10, and 20 min, aliquots were removed and diluted 1:7 into a chilled potassium phosphate buffer (100 mM, pH 7.4) solution containing EDTA (0.1 mM), dithiothreitol (0.1 mM), sodium cholate (1% w/v), and glycerol (20% v/v) and set on ice. These mixtures were then reduced with a few grains of sodium dithionite and assayed for cytochrome P450 content using a Varian Cary 3E double-beam UV-VIS spectrophotometer. The reduced solution was split into matched reference and sample cuvettes, and CO was gently bubbled into the sample cuvette for approximately 1 min. A difference spectrum was recorded, and P450 content was calculated based on an extinction coefficient of 91 mM−1cm−1 after full development of the spectrum (A448–A490).
Partition Ratio Experiments.
The inactivation assay consisted of purified expressed P450 2A6 (50 pmol), rat P450 reductase (100 pmol), human cytochromeb5 (50 pmol), and 8-MOP (10 μM) in potassium phosphate buffer (25 mM, pH 7.4). Reaction was initiated by the addition of buffer (control) or NADPH (1 mM) in buffer and terminated by the addition of 50 μl of 6 N HClO4 after a 60-min incubation at 30°C to ensure complete P450 inactivation. After centrifugation at 2500 rpm for 10 min, an aliquot was injected onto an HPLC equipped with the reversed phase C8 HPLC column described above and monitored by UV at 320 nm. Separation of the parent compound from its metabolites was achieved using 10 mM H3PO4 (pH 2.5):ACN (95:5) with a gradient elution of 5–45% ACN over 30 min. Under these conditions, the parent compound eluted at 23 min. A partition ratio for the 8-MOP-mediated inactivation of P450 2A6 was calculated as the ratio of the amount of 8-MOP consumed (relative to control) to the amount of spectrally detectable P450 2A6 present in the incubation. There was no detectable degradation of 8-MOP in these experiments in the absence of enzyme; however, MeOH solutions of 8-MOP have been found to degrade if kept at room temperature for a few days. Therefore, all of these experiments were performed using freshly prepared MeOH solutions of 8-MOP.
Results
Determination of Coumarin-P450 2A6 Km and Vmax Values.
To avoid substrate depletion, preliminary experiments were carried out to estimate the amount of active P450 2A6 (measured as coumarin 7-hydroxylase activity) present in each human microsomal sample. Eadie-Hofstee plots were used to estimate the values of the kinetic constants for 12 different human liver microsomal preparations, and in each case, single enzyme kinetics was observed. TheKm and Vmaxvalues for P450 2A6 activity were then determined for the 12 different human liver microsomal preparations using nonlinear regression analysis of the rate data (table 1). A (± SD,N = 12) Km of 0.40 (±0.13) μM and Vmax of 6.34 nmol/nmol P450/min (36-fold variation) was found for coumarin 7-hydroxylase activity in these microsomes.
Kinetic parameters for the 7-hydroxylation of coumarin in a panel of 12 human liver microsomal preparations1-a
Optimization of Purified Recombinant P450 2A6 Activity.
The coumarin 7-hydroxylation activity of the purified recombinant enzyme was optimized by varying the molar ratio of P450 2A6, rat P450 reductase, and human cytochrome b5 present in the incubation. The ionic strength of the incubation and amount of phospholipid included were also varied. Results of these experiments (table 2) indicated that a molar ratio of P450 2A6:rat P450 reductase of 1:2 in 25 mM potassium phosphate buffer (pH 7.4) containing 25 μg of DLPC was suitable for reconstituting P450 2A6 activity. Optimal activity was achieved by the addition of human cytochrome b5 in a 1:1 molar ratio with P450 2A6. It should be noted that the reconstituted system yielded a single coumarin metabolite that had an identical HPLC retention time to the 7-hydroxycoumarin standard.
Optimization of purified cDNA-expressed P450 2A6 coumarin 7-hydroxylase activity
Time-Dependent Inhibition of Human P450 2A6 by 8-MOP.
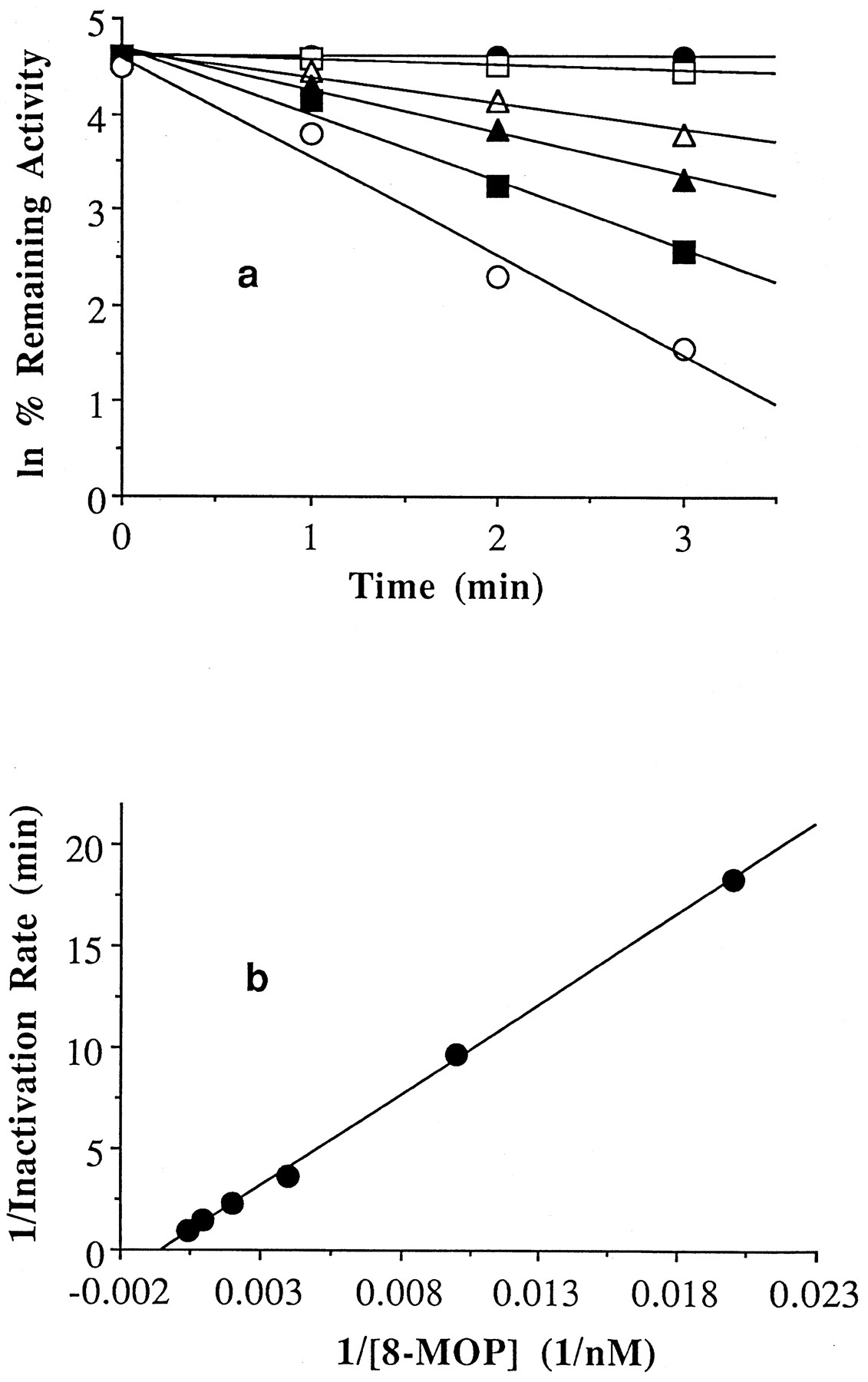
8-MOP (0–2.5 μM) was incubated with human liver microsomes (HL109) or purified P450 2A6 and the NADPH-GS to determine its ability to inhibit P450 2A6 by mechanism-based inactivation. At various times points after starting an incubation, a small aliquot from each was transferred to a vial containing coumarin and the NADPH-GS to determine the amount of P450 2A6 activity remaining. To properly determine enzyme inactivation and eliminate any contribution from competitive inhibition to observed inactivation, all of the suspected inactivator must be displaced from the enzyme active site. To achieve this condition, only 5–12% of the inactivation assay was transferred to the activity assay (to dilute the inhibitor), whereas the substrate, coumarin, was used at a relatively high concentration (80 timesKm ). Minimal carryover was observed (i.e. little contribution from competitive inhibition apparent at time zero) using the assay conditions described (fig.2a). Over the time course of the incubations at 30°C, approximately 5% of the coumarin 7-hydroxylase activity present in microsomes was lost in the absence of 8-MOP, whereas, over the same time span, 20% of the activity was lost in the reconstituted system. Moreover, the noninactivator-mediated loss in activity observed in the reconstituted system was found to be significantly greater at 37°C. The reason for the much greater loss of P450 2A6 activity with the reconstituted system is currently under investigation. Inhibition of P450 2A6 activity by 8-MOP was found to be time and concentration dependent in both the microsomal (fig.2a) and reconstituted systems (data not shown). The reversible binding constant (KI ) for 8-MOP with P450 2A6 was determined from nonlinear regression analysis of the data to be 1.9 and 0.8 μM for the microsomal (fig. 2b) and reconstituted systems, respectively. The inactivation rate constant was determined to be 2.1 min−1 and 1.0 min−1 for the microsomal (fig. 2b) and reconstituted systems, respectively.
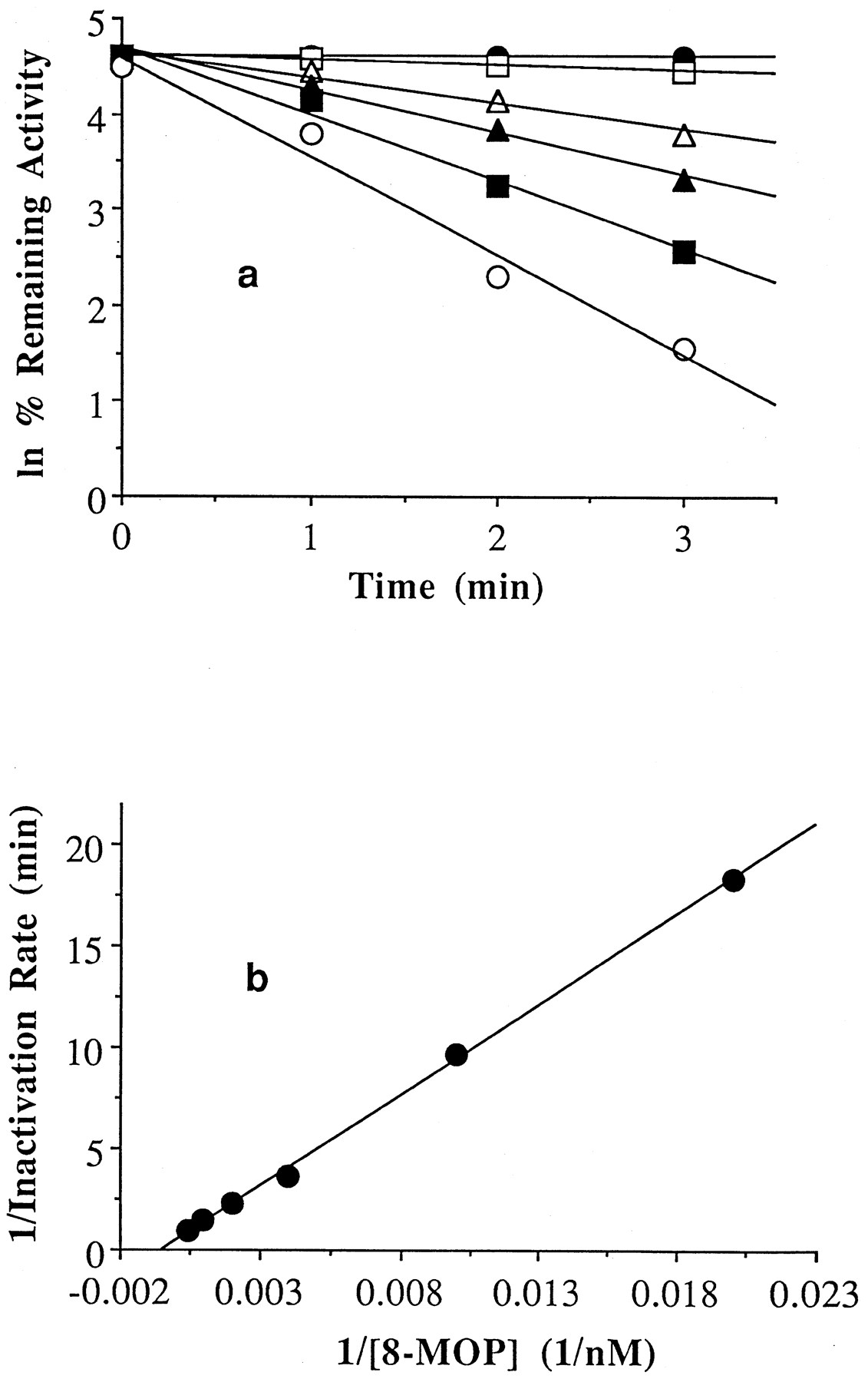
8-MOP mediated inactivation of P450 2A6 activity in human liver microsomes (HL109) in the presence of an NADPH-generating system (a) and double-reciprocal plot of the relationship between inactivation rate and 8-MOP concentration (b).
The concentrations of 8-MOP present in the inactivation assay were 0 μM (•), 0.05 μM (□), 0.25 μM (▵), 0.5 μM (▴), 1 μM (▪), and 2.5 μM (○). The reversible binding constant (KI) and the rate constant for inactivation (kinact) associated with microsomal P450 2A6 and 8-MOP were calculated (41) using nonlinear regression to be 1.8 μM and 2 min−1, respectively. The rate of turnover for the uninhibited reaction in microsomes (HL109) was 5.21 nmol/nmol P450/min.
NADPH Dependence, Effect of Trapping Agents, and Effect an Alternate Inhibitor on Inactivation.
The dependence of P450 2A6 inactivation on NADPH was tested, and enzymatic processing of 8-MOP was found to be a necessary prerequisite (table 3). Various trapping agents were tested for protective effects against inactivation to determine if the inactivating event is confined to the active site of the enzyme. In these studies, a concentration of 8-MOP (2.5 μM) and an inactivation time (3 min) was chosen so that approximately 10% of the enzyme activity remained. The nucleophilic trapping agents glutathione,N-acetylcysteine, and sodium cyanide failed to protect P450 2A6 from inactivation. The free aldehyde trapping agents methoxylamine and semicarbazide and the free iron scavenger deferoxamine also failed to protect P450 2A6 from inactivation. Similarly, superoxide dismutase and catalase, included to assess the contribution of reactive oxygen species to the inactivation of the enzyme, were equally ineffective as protectants. Addition of potassium ferricyanide to the inactivation mixture did not reverse P450 2A6 inactivation. Finally, the inactivated enzyme mixture was dialyzed extensively to remove any free or noncovalently bound 8-MOP from the mixture. Pilocarpine, which has been reported (46) to be a P450 2A6 inhibitor (Ki ≈ 1 μM), was used to protect the enzyme from inactivation by 8-MOP (table 3). As expected, the protective effect of pilocarpine on P450 2A6 inactivation was concentration dependent (fig. 3).
Effect of NADPH and various trapping and protecting agents on 8-MOP mediated microsomal P450 2A6 inactivation

Time-dependent loss of microsomal P450 2A6 activity mediated by 2.5 μM 8-MOP in the presence of different concentrations of the P450 2A6 inhibitor pilocarpine.
(•) represents values derived from experiments performed in the absence of 8-MOP. The concentrations of pilocarpine present in the inactivation assay with 8-MOP were 10 μM (□), 1 μM (▪), and 0 μM (○).
Effect of 8-MOP on Cytochrome P450 Spectral Content.
These studies were carried out with cDNA-expressed P450 2A6 and 8-MOP in a molar ratio of 1:100 for various time periods up to 20 min at 30°C. Human cytochrome b5 was omitted from these studies to ensure that any spectral differences observed were attributable only to changes in P450 2A6. At each time point, an aliquot of the incubation mixture was removed and diluted in a chilled buffer solution containing cholate (1% w/v) and glycerol (20% v/v). After 20 min, only 31% of the spectrally detectable P450 remained (table 4). A similar loss in spectral P450 content was not observed in the absence of either NADPH or 8-MOP after 20 min (results not shown).
Loss of spectrally detectable purified P450 2A64-a
Partition Ratio Experiments.
Incubations of 8-MOP with cDNA-expressed P450 2A6 (50 pmol) and 8-MOP (10 nmol) were carried out in the absence and presence of NADPH (1 mM) for an extended period of time to ensure complete inactivation of P450. The loss in P450 was coincident with a loss of 1050 pmol (11%) of 8-MOP. These values yielded a calculated P450 substrate depletion-based partition ratio of 21.
Effects of 8-MOP on P450 1A2, 2C9, 2C19, 2D6, 2E1, and 3A4 Activities.
The effect of 8-MOP (10 μM) on the activity of P450 1A2, 2C9, 2C19, 2D6, 2E1, and 3A4 was examined at 0, 10, and 20 min after initiation of the reaction with the NADPH-GS (fig. 5). Possible 8-MOP mediated inactivation of these P450s was tested using known selective P450 isoform marker activities. Preincubation with 8-MOP for 10 and 20 min resulted in no significant time-dependent loss of P450 1A2, 2D6, 2C9, or 3A4 activities, although a small amount of competitive inhibition was observed for these isoforms. In contrast, under similar conditions loss of P450 2C19 and P450 2E1 activity was time dependent. The loss of 30% of P450 2C19 and 50% of P450 2E1 activity required 10 and 20 min, respectively, at 10 μM 8-MOP, whereas only 3 min was required to inactivate greater than 90% of the P450 2A6 activity at 2.5 μM 8-MOP. The inactivation of P450 2C19 and 2E1 could be minimized by decreasing the concentration of 8-MOP to 2.5 μM (fig. 5, for P450 2E1 data) or by decreasing the time of exposure to the inactivator.

Effect of 8-MOP on P450 1A2, 2A6, 2C9, 2D6, 2E1, and 3A4 activities in human liver microsomes.
Human liver microsomes were preincubated with 0 (control,diagonal hatched bars), 2.5 (*), or 10 μM 8-MOP for 0 (shaded bars), 10 (solid bars), and 20 min (vertical hatched bars) in the presence of an NADPH-GS at 37°C. In the case of P450 2A6, the preincubation times were 0 (shaded bar) and 3 min (solid bar). The following human liver samples were used for the various activity determinations: coumarin 7-hydroxylation (P450 2A6), HL109; chlorzoxazone 6-hydroxylation (P450 2E1), HL119; (S)-mephenytoin 4′-hydroxylation, HL141; (R)-warfarin 10-hydroxylation (P450 3A4), HL109; (R)-warfarin 6-hydroxylation (P450 1A2), HL109; (S)-warfarin 7-hydroxylation (P450 2C9), HL109; and dextromethorphan O-demethylation (P450 2D6), HL119.
Discussion
A major goal of this investigation was to firmly establish aKm for the P450 2A6 catalyzed 7-hydroxylation of coumarin. In the course of these studies, we discovered that ratios of coumarin to microsomal P450 of less than 20 resulted in significant substrate depletion. Substrate depletion is a factor that must be addressed if significant errors in the determinations of substrate and inhibitor kinetic constants are to be avoided. To this end, relatively small amounts of human microsomal P450 (0.5–5 pmol) were used together with a relatively short, but manageable, reaction time (7.5 min) to generate sufficient 7-hydroxycoumarin for reliable quantitation. An assay limit (signal:noise, 10:1) of less than 10 pmol was necessary to ensure avoidance of error in product formation (less than 10% substrate turnover) at the lowest substrate (coumarin) concentration used (47). To achieve the required sensitivity, an HPLC-fluorescence assay was developed that had a detection limit of 1 pmol and the added advantages that 1) extraction was not necessary and 2) more than ten samples per hour could be processed.
The mean Km andVmax for P450 2A6 catalyzed coumarin 7-hydroxylation were determined in a panel of 12 human livers using nonlinear regression analysis and the Michaelis-Menten equation (table1). The reaction is so highly favored that it was possible (and necessary to avoid substrate depletion) to use very small amounts of microsomal P450 (0.5–5 pmol of enzyme produced a product signal 10 times greater than noise) to determine these kinetic constants. The large variation in Vmax is consistent with the large interliver variation in coumarin 7-hydroxylation activity reported previously (1, 2, 48), and the Km value agrees with that determined using cDNA-expressed P450 2A6 (10) and also with that reported in an earlier study (1). However, significantly higher Km values (1.2–10 μM) have been reported in other studies (2, 15, 49), which might be accounted for by errors arising from substrate depletion.
IC50 and Ki values associated with the 8-MOP inhibition of P450 2A6 also vary significantly. 8-MOP seems to be a potent inhibitor of P450 2A6, with an IC50 of 0.3–5.4 μM, in human liver microsomes (26, 28, 29), or an IC50 greater than 50 μM, when the enzyme has been expressed from a baculovirus system (not purified) (50), or a Ki of 1.5 μM (27), in human liver microsomes. Thus, a second goal of this study was to establish the nature of 8-MOP inhibition of P450 2A6. If it were found to be a selective mechanism-based inactivator of human P450 2A6, as has been reported for the rodent (19-23), it would provide an explanation for the seeming variability in inhibitor potency.
The time and concentration dependence of P450 2A6 catalyzed formation of 7-hydroxycoumarin on 8-MOP were readily apparent from a plot of the rate data (fig. 2a). In both the microsomal and reconstituted systems, the data could be fitted to a straight line in a double-reciprocal plot of inactivation rates vs. 8-MOP concentration (fig. 2b). The concentration necessary to significantly inactivate P450 2A6 was within the peak plasma concentration range of 0.7–5.3 μM observed after a normal oral dose of 8-MOP (34, 35). The discrepancy in KI values between purified and microsomal systems (0.8 and 1.9 μM, respectively) is not appreciable. However, the discrepancy inkinact between systems (1.0 min−1 and 2.1 min−1, respectively) is probably the result of the difficulty in generating an efficient P450 2A6 reconstituted system, as has been previously reported for this enzyme (2). This could also explain the apparent fragility of the reconstituted system as noted underResults.
8-MOP mediated P450 2A6 inactivation was dependent on NADPH and could not be prevented by the addition of various nucleophiles, reactive oxygen species scavengers, or a free iron chelator (table 3). Inactivation could not be reversed by addition of potassium ferricyanide or by extensive dialysis. However, the addition of pilocarpine, the P450 2A6 competitive inhibitor, was able to prevent inactivation (table 3, fig. 3). These results suggest that 1) inactivation requires catalytic processing by the P450, 2) the inactivating event is confined to the active site of the enzyme, 3) the inactivator-enzyme complex is not readily reversible, and 4) inactivation is not a consequence of reaction with reactive oxygen species generated by uncoupling of P450.
With a mechanism-based inactivator, a loss of catalytic activity should be accompanied by a loss in spectral P450 2A6 content. The results clearly indicate that the loss in catalytic activity is accompanied by a corresponding loss in spectral P450 2A6 content (table 4 and fig.4). The decrease in P450 2A6 spectral content, as measured by the Soret maximum at 448 nm, was associated with formation of a chromophoric species with a maximum absorbance at 417 nm. The structural implications of the chromophoric change are unknown, but possibilities are that the 417 nm chromophore may represent a species resulting from denaturation of the apoprotein, modification of the heme, or covalent binding of 8-MOP to the active site of P450 2A6 in a manner that prevents CO binding.

Destruction of purified cDNA-expressed spectral P450 2A6 content following incubation with 8-MOP and NADPH in a reconstituted system.
P450 2A6 was incubated with 10 μM 8-MOP for various time periods (0–20 min) at 30°C, and the reaction was quenched by diluting an aliquot 1:7 with a chilled buffer solution containing 1% cholate (w/v) and 20% glycerol (v/v). Absorbance maxima were observed at 417 and 448 nm.
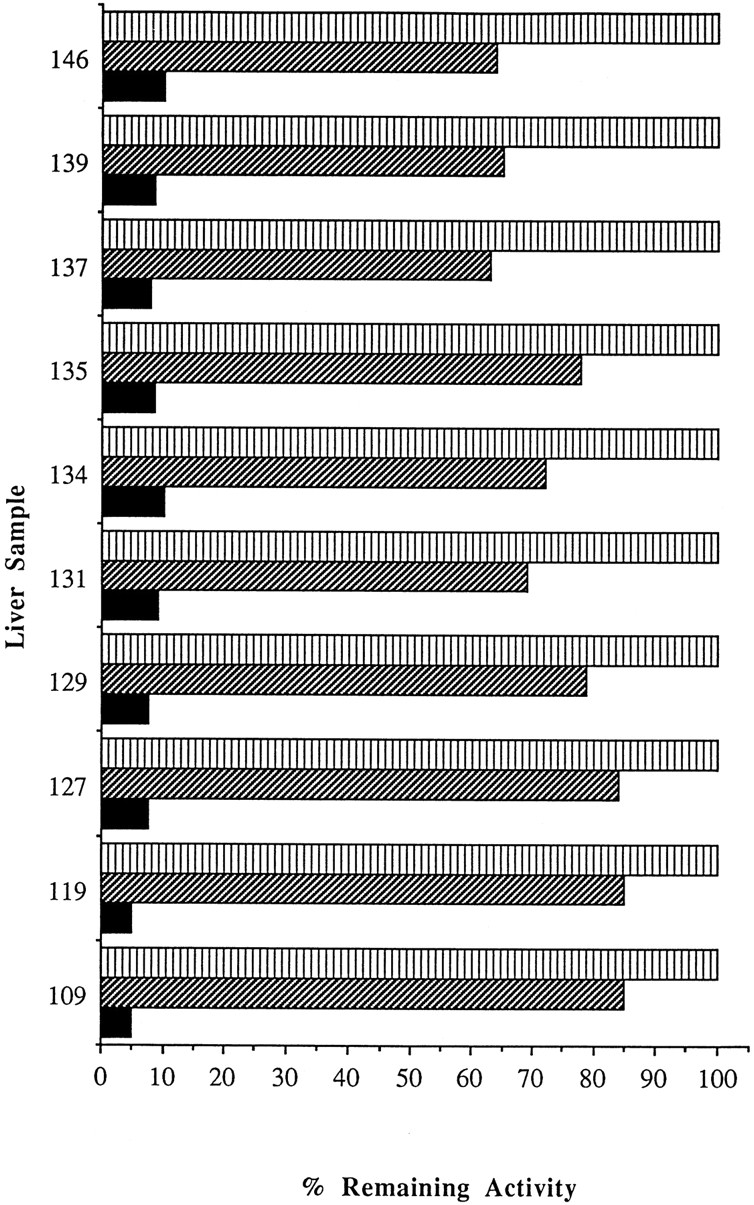
The ability of 8-MOP to inhibit P450 1A2, 2C9, 2C19, 2D6, 2E1, and 3A4 (fig. 5) by mechanism-based inactivation was investigated using isoform-selective substrates. Inhibition of P450 2C19 and 2E1 was time dependent but weak, whereas inhibition of P450 1A2, 2C9, 2D6, and 3A4 was competitive and relatively insignificant (2–24%) when compared with the inactivation of P450 2A6 activity. 8-MOP was found to inactivate P450 2A6 activity in nine other human liver microsomal preparations by greater than 90% at 2.5 μM and an incubation time of only 5 min (fig. 6). These results suggest that 8-MOP is a highly selective enzyme inactivator that could provide a precise and effective means of removing P450 2A6-dependent activity from human liver microsomal preparations.

Inactivation of P450 2A6 by 8-MOP in 10 different human liver microsomal preparations.
P450 2A6 activity was tested after exposure to 0 μM 8-MOP for 5 min (vertical hatched bar) and 2.5 μM 8-MOP for 0 (diagonal hatched bar) and 5 min (solid bar) at 37°C. The range of P450 2A6 activities in this panel of 10 human liver microsomes was approximately 21-fold.
The partition ratio of a mechanism-based inactivator defines the stoichiometry of inactivation relative to turnover and thus is a useful measure of inhibitory efficiency. Relative to furafylline (partition ratio, 3.8–5.6) (51) and midazolam (partition ratio, ∼300) (52), 8-MOP (partition ratio, 21) can be classified as a moderately efficient inactivator. In contrast, an NIH 3T3 cell line expressing P450 2A6 has been reported to be unable to metabolize 8-MOP (29). However, the apparent sensitivity of P450 2A6 catalytic efficiency to the relative amounts of supporting enzyme activity (P450 reductase and cytochromeb5) as demonstrated in the present study suggests that expressed P450 2A6-dependent 8-MOP metabolism in the NIH 3T3 cell line might be compromised if the cells are deficient in supporting enzyme.
In summary, the kinetic parameters for the P450 2A6 mediated coumarin 7-hydroxylation have been determined in a panel of 12 human liver microsomal preparations under conditions designed to exclude the possibility of substrate depletion. The high variability inVmax (36-fold) is consistent with the known polymorphic and inducible expression of the enzyme. The criteria of time, concentration, and NADPH dependence that define mechanism-based inactivation were met by the 8-MOP inactivation of P450 2A6. Inactivation was irreversible, unaffected by a variety of trapping agents, associated with a loss of spectroscopically detectable cytochrome P450, and occurred at concentrations and incubation times at which other human liver P450 activities were relatively unaffected. Thus, all the evidence indicates that 8-MOP is a potent mechanism-based inactivator and suggests that in vitro it could provide an effective method for assessing P450 2A6 content in microsomes. However,in vivo, because it is capable of inactivating human P450 2A6 at physiologically relevant concentrations, it carries the potential of causing a serious drug-drug interaction with any drug, compound, or toxin whose clearance is largely dependent on P450 2A6. Whereas the present studies do not address the mechanism of 8-MOP-dependent P450 2A6 inactivation, precedence (53-55) suggests that a likely possibility is a reactive intermediate generated by initial oxidation of the furan ring. Indirect evidence that supports this possibility is the fact that coumarin is not a mechanism-based inactivator of P450 2A6. Because the coumarin part of the structure of 8-MOP can be eliminated as a site of activation, the remaining possibilities are limited and are currently being explored.
Footnotes
-
Send reprint requests to: Dr. William F. Trager, Department of Medicinal Chemistry, Box 357610, University of Washington, Seattle, WA 98195. E-mail: trager{at}u.washington.edu.
-
This research was supported by National Institutes of Health Grant GM32165 (to W.F.T.).
- Abbreviations used are::
- P450
- cytochrome P450
- HPLC
- high pressure liquid chromatography
- ACN
- acetonitrile
- DLPC
- L-d-phosphatidylcholine, dilauryl
- 8-MOP
- 8-methoxypsoralen
- NADPH-GS
- NADPH-generating system
- Received June 25, 1997.
- Accepted September 4, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}