


Abstract
In primary human and porcine hepatocyte cultures, we investigated the relationship between metabolism and cytotoxicity of troglitazone. Treatment of human hepatocytes for 2 h with 10, 20, 25, 35, and 50 μM troglitazone in protein-free medium resulted in concentration-dependent decreases in total protein synthesis. Decreases at 10 and 20 μM were reversible by 24 h, however protein synthesis did not recover at concentrations ≥25 μM. Troglitazone at 50 μM caused cellular death. In porcine hepatocytes, 100 μM troglitazone was lethal, whereas at 50 μM, protein synthesis completely recovered by 24 h. Recovery in protein synthesis was associated with metabolism of parent drug, whereas toxicity correlated (r2 = 0.82) with accumulation of unmetabolized troglitazone. By 1 h, in human hepatocytes, troglitazone was metabolized to similar amounts of sulfate and quinone metabolites with little glucuronide detected. In contrast, porcine hepatocytes metabolized troglitazone to the similar amounts of glucuronide and the quinone metabolites with little sulfate detected. Exposure of human hepatocytes to a combination of 10 μM troglitazone and 10 μM 2,4-dichloro-4-nitrophenol resulted in a 70% decrease in protein synthesis, associated with 90% inhibition in the formation of troglitazone sulfate, a 4-fold increase in unmetabolized troglitazone, and no effect on formation of the quinone metabolite. Treatment with a combination of acetaminophen or phenobarbital with 20 μM troglitazone resulted in sustained decrease in protein synthesis associated with inhibition of sulfation and accumulation of troglitazone. These results suggest that inhibition of troglitazone sulfation may result in increased hepatotoxicity due to exposure to parent drug, or increased metabolism by alternate pathways.
Troglitazone (TRO)1 (Rezulin; Warner-Lambert Company) was the first member of the thiazolidinedione chemical series developed to treat type II diabetes. It has a novel mechanism of action, lowering blood glucose levels through increased glucose uptake by skeletal muscles, decreased hepatic glucose production, and an increased sensitivity to insulin (Fujiwara et al., 1988, 1995; Ciaraldi et al., 1990). However, a rare hepatic injury has been associated with TRO therapy. During clinical trials, 1.9% of patients experienced increases in alanine aminotransferase levels greater than 3 times the upper limit of normal. Fulminant hepatic failure in single patients was reported by several authors (Gitlin et al., 1998; Neuschwander-Tetri et al., 1998; Shibuya et al., 1998). The mechanism responsible for TRO-induced liver failure, however, is not currently known.
TRO is metabolized to sulfate, quinone, and glucuronide derivatives. TRO sulfate and TRO quinone account for about 70 and 10% of the metabolites detected in human plasma, respectively (Loi et al., 1997,1999a). Only 3% of orally administered TRO is recovered in urine (Rezulin package insert, 1998; Loi et al., 1999b). About 85% of the drug administered to human is recovered in feces, suggesting that the major route of excretion is bile (Rezulin package insert, 1998; Loi et al., 1999b). TRO sulfate is the major metabolite detected in the bile of dogs and rats (Kawai et al., 1997). Thus conjugation of TRO with sulfate represents the main metabolic pathway responsible for TRO elimination. Pharmacokinetic studies in normal volunteers and a limited number of diabetic patients revealed no differences in TRO metabolism (Loi et al., 1997), suggesting that diabetic patients, in general, are not metabolically predisposed to TRO toxicity. Because therapy for diabetes often involves multiple medications, it is also important to determine whether hepatotoxic risk is due to TRO alone or in combination with other medications. TRO or its metabolites, if accumulated in the liver over time, may either directly cause hepatotoxicity or affect metabolism and excretion of other drugs or endogenous substrates, thus indirectly resulting in liver damage.
In this study, using cultured human and porcine hepatocytes, we investigated the role of TRO metabolism in cytotoxicity. In addition, we investigated whether the low sulfation capacity of porcine hepatocytes relative to human cells contributes to TRO toxicity.
Materials and Methods
Chemicals.
Williams E culture medium (HMM) and medium supplements, dexamethasone and insulin, were obtained from BioWhittaker (Walkersville, MD). Penicillin G/streptomycin was obtained from Life Technologies Laboratories (Grand Island, NY). Troglitazone was provided by Dr. J. R. Koup, Parke-Davis Pharmaceutical Research (Ann Arbor, MI). Phenobarbital, 2,6-dichloro-4-nitrophenol, pentachlorophenol, and MTT were obtained from Sigma (St. Louis, MO). Leucinel-14C was obtained from NEN Life Science Products (Boston, MA). Falcon culture dishes (60-mm and six-well) were obtained from Becton Labware (Franklin Lakes, NJ).
Hepatocyte Cultures and Treatment Protocol.
Human hepatocytes were isolated from livers not used for whole organ transplant. Porcine hepatocytes were isolated from male Hanford miniature pigs. Hepatocytes were isolated by three-step collagenase perfusion as described previously (Strom et al., 1996, 1998). The viability of cells obtained, as measured by trypan blue exclusion test, ranged from 74 to 90%. Hepatocytes were plated in Williams E medium supplemented with 10−7 M dexamethasone, 10−7 M insulin, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 10% bovine calf serum. Hepatocytes (3 × 106/plate or 2 × 106/well) were plated on 60-mm or 6-well culture plates previously coated with type I (rat-tail) collagen. Cells were allowed to attach for 4 to 6 h in 37°C, at which time the medium was replaced with serum-free medium with the supplements listed above and changed every 24 h thereafter. After 96 h in culture, cells were treated with TRO (1–100 μM) for 2 or 24 h. Where indicated, cells received TRO in combination with 10 μM DCNP dissolved in sterile water or 5 μM PCP dissolved in DMSO. A combination of PB (2 mM) or APAP (5 mM) dissolved in culture media with TRO was added to some hepatocytes for 2 or 24 h. Concentrated stocks of TRO were prepared in DMSO. The final concentration of DMSO in culture medium was 0.1%.
Sample Analysis.
Sulfate, glucuronide, and quinone metabolites of TRO and parent drug were measured in culture media. After 96 h in culture, the medium was replaced with fresh Williams E medium containing TRO at concentrations indicated in the figure legends. Aliquots (500 μl) of the media were removed after 1 or 23 h of incubation at 37°C and stored at −20°C. TRO and its metabolites were measured by liquid chromatography-tandem mass spectrometry. Samples were extracted by adding the following to 1.5-ml polypropylene microcentrifuge tubes: 300 μl of hepatocyte culture media, 30 μl of working standard solution to standard samples, 10 μl of internal standard solution, 600 μl of acetonitrile, and 10 μl of acetic acid (0.1%). After vortexing thoroughly and centrifuging at 14,000 rpm for 15 min, 500 μl of the supernatant was transferred to a 96-well polypropylene autosampler plate and evaporated to dryness at 40°C under nitrogen. Finally, samples were reconstituted in 150 μl of acetonitrile/2 mM ammonium acetate (60:40), and 7.5 μl was injected into the liquid chromatography-tandem mass spectrometry. The LC system consisted of a Perkin-Elmer (Norwalk, CT) Series-200 autosampler and pump (flow rate 0.2 ml/min). The analytical column was a Supelco (Bellefonte, PA) Discovery RP Amide C16 (2.1 × 100 mm, 5 μ). The mobile phase consisted of acetonitrile/2 mM ammonium acetate, 0.05% acetic acid (60:40, v/v). Final chromatographic retention times for TRO, metabolites, and PD 166793 (internal standard) were between 3.0 and 8.0 min. All measurements were made with a Micromass (Manchester, UK) Quattro II tandem quadrupole mass spectrometer, set to electrospray negative ionization mode, with MassLynx version 3.1 operating software. For analytes of interest, parent-to-daughter ion transitions were established through direct infusion of each compound into the mass spectrometer. The following ion transitions were obtained: TRO (440.0 → 397.1), quinone metabolite (456.0 → 179.0), sulfate metabolite (519.7 → 439.9), glucuronide metabolite (615.8 → 439.9), and PD 166793 internal standard (409.8 → 78.8). Sensitivity was then optimized for each compound by varying cone voltage and collision energy in the multiple reaction monitoring mode and maximizing ion intensity. A single 11-point standard curve was prepared by diluting standards in Williams E medium. The assay standards were injected twice, once at the beginning and once at the end of the sample run.
Toxicity Assays.
Toxicity was determined by 1) the measurement of total protein synthesis by pulse-labeling hepatocytes for 1 h with [14C]leucine, as described previously (Kostrubsky et al., 1997), 2) reduction of MTT, using a protocol described by the manufacturer (Sigma), and 3) microscopic examination of the hepatocytes. In the text, incubation time refers to total time including the presence of [14C]leucine.
Additional Assays.
Proteins were determined by the procedure of Lowry et al. (1951).
Statistical Analysis.
Results were analyzed by a two-factor ANOVA. A P < .05 was interpreted as the level of statistical significance.
Results
Toxicity of TRO in Cultured Human Hepatocytes.
Hepatocytes prepared from four donors were treated with TRO at 10, 20, 25, 35, and 50 μM for 2 or 24 h, and total protein synthesis was determined (Fig. 1A). TRO produced a concentration-dependent decrease in protein synthesis by 2 h. However, recovery of protein synthesis occurred with 24-h exposure to TRO at concentrations up to 20 μM. Concentrations equal to or exceeding 25 μM resulted in sustained inhibition of protein synthesis. MTT conversion also was measured. Figure 1B shows that treatment of hepatocytes for 24 h with 35 or 50 μM TRO resulted in 15 and 50% decreases in MTT reduction, respectively, compared with untreated cells. TRO at 50 μM was lethal to the cells as judged by microscopic examination of the hepatocytes.
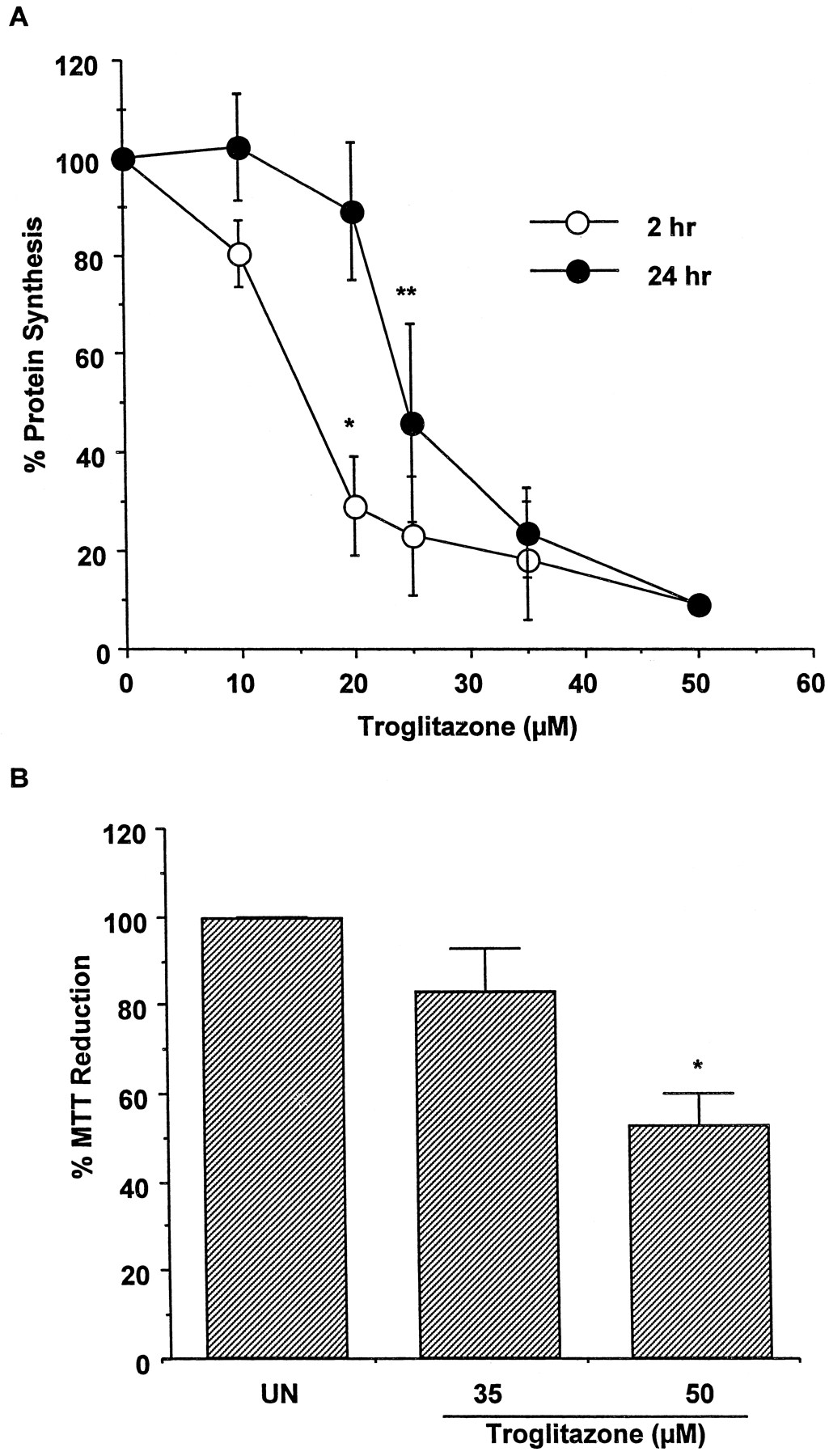
Effect of troglitazone on protein synthesis and MTT reduction in human hepatocytes.
A, hepatocytes from four human donors were treated for 2 or 24 h with 10, 20, 25, 35, and 50 μM TRO. The protein synthesis was determined by a pulse labeling with [14C]leucine for 1 h, as described under Materials and Methods. Each value is expressed as a percentage of the value in untreated cells and represents the mean of duplicate treatments of hepatocytes from each of four donors, with the S.D. indicated by the vertical bars. ∗, significantly different from cells treated with 10 and 20 μM TRO for 2 or 24 h, with P < .001. ∗∗, significantly different from cells treated with 20 μM TRO for 24 h, with P < .01. B, hepatocytes from two human donors were treated for 24 h with 35 and 50 μM TRO. At the end of this time, the reduction of MTT was measured. Each value is expressed as a percentage of the value in untreated cells and represents the mean of duplicate treatments of hepatocytes from each of two donors, with the S.D. indicated by the vertical bars. ∗, significantly different from the untreated (UN) and the cells treated with 35 μM TRO, with P < .001.
Because TRO sulfate is the major metabolite detected in patients (Loi et al., 1997, 1999a), we hypothesized that inhibiting TRO sulfation might result directly, or indirectly, in greater toxicity. To inhibit TRO conjugation, APAP and PB, drugs whose metabolism involves sulfation, were used. Under the current assay conditions, APAP (5 mM) was both glucuronidated and sulfated by cultured human hepatocytes (results not shown), and PB has been reported to be metabolized to a sulfoconjugated derivative in rat hepatocytes (Verite et al., 1996). Figure 2A shows that coincubation of APAP or PB with 10 μM TRO resulted in about 70% inhibition of protein synthesis by 2 h, compared with untreated cells. In addition, treatment of four different hepatocyte cultures with 20 μM TRO in combination with APAP or PB for 24 h resulted in a sustained decrease in protein synthesis that in two cultures was associated with cell death (results not shown). To further assess the effect of inhibiting TRO sulfation on toxicity, hepatocytes were treated for 2 h with a combination of 10 μM TRO and the known inhibitors of sulfation, DCNP or PCP (Boles and Klaassen, 1999; Raftogianis et al., 1999). Both inhibitors significantly potentiated the toxicity of TRO as shown in Fig. 2B.

Effect of cotreatment of troglitazone with APAP, PB, DCNP, or PCP on protein synthesis.
A, hepatocytes from four human donors were treated for 2 h with a combination of 10 μM TRO with 5 mM APAP or 2 mM PB. The protein synthesis was determined by a pulse labeling with [14C]leucine for 1 h as described underMaterials and Methods. Each value is expressed as a percentage of the value in untreated cells and represents the mean of duplicate treatments of hepatocytes from each of four donors, with the S.D. indicated by the vertical bars. ∗, significantly different from cells treated with TRO and PB alone, with P < .001. ∗∗, significantly different from cells treated with TRO and APAP alone, with P < .001 andP < .01, respectively. UN, untreated cells; ■, none; ▨, PB; ▪, APAP. B, hepatocytes from three human donors were treated for 1 h with a combination of 10 μM TRO with 10 μM DCNP or 5 μM PCP. The protein synthesis was determined by a pulse labeling with [14C]leucine for 1 h as described under Materials and Methods. Each value is expressed as a percentage of the value in untreated cells and represents the mean of duplicate treatments of three donors, with the S.D. indicated by the vertical bars. ∗, significantly different from cells treated with DCNP with P < .05 or TRO alone withP < .01. ∗∗, significantly different from cells treated with PCP or TRO alone, with P < .001. UN, untreated cells; ■, none; ▨, DCNP; ▪, PCP.
Toxicity of TRO in Cultured Porcine Hepatocytes.
Because pigs are reported to be deficient in sulfation of xenobiotics (Jakoby, 1980), we investigated whether pig hepatocytes were more susceptible to TRO toxicity than human hepatocytes. Conversely, the pig may compensate for the lack of sulfation by increased glucuronidation. The toxicity of TRO to porcine cells is shown in Fig.3. Similar to human hepatocytes, pig cells experienced a transient decrease in protein synthesis after a 2-h treatment with increasing concentrations of TRO. However, only a 30% decrease in protein synthesis was detected at 20 μM TRO, in comparison with a 70% decrease in human hepatocytes (Fig. 1A). A 24-h exposure of TRO resulted in complete recovery of protein synthesis at up to 50 μM TRO. Cellular death was associated with 90% inhibition in protein synthesis detected at 100 μM TRO (Fig. 3). Thus, porcine hepatocytes are resistant to TRO toxicity at concentrations found to be toxic to the human cells.

Effect of troglitazone on protein synthesis in porcine hepatocytes.
Hepatocytes from two pigs were treated for 2 or 24 h with 10, 20, 25, 35, 50, 75, and 100 μM TRO. The protein synthesis was determined by a pulse labeling with [14C]leucine for 1 h as described under Materials and Methods. Each value is expressed as a percentage of the value in untreated cells and represents the mean of duplicate treatments of hepatocytes from each of two pigs, with the S.D. indicated by the vertical bars. ○, 2 h; ●, 24 h.
Metabolism of TRO in Cultured Human Hepatocytes.
Metabolism of TRO at concentrations causing no significant decrease in protein synthesis (1, 5, and 10 μM) is shown in Fig.4A. The major metabolites detected were sulfate and quinone derivatives of TRO (Fig. 4A). Only a small increase in TRO sulfate and a large increase in unmetabolized parent TRO were detected in cells exposed to 10 μM TRO relative to cells treated with 5 μM TRO, suggesting that sulfation at 10 μM was close to the saturation of this pathway. In addition to treatment with 10 μM TRO, hepatocytes from a different donor were also exposed to 25 and 35 μM TRO, concentrations causing a sustained inhibition of protein synthesis (Figs. 4B and 1A). Sulfation and formation of the quinone metabolite were saturated at 10 μM TRO. Accordingly, concentration-dependent increases in unmetabolized drug were observed after treatment with 25 and 35 μM TRO. Only a trace level of TRO glucuronide was detected at any concentration of TRO. There was a correlation (r2 = 0.82) between the increase in parent TRO and the decrease in protein synthesis after 2 h in culture.
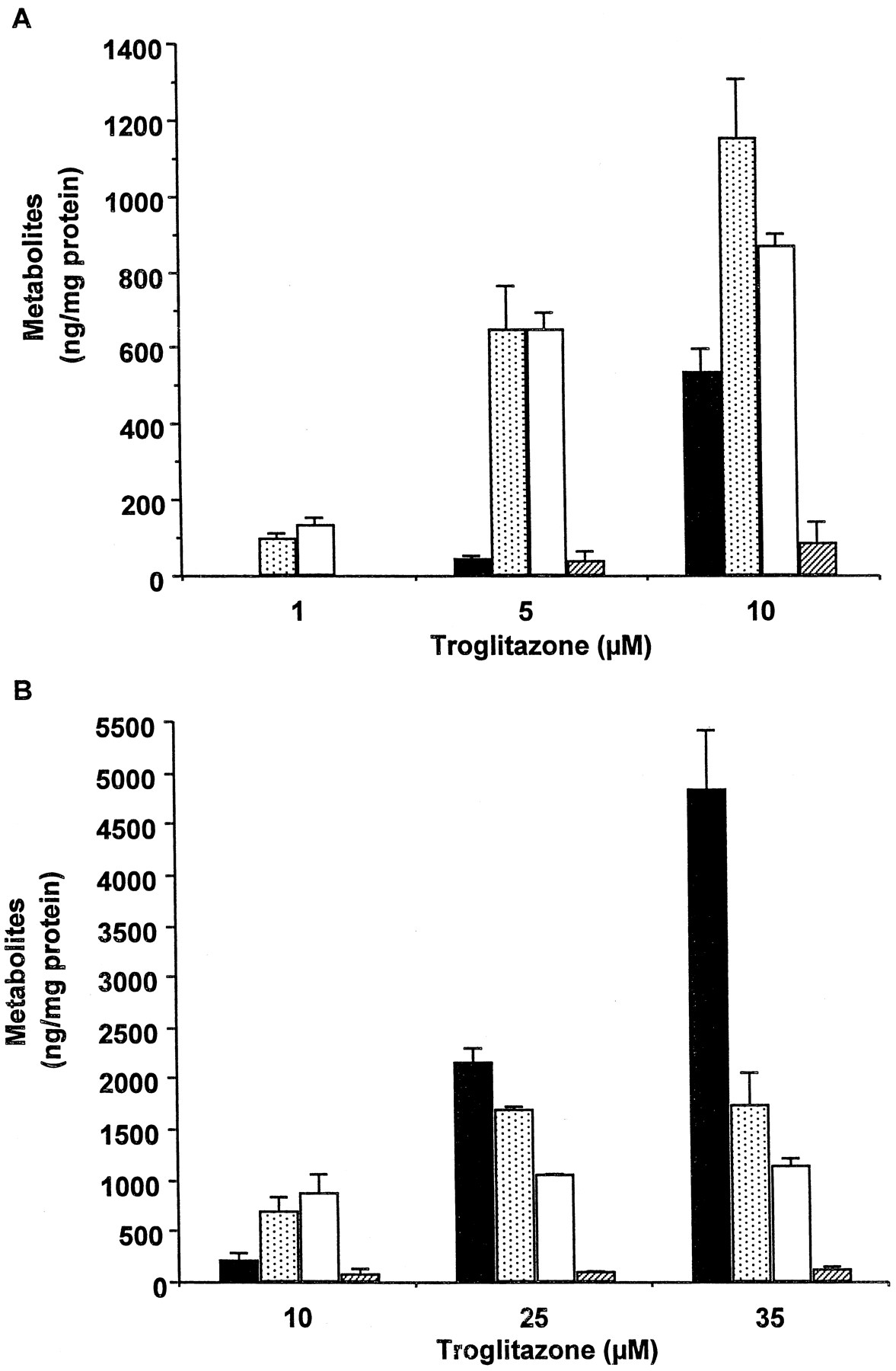
Metabolism of troglitazone in human hepatocytes.
Human hepatocytes from a single donor were treated for 1 h with 1, 5, and 10 μM TRO (A) or hepatocytes from a different donor were treated with 10, 25, and 35 μM TRO (B). At the end of this time, aliquots of the medium were removed, and TRO metabolites and parent drug were determined as described under Materials and Methods. Each value represents the mean of duplicate treatments with the range indicated by the vertical bars. ▪, troglitazone; ░, troglitazone quinone; ■, troglitazone sulfate; ▨, troglitazone glucuronide.
Because treatment with 20 μM TRO alone for 24 h caused no toxicity, but coincubation of TRO with APAP or PB resulted in sustained decrease in protein synthesis, the association with inhibition of sulfation and accumulation of parent TRO was examined. Figure5A shows that in the absence of APAP or PB, 90% TRO was metabolized by 24 h to the sulfate derivative, with little quinone or glucuronide observed. There was no parent drug detected at this time. In contrast, coincubation of TRO with APAP or PB resulted in a 50 and 25% decrease in sulfation of TRO, respectively, associated with accumulations of unmetabolized drug and its quinone derivative.

Effect of APAP, PB, and DCNP on metabolism of troglitazone in human hepatocytes.
A, human hepatocytes were treated for 24 h with 20 μM TRO alone or in combination with 5 mM APAP or 2 mM PB. At the end of this time, aliquots of the medium were removed, and TRO metabolites and parent drug were determined as described under Materials and Methods. Each value represents the mean of duplicate treatments with the range indicated by the vertical bars. ▪, troglitazone; ░, troglitazone quinone; ■, troglitazone sulfate; ▨, troglitazone glucuronide. B, human hepatocytes were treated for 1 h with 10 μM TRO alone or in combination with 10 μM DCNP. At the end of this time, aliquots of the medium were removed, and TRO metabolites and parent drug were determined as described under Materials and Methods. Each value represents the mean of duplicate treatments with the range indicated by the vertical bars. ▪, troglitazone; ░, troglitazone quinone; ■, troglitazone sulfate; ▨, troglitazone glucuronide.
A 1-h exposure to the combination of TRO (10 μM) and DCNP resulted in 90% inhibition of TRO sulfation, with an increase in unmetabolized parent drug and no significant increase in formation of TRO quinone (Fig. 5B). As shown in Fig. 2B, the inhibition of sulfation was associated with about 70% decrease in protein synthesis. Thus, it appeared that the inability of hepatocytes to metabolize TRO due to inhibition of sulfation resulted in increased toxicity.
Metabolism of TRO in Cultured Porcine Hepatocytes.
In contrast to sulfation of TRO in human hepatocytes, Fig.6A shows that porcine cells primarily glucuronidated TRO, with less than 10% TRO sulfate formed. There were no increases in the total amount of glucuronide and quinone formed at TRO concentrations exceeding 20 μM. In contrast, there was a concentration-dependent increase in unmetabolized TRO. Accumulation of parent drug correlated with the decrease in protein synthesis withr2 = 0.9 (Fig. 3). Thus, as observed in human hepatocytes, the development of toxicity in pig hepatocytes was associated with the parent drug and not with its quinone metabolite.

Metabolism of troglitazone in porcine hepatocytes.
Porcine hepatocytes were treated for 1 h (A) or 24 h (B) with 10, 20, 25, 35, and 50 (A) or 50 and 100 (B) μM TRO. At the end of this time, aliquots of the medium were removed, and TRO metabolites and parent drug were determined as described under Materials and Methods. Each value represents the mean of duplicate treatments with the range indicated by the vertical bars. ▪, troglitazone; ■ troglitazone sulfate; ░, troglitazone quinone; ▨, troglitazone glucuronide.
Analyses of culture media after a 24-h incubation detected unmetabolized TRO in cells treated with 100 μM TRO, a concentration lethal to porcine hepatocytes (Figs. 6B and 3). In these cells, the quantity of TRO quinone detected was similar to that detected by 1 h in cells treated with lower concentrations of TRO (Fig. 6A). These concentrations caused no sustained decrease in protein synthesis by 24 h (Fig. 3). In contrast, TRO was undetectable in cells treated with 50 μM (Fig. 6B) and showed no decrease in protein synthesis by 24 h (Fig. 3).
Discussion
In this study, the metabolism and toxicity of TRO were compared in human and porcine hepatocytes to investigate whether timely clearance of TRO is an important factor in preventing toxicity. Analysis of media from human hepatocyte cultures revealed a similarity between metabolic profiles of TRO in vitro and the plasma of patients taking this drug. The majority of drug was metabolized to TRO sulfate by 24 h, with approximately 10% detected as combined glucuronide and quinone metabolites (Fig. 5A). These data are in agreement with the elimination half-life of 24, 36, and 23 h for TRO, TRO sulfate, and TRO quinone metabolites, respectively (Loi et al., 1997), suggesting that data from cultures of human hepatocytes can be extrapolated for in vivo interpretation. Disappearance of TRO quinone by 24 h may be explained by its further metabolism to the quinone sulfate (Kawai et al., 1997). In addition, nontoxic concentrations of TRO used in this study are close to those found in human plasma. Concentrations of 3.6 and 6.3 μM were reported as the maximum plasma concentration in humans taking TRO at therapeutic doses of 400 and 600 mg/day, respectively (Loi et al., 1999a).
We hypothesized that inhibition of protein synthesis as early as 2 h, as well as sustained decrease in protein synthesis by 24 h, resulted from the accumulation of the parent drug. Accumulation of unmetabolized TRO could be due to an increase in treatment concentrations or inhibition of TRO sulfation. Increase in parent drug may directly result in toxicity or cause increased conversion of parent to an as yet unidentified minor reactive metabolite responsible for toxicity.
We observed that hepatocytes, which microscopically appeared to be severely compromised by 24 h, still had about half the MTT reduction capability of untreated hepatocytes at 50 μM TRO. Measurement of MTT reduction at early times produced no detectable changes, implying that MTT is not a sensitive measure of cell toxicity. This observation is supported by the finding that reduction of MTT is not specific for mitochondria but also occurs in lysosomes and is supported by multiple intracellular substrates (Liu et al., 1997). Thus reduction still would be possible in a compromised cellular environment. However, decreases in protein synthesis appeared to be a more sensitive index of toxicity, with sustained decreases associated with cell death.
We found that increases in toxicity correlated with increases in unmetabolized TRO. Furthermore, recovery in protein synthesis was associated with a complete disappearance of parent TRO. In addition, inhibiting sulfation with DCNP, APAP, or PB resulted in cytotoxicity. The inhibitory effects of APAP and PB were also associated with increases in TRO quinone formation, possibly due to inhibition of TRO quinone sulfation (Fig. 5A), suggesting that TRO quinone may contribute to the overall toxicity. However, treatment with DCNP and TRO caused no changes in TRO quinone but resulted in decreased protein synthesis. In addition, the quantities of troglitazone quinone detected in pig hepatocytes were similar under both toxic and nontoxic conditions. These findings suggest that the parent drug either directly or indirectly (through the formation of a toxic intermediate), rather than quinone metabolite, is responsible for toxicity.
Porcine hepatocytes were resistant to TRO toxicity at concentrations toxic to the human cells. Although proved to be deficient in sulfating TRO, porcine cells compensated this pathway with glucuronidation. Therefore, resistance to TRO toxicity relative to human cells may in part be explained by a higher capacity of pig cells to conjugate TRO.
Using cultured human hepatocytes prepared from different donors, we found that absolute decreases in protein synthesis, as well as morphological changes in response to TRO alone or its combinations with APAP or PB, would vary from culture to culture. It may, in part, be attributed to different rates of TRO sulfation in different donors. In fact, we found more than 5-fold variation in the rate of 4-methylumbelliferone sulfation between different human hepatocyte cultures (results not shown).
It is most likely that in the clinical situation, a combination of several factors rather than a single event results in hepatic failure. The present study, although limited to the role of conjugation in TRO toxicity, may suggest some possible explanations for TRO-induced hepatotoxicity in humans as a result of prolonged exposure of hepatocytes to parent drug. Increases in liver enzymes and liver failure have been reported to occur between 17 and 287 days (mean, 147) after the beginning of therapy (Gitlin et al., 1998; Neuschwander-Tetri et al., 1998; Shibuya et al., 1998; Watkins and Whitcomb, 1998), suggesting that a combination of several mechanisms could be involved. Inhibition of TRO sulfation with competitive drug or deficiency in sulfotransferase activity in a small number of patients may result in accumulation of TRO in liver with time and developing hepatotoxicity. One distinguishing feature of TRO metabolism is that the majority of the drug is excreted into the bile as TRO sulfate (Kawai et al., 1997). Therefore, another possibility is a deficiency in transport of TRO sulfate across the bile canalicular membrane, resulting in increased intracellular TRO concentration through the action of hepatic sulfatases. Similar to inhibition of TRO sulfation, inhibition in biliary transport may be due to the competition from other substrates or to genetic polymorphism of specific transmembrane transporter. The existence of TRO sulfate transporter is supported by the finding in perfused rat liver where decreased biliary excretion of [14C]TRO sulfate was found when TRO glucuronide was also included in the perfusion buffer (Parke-Davis, data on file). Indirectly, the possibility of involvement of biliary transport with TRO-associated hepatotoxicity is supported by the presence of jaundice and an increased level of bilirubin in patients who experienced TRO-associated hepatotoxicity. TRO sulfate may compete for the excretion with bilirubin glucuronide for the canalicular organic anion transporter resulting in hyperbilirubimia. Diabetic patients with a history of cholestasis may have a decreased capacity to excrete TRO sulfate into the bile and therefore be at increased risk of developing hepatotoxicity. In addition, cholestasis has been observed during histological examination of livers from patients experiencing liver failure (Gitlin et al., 1998; Shibuya et al., 1998; Med Watch reports provided to Parke-Davis). Alternatively, toxicity may result not from TRO itself, but rather from the accumulation of endogenous substances or other drugs due to the inhibition of their metabolism or excretion by high levels of TRO or TRO sulfate. Thus, multiple factors including competition for sulfation, genetic polymorphism, and deficiency in biliary transport are possible reasons for TRO-induced hepatotoxicity.
In conclusion, we demonstrated that TRO metabolism in cultured human hepatocytes was similar to the metabolism observed in humans. Increases in TRO toxicity noted in cultured human and pig hepatocytes correlated with the accumulation of unmetabolized TRO. Accordingly, disappearance of TRO resulted in hepatocyte recovery. In addition, inhibition of TRO sulfation resulted in cytotoxicity associated with the accumulation of parent drug. In contrast, glucuronidation was the major route of TRO metabolism in cultured porcine hepatocytes. Due to the higher capacity of this pathway, porcine hepatocytes were resistant to TRO toxicity at concentrations lethal to human cells.
Acknowledgment
We thank Dr. Michael Bleavins for help in reviewing the manuscript.
Footnotes
-
Send reprint requests to: Dr. V. Kostrubsky, Pfizer Global Research & Development, Department of Drug Safety Evaluation, 2800 Plymouth Rd., Ann Arbor, MI 48105. E-mail:vsevolod.kostrubsky{at}pfizer.com
- Abbreviations used are::
- TRO
- troglitazone
- APAP
- acetaminophen
- PB
- phenobarbital
- DCNP
- 2,6-dichloro-4-nitrophenol
- PCP
- pentachlorophenol
- DMSO
- dimethyl sulfoxide
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Received March 20, 2000.
- Accepted June 30, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}