


Abstract
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone and its major metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), are potent lung carcinogens in animals. UGT-mediatedO-glucuronidation of NNAL is an important detoxification pathway for these carcinogens. To better characterize this pathway in humans, we screened a series of UGT-overexpressing cell lines and baculosome preparations for their ability toO-glucuronidate NNAL and examined multiple human liver and lung specimens for NNAL-glucuronidating activity and their levels of expression of NNAL-glucuronidating UGTs. Human liver microsomal fractions exhibited significant levels of NNAL-glucuronidating activity, with the NNAL-Gluc II diastereomer formed at a rate 3.4 times that observed for NNAL-Gluc I. As with liver microsomal fractions, NNAL-Gluc II was the major diastereomer formed by homogenates from UGT2B7-overexpressing HK293 cells or UGT2B7-overexpressing baculosomes; the major diastereomer formed by homogenates from UGT1A9-overexpressing V79 cells was NNAL-Gluc I. No significantO-glucuronidating activity of NNAL was detected in UGT1A1-, UGT1A4-, UGT1A6-, UGT2B4-, or UGT2B15-overexpressing HK293 or V79 cell homogenates, or in UGT1A1-, UGT1A3-, UGT1A7-, or UGT1A10-overexpressing baculosomes. Significant levels of UGT2B7 mRNA were detected by reverse transcriptase-polymerase chain reaction in human liver and at low levels in human lung specimens. UGT1A9 mRNA was detected in liver but not in lung. These results suggest that although both UGT2B7 and UGT1A9 play an important role in the overall glucuronidation of NNAL in humans, UGT2B7 potentially plays an important role in the detoxification of NNAL in the lung.
One of the most abundant potent procarcinogens in tobacco and tobacco smoke is the nicotine-derived tobacco-specific nitrosamine, NNK1 (Hecht and Hoffmann, 1989; Hecht, 1998). In mainstream cigarette smoke, the total level of NNK is 3 to 15 times that of another major potent carcinogen in tobacco smoke, benzo[a]pyrene (Adams et al., 1987). NNK induces predominantly lung adenocarcinomas in rodents independent of the route of administration (Hecht, 1998). In the Fischer 344 rat, NNK induces pancreatic tumors (Rivenson et al., 1988) and, when applied together with the related tobacco-specific nitrosamine,N′-nitrosonornicotine, oral cavity tumors (Hecht et al., 1986). The cumulative dose of 1.8 mg of NNK per kg of body weight that induces lung tumors in rats (Belinsky et al., 1990) is similar to the cumulative lifetime dose of 1.6 mg of NNK per kg of body weight estimated for the average American who smoked 2 packs a day for 40 years (Hecht and Hoffmann, 1989; Hecht, 1998). NNK is therefore considered to be a likely causative agent for cancers of the lung, oral cavity, and pancreas in humans (Hecht and Hoffmann, 1989).
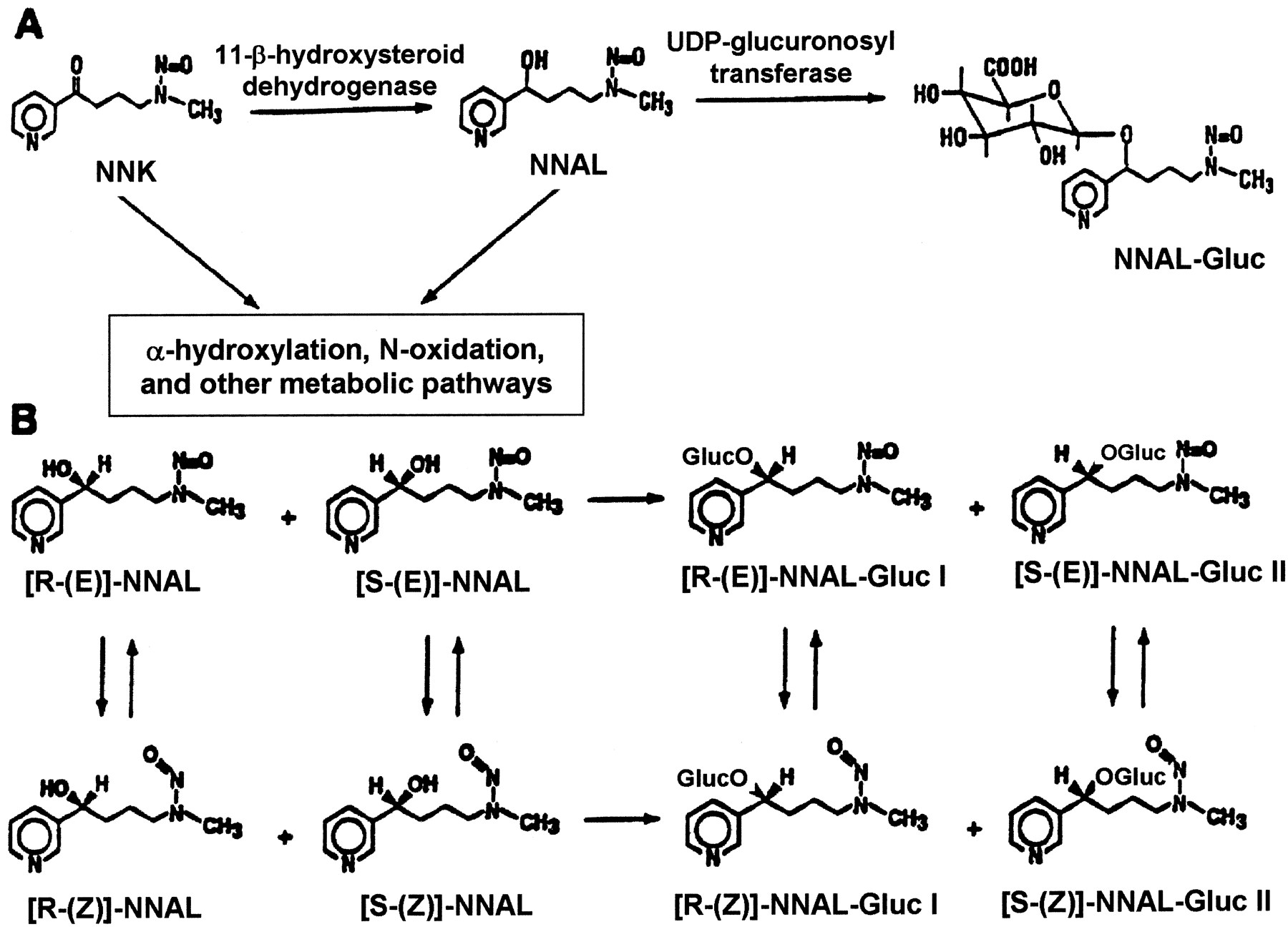
The major metabolic pathway for NNK in most tissues is conversion to NNAL by carbonyl reduction (Fig. 1). NNK reduction to NNAL occurs in rodents, monkeys, and humans (Carmella et al., 1993; Hecht et al., 1993; Hecht, 1998), with estimates that between 39 and 100% of the NNK dose is converted to NNAL in smokers (Carmella et al., 1993). NNAL is activated via pathways similar to those observed for NNK and, like NNK, is a potent lung and pancreatic carcinogen in rodents (Rivenson et al., 1988; Hecht, 1998). In a reaction likely to be catalyzed by the UGT family of enzymes, NNAL is also metabolized to its O-glucuronide, NNAL-Gluc (Fig. 1) (Morse et al., 1990; Carmella et al., 1993; Hecht et al., 1993;Hecht, 1998). Because NNK is metabolized to both the (R)- and (S)-enantiomers of NNAL, two diastereomericO-glucuronides of NNAL are formed, NNAL-Gluc I and NNAL-Gluc II (Fig. 1). In a recent addendum to previous studies, NNAL-Gluc I and NNAL-Gluc II were described as being formed from (R)-NNAL and (S)-NNAL, respectively (Hecht et al., 1997, 2000). NNK-treated rats form predominantly NNAL-Gluc I (Hecht et al., 1997;Murphy et al., 1997; Ren et al., 1999) whereas the major diastereomer observed in the urine of human snuff dippers is NNAL-Gluc II (Murphy et al., 1994). Both NNAL and NNAL-Gluc are excreted in the bile of rats (Shulze et al., 1992) and the urine of animals, smokers, and individuals exposed to sidestream smoke (Morse et al., 1990; Murphy et al., 1994; Hecht et al., 1994; Carmella et al., 1995; Richie et al., 1997; Parsons et al., 1998). Although the formation of NNAL is clearly not a detoxification pathway for NNK, the glucuronidation of NNAL appears to be an important mechanism for NNK detoxification. Recent studies have demonstrated that the glucuronidation of (R)-NNAL was significantly greater than that of (S)-NNAL after injection into A/J mice, a pattern consistent with the higher tumorigenicity exhibited by (S)-NNAL in the same experiments (Upadhyaya et al., 1999; Hecht, 2000). In contrast to the high tumorigenicity exhibited by both (R)- and (S)-NNAL, NNAL-Gluc is nontumorigenic after subcutaneous injection into A/J mice (Upadhyaya et al., 1999; Hecht, 2000). In addition, Kim and Wells (1996) reported that skin fibroblasts from UGT family 1-deficient rats were more sensitive to NNK-mediated cytotoxicity. Significant interindividual variability in the ratio of NNAL-Gluc/NNAL excreted in human urine has been observed, suggesting that individuals may differ greatly in their ability to detoxify NNK (Carmella et al., 1995). It has been hypothesized that the ability of an individual to glucuronidate NNAL may directly affect their susceptibility to lung and potentially other tobacco-related cancers.

Simplified schematic diagram of NNK metabolism to NNAL-Gluc (A) and structures of NNAL and NNAL-Gluc rotamers and enantiomers (B).
UGTs are a superfamily of enzymes that glucuronidate many xenobiotics and endogenous compounds (Tephly and Burchell, 1990). Based upon differences in sequence homology and substrate specificity, two families of UGTs (UGT1 and UGT2) have been identified in several species, each containing several highly homologous UGT genes. Recent studies have demonstrated that O-glucuronidation of NNAL is catalyzed primarily by UGT2B1 in the rat, an activity that is inducible by both phenobarbital and 3,5-di-tert-butyl-4-hydroxytoluene (Ren et al., 1999). The diastereomeric pattern of NNAL-Gluc formation in UGT2B1-overexpressing cell homogenates was similar to that observed for rat liver microsomes (Ren et al., 1999) and in urine from NNK-treated rats (Hecht et al., 1997; Murphy et al., 1997). The goal of the present study was to elucidate the major UGTs responsible forO-glucuronidation of NNAL in humans.
Materials and Methods
Chemicals and Materials.
Imipramine, 1-naphthol, 4-hydroxyacetophenone, clofibric acid, androsterone, 4-NP, hyodeoxycholic acid, UDPGA,dl-2-lysophosphatidylcholine palmityl C16:0, and β-glucuronidase were purchased from Sigma Chemical Co. (St. Louis, MO). Bilirubin was obtained from Fluka Chemicals (Ronkonkoma, NY). [14C]UDPGA (specific activity: 318 mCi/μmol) was obtained from ICN Pharmaceuticals (Costa Mesa, CA) and [3H]NNK (specific activity: 2.4 Ci/mmol) and unlabeled NNK were purchased from Chemsyn Scientific (Lenexa, KS). [3H]NNAL was purified from [3H]NNK by purification through HPLC after reduction of [3H]NNK with NaBO4. Briefly, [3H]NNK preparations were dried, resuspended in 1 ml of methanol, and incubated with 5 mg of NaBO4 for 30 min at room temperature. Samples were then dried, resuspended in H2O, filtered (0.2 μm), and injected on HPLC columns using the elution program described below. At the appropriate retention time (i.e., 55–62 min), [3H]NNAL was collected directly from the HPLC column, purified through a Sep-Pak C18 column (Waters Associates, Milford, MA), and resuspended in methanol for increased stability and long-term storage. Immediately before individual experiments, [3H]NNAL aliquots were dried and resuspended in H2O. The purity of all radiolabeled compounds was greater than 98% as determined by HPLC analysis. Dulbecco's modified Eagle's medium was obtained from Mediatech (Herndon, VA), and fetal bovine serum, genticin, and Maloney murine leukemia virus reverse transcriptase were purchased from Life Technologies (Grand Island, NY). Baculosome preparations overexpressing UGTs 1A1, 1A3, 1A7, 1A10, and 2B7 were purchased from PanVera Corporation (Madison, WI). Internal standards for NNAL glucuronidation studies, including 4-hydroxy- 1-(3-pyridyl)-1-butanone, 4-(methylnitrosamino)-1-(N-oxy-3-pyridyl)-1-butanone (NNK-N-oxide), 4-(methylnitrosamino)-1-(N-oxy-3-pyridyl)-1-butanol (NNAL-N-oxide), 4-hydroxy-1-(3-pyridyl)-1-butanol (diol), and 4-oxo-4-(3-pyridyl)-butyric acid (keto acid) were kind gifts from Shantu Amin at the American Health Foundation (Valhalla, NY). The mammalian expression vector pCI-neo, Taq DNA polymerase, T4 DNA ligase, and DNA polymerase I (Klenow fragment) were purchased from Promega Corporation (Madison, WI), and restriction enzymes were purchased from Boehringer Mannheim (Indianapolis, IN). Oligonucleotide primers were purchased from Integrated DNA Technologies, Inc. (Coralville, IA).
Tissue Samples and Microsome Preparation.
Normal human liver and lung sections (2–6 g) were obtained from the Cooperative Tissue Research Network (Eastern Division, Philadelphia, PA), the International Institute for the Advancement of Medicine (Scranton, PA), or the tissue procurement facilities at either the H. Lee Moffitt Cancer Center (Tampa, FL) or the University of Minnesota Cancer Center (Minneapolis, MN), with all specimens obtained from different subjects. Microsomes for examining NNAL-glucuronidating activity and total RNA for examining UGT expression were prepared for one liver specimen (no. 61297) and for all of the lung specimens examined in this study. Liver specimens 61297 and 1049G were obtained during surgery for hepatectomies whereas lung specimen 99604 was obtained via organ donation; all three specimens were from subjects who had never smoked. Normal lung specimens 17682, 17702, and SEM were all obtained during lung cancer surgery, with specimens 17682 and 17702 obtained from smokers (no data were available for specimen SEM). Samples obtained via organ donation were isolated and quick-frozen at −70°C within 24 h postmortem whereas specimens obtained during cancer surgery were quick frozen (−70°C) within 2 h postsurgery. Two of the liver microsomal fractions analyzed in this study (specimens 22 and 109) were kind gifts from Peter F. Guengerich (Vanderbilt University, Nashville, TN) and were obtained as microsomal fractions without corresponding RNA for expression analysis or corresponding subject smoking information. All of the lung samples were from the lower peripheral bronchia. Because of low quantities of tissue, only total RNA (and not microsomes) was purified from liver specimen number 1049G. Liver and lung microsomes were prepared through differential centrifugation as previously described (Coughtrie et al., 1987) and were stored (10–20 mg protein/ml) at −70°C in 100-μl aliquots, with total protein concentrations measured using the BCA assay (Pierce Corp., Rockford, IL).
Cell Lines and Cell Homogenate Preparation.
HK293 (human embryonic kidney fibroblast) cells and HK293 cell lines overexpressing UGT1A1, UGT1A4, UGT2B1, UGT2B7, or UGT2B15 were kindly provided by Tom Tephly (University of Iowa, Iowa City, IA) (Green et al., 1994, 1995; Pritchard et al., 1994; Coffman et al., 1995, 1998;King et al., 1997), V79 (Chinese hamster fibroblast) cells and V79 cells overexpressing UGT1A6 and UGT1A9 were kindly provided by Dr. Brian Burchell (University of Dundee, Scotland, UK) (Ebner and Burchell, 1993), and the p91023(B) expression vector carrying human UGT2B4 cDNA (Jin et al., 1993) was kindly provided by Peter Mackenzie (Flinders University, Adelaide, Australia). A HK293 cell line overexpressing UGT2B4 was generated by stable transfection using the calcium phosphate procedure as previously described (Ausubel et al., 1995). Briefly, UGT2B4 cDNA was excised from the p91023(B) vector by digestion with SpeI, blunt-ended using the Klenow fragment, and subcloned into the SmaI site of the pCI-neo mammalian expression vector. Insert orientation and sequence were confirmed by restriction enzyme digestion and dideoxy sequencing (Sanger et al., 1977). The pCI-neo/UGT2B4 construct was transfected into HK293 cells via calcium phosphate precipitation, and at 72 h post-transfection, cells were passaged and subsequently grown in genticin (700 μg/ml of medium) for the selection of genticin-resistant colonies. The selective medium was changed every 3 to 4 days and individual genticin-resistant colonies were selected after 2 weeks in culture.
All V79 and HK293 cell lines were grown to 80% confluence in Dulbecco's modified Eagle's medium supplemented with 4.5 mM glucose, 10 mM HEPES, 10% FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin and maintained in 700 μg/ml of genticin for selection of UGT overexpression in a humidified incubator in an atmosphere of 5% CO2. Cells were suspended in Tris-buffered saline (25 mM Tris base, 138 mM NaCl, 2.7 mM KCl; pH 7.4) and subjected to three rounds of freeze-thaw before gentle homogenization. Cell homogenates (5–30 mg/ml) were stored at −70°C in 100-μl aliquots. Total cell homogenate protein concentrations were determined using the BCA assay as described above.
NNAL Glucuronidation Assays.
The rate of NNAL glucuronidation by liver and lung microsomes was determined using the following conditions: microsomal protein (0.1–2.5 mg) was incubated in 50 mM Tris-HCl (pH 7.5)/10 mM MgCl2 (100–800 μl volume) containingdl-2-lysophosphatidylcholine palmityl C16:0 (20–500 μg), UDPGA (4 mM), NNAL (1–5 mM), and either [14C]UDPGA (2 μCi) or [3H]NNAL (10 μCi) at 37°C for either 40 min (liver) or 2 h (lung). Reactions were terminated by the addition of
Glucuronidation Assays for Other Substrates.
UGT activity toward 4-NP in both liver and lung microsomes and in UGT-overexpressing baculosomes was determined using radioflow-HPLC as described above. The analysis conditions of 4-nitrophenyl-O-glucuronide formation were as follows: enzyme assay mixtures (100 μl final volume) of 50 mM Tris-HCl (pH 7.5)/10 mM MgCl2 containing 0.1 μg/μldl-2-lysophosphatidylcholine palmital C16:0 (unless otherwise indicated in the text), 50 μg of microsomal protein or 200 μg of baculosomal protein, 2 mM14C-UDPGA (2 μCi/reaction), and 2 mM 4-NP, were incubated at 37°C for 2 h. Reactions were terminated by the addition of
The glucuronidation of 4-NP and other aglycones in cell homogenates and androsterone in baculosomes, was determined by TLC analysis as described by Bansal and Gessner (1980). Enzyme assay mixtures (100 μl final volume) of 50 mM Tris-HCl (pH 7.5)/10 mM MgCl2 containing 10 μg ofdl-2-lysophosphatidylcholine palmital C16:0, 250 μg of cell homogenate, or 100 μg of baculosomes, 2 mM [14C]UDPGA (0.5 μCi/reaction), and 67.5 μM to 16.8 mM aglycone were incubated at 37°C for up to 2 h. Reactions were terminated by the addition of 2 volumes of ice-cold 100% ethanol and then centrifuged at 17,000g for 10 min. Supernatants were collected, dried, and resuspended in 10 to 20 μl of water. Aliquots were applied to TLC plates and developed in organic solvent as described previously (Bansal and Gessner, 1980). [14C]4-nitrophenyl-O-glucuronide, previously purified by HPLC, was used as a TLC reference. The chromatograms were air dried and exposed for autoradiography for 1 to 2 weeks. Quantification was performed by densitometric readings of autoradiographs using computer analysis (Photoshop, NIH Image 1.61 analysis system for Macintosh). As with other glucuronidation analysis, glucuronide formation was confirmed by treatment with E. coli β-glucuronidase as described above.
RT-PCR Analysis.
Total RNA was isolated from normal liver and lung sections (0.5 g) as well as from UGT1A9- and UGT2B7-overexpressing cells (108 cells were used for each cell line) using the guanidinium isothiocyanate/cesium chloride method and treatment with DNase I as previously described (Ausubel et al., 1995). Total RNA specimens were stored at −70°C in individual aliquots.
RT was performed in 20-μl total volumes using 3 μg of total RNA, 200 U of Maloney murine leukemia virus reverse transcriptase, and 0.5 μg of oligo(dT)12–18 primer as outlined in the manufacturer's protocol (Life Technologies). PCRs were performed as reported previously (Belanger et al., 1995; Strassburg et al., 1997) with minor modifications. Each reaction (100 μl volume) contained 5 μl of the RT reaction mix described above, 2.4 mM MgCl2, 50 mM KCl, 20 mM Tris-Cl (pH 8.0), 0.2 mM levels of each dNTP, 400 pmol of sense and antisense UGT primers, and 2.5 U of Taq DNA polymerase. Reactions were incubated in a Perkin-Elmer 9600 thermocycler (Perkin-Elmer Corp., Foster City, CA) for 1 cycle of 94°C for 3 min, up to 40 cycles of 94°C for 1 min, 64°C for 1 min, and 72°C for 1 min, followed by 1 cycle at 72°C for 7 min. Where indicated, human β-actin sense and antisense primers (40 pmol for each primer; see Strassburg et al., 1997 for primer sequences) were added after 6 (for RT-PCRs using liver RNA) or 9 (for RT-PCRs using lung RNA) cycles as an internal control for PCR amplification. Reactions without template and reactions using total RNA purified from either UGT1A9- or UGT2B7-overexpressing cell lines were included as controls in all RT-PCR experiments. All PCRs were performed in triplicate, and 20% of each PCR was resolved by electrophoresis in 8% polyacrylamide gels containing 1 μg/ml of ethidium bromide dye. Gels were photographed over UV light and DNA bands corresponding to UGT1A9, UGT2B7, and β-actin were scanned and analyzed using a photoimager (Alpha Innotech Corp., San Leandro, CA). The sequences of all PCR-amplified bands were confirmed by dideoxy sequencing (Sanger et al., 1977). For semiquantitative RT-PCR, 15-μl aliquots were removed at specific cycles as indicated in the text.
Statistical Analysis.
The Student's t test (two-tailed) was used for all comparative analyses. The correlation coefficient (r2) from linear regression analysis of Lineweaver-Burk plots was used to determine theKm and Vmax of UGT1A9- and UGT2B7-induced glucuronidation of NNAL.
Results
NNAL Glucuronidation in Microsomes from Human Tissues.
Previous studies have demonstrated that the major NNAL-Gluc diastereomer observed in the urine of snuff users was NNAL-Gluc II (Murphy et al., 1994). As illustrated in Fig.2 for HLiM (B and C), the glucuronides of (R)- and (S)-NNAL separate under the HPLC conditions of our analysis. As reported in previous studies (Murphy et al., 1994), NNAL-Gluc eluted between twoN-oxidation metabolites of NNAL and NNK, NNAL-N-oxide and NNK-N-oxide (between 31–38 min). The pair of early-eluting peaks (peaks 1 and 2) with a retention time of 31 to 34 min correspond to the E and Zisomers of NNAL-Gluc I [(R)-NNAL-Gluc], whereas peaks 3 and 4 (retention time of 34–38 min) correspond to the E andZ isomers of NNAL-Gluc II [(S)-NNAL-Gluc; see Fig. 1] (Hecht et al., 1997; Hecht, 2000). These peaks were observed using [3H]NNAL (Fig. 2) or [14C]UDPGA (results not shown) as the radiolabeled substrate. When this entire region was collected and treated with β-glucuronidase, the only product was NNAL (Fig. 2D), confirming that these peaks correspond to glucuronidated NNAL conjugates. The level of total NNAL-Gluc formation in the three liver microsomal specimens examined ranged from 0.6 to 2.6 nmol of NNAL-Gluc formed per mg of microsomal protein. All liver microsomes analyzed in this study preferentially catalyzed the glucuronidation of (S)-NNAL (i.e., the formation of NNAL-Gluc II) (Table1). The ratio of NNAL-Gluc II/NNAL-Gluc I formed in liver microsomes averaged 3.4:1.

HPLC analysis of NNAL-Gluc formation by human liver microsomes.
Human liver microsomes (100 μg of protein) were incubated with 1 mM [3H]NNAL (10 μCi) and 4 mM UDPGA as described underMaterials and Methods. A, NNK metabolite standards (3–10 μg each metabolite). B, [3H]NNAL metabolites formed by human liver microsomal sample no. 22. C, [3H]NNAL metabolites formed by human liver microsomal sample no. 109 D, analysis of the β-glucuronidase-treated [3H]NNAL-Gluc fraction (30–37 min, from human liver microsomal sample 109).
Glucuronidation of 4-NP and NNAL in human liver and lung microsomes
No detectable levels of NNAL glucuronidation were observed in four microsomal samples prepared from individual lung specimens, even when up to 2.5 mg of lung microsomal protein was used in the glucuronidation assay (Table 1). As 4-NP is glucuronidated by multiple UGT family 1 and family 2 enzymes (Tephly and Burchell, 1990), 4-NP was used as a control substrate to assay for activity in these lung microsomal samples. Detectable levels of 4-NP-glucuronidating activity were observed for all lung microsomal preparations (Table 1); however, these levels were markedly lower (mean = 260-fold) than that observed for the liver microsomal sample tested in these studies (see Table 1; only one liver microsome was tested for 4-NP glucuronidation activity because of insufficient microsome quantity for the two remaining liver specimens).
NNAL Glucuronidation in UGT-Overexpressing Cell Homogenates and Baculosomes.
To elucidate whether any previously cloned human UGTs exhibit activity against NNAL, we performed a comprehensive screening of known human UGTs for their activity against NNAL. Before determining which, if any, of these previously cloned human UGTs catalyzed theO-glucuronidation of NNAL, homogenates from each of the UGT-overexpressing HK293 or V79 cell lines were analyzed for glucuronidation of previously characterized substrates. Saturating concentrations of substrate, equivalent to 10 times the publishedKm were used for these analyses. These activities for seven human UGTs and the rat UGT2B1 are listed in Table2. We previously reported that UGT2B1 was a catalyst of NNAL O-glucuronidation (Ren et al., 1999). All homogenates glucuronidated the chosen substrates, with activities ranging from 13-fold less (UGT2B4) to 30-fold greater (UGT1A6) than that observed for the glucuronidation of clofibric acid by UGT2B1. Knowing these relative activities provided some measure by which to compare the extent of NNAL O-glucuronidation by each of the expressed UGT.
UGT-overexpressing homogenate activities
As reported previously (Ren et al., 1999), rat UGT2B1 catalyzed theO-glucuronidation of (S)-NNAL significantly more efficiently than the glucuronidation of (R)-NNAL. This resulted in the formation of NNAL-Gluc II and NNAL-Gluc I in a ratio of 0.2:1.0 (Table 2). Of the seven human UGT-overexpressing cell lines screened in this study, only homogenates from UGT1A9- and UGT2B7-overexpressing cell lines glucuronidated NNAL (Table 2). As shown by radioflow-HPLC, the relative amounts of the diastereomers of NNAL-Gluc formed by homogenates from cells overexpressing UGT1A9 (Fig.3B) was different from that of homogenates from cells overexpressing UGT2B7 (Fig. 3D). The major glucuronide product of UGT1A9-catalyzed NNAL metabolism eluted at 31 to 34 min (peaks 1 and 2), which corresponds to the two rotamers (i.e.,E and Z; see Fig. 1) of the glucuronide of (R)-NNAL (NNAL-Gluc I). A smaller amount of the glucuronide of (S)-NNAL (peaks 3 and 4, corresponding to the two rotamers of NNAL-Gluc II) was also observed. The ratio of NNAL-Gluc II/NNAL-Gluc I observed in UGT1A9-overexpressing cell homogenates was 0.6:1 (Table 2). The estimated Km for the glucuronidation of NNAL by UGT1A9 was 23.1 mM with aVmax/Km of 0.79.

HPLC analysis of NNAL-Gluc formation in UGT-overexpressing V79 and HK293 cell lines and UGT-overexpressing baculosomes.
Cell (2.5–5 mg of protein) or baculosome (0.1 mg of protein) homogenate was incubated at 37°C for 2 h with 10 mM NNAL and 2 mM 14C-UDPGA as described under Materials and Methods. A, NNK metabolite standards (3–10 μg each metabolite). B, 14C-labeled metabolites from incubations using homogenates from UGT1A9-overexpressing V79 cells. C,14C-labeled metabolites from incubations using homogenates from UGT1A9-overexpressing cells shown in B after subsequent incubation with β-glucuronidase. D, 14C-labeled metabolites from incubations using homogenates from UGT2B7-overexpressing HK293 cells. E, 14C-labeled metabolites from incubations using homogenates from UGT2B7-overexpressing cells shown in D after subsequent incubation with β-glucuronidase. F,14C-labeled metabolites from incubations using baculosome homogenates overexpressing UGT2B7. G, 14C-labeled metabolites from incubations using baculosome homogenates overexpressing UGT2B7 shown in F after subsequent incubation with β-glucuronidase.
The major UGT2B7-catalyzed product of NNAL glucuronidation was the (S)-glucuronide (peaks 3 and 4, Fig. 3D). A small amount of NNAL-Gluc I [the glucuronide of (R)-NNAL] was also formed by this UGT enzyme (peaks 1 and 2). The ratio of NNAL-Gluc II/NNAL-Gluc I was 4.3:1 for UGT2B7 (Table 2). The 14C-labeled products formed by homogenates from both UGT2B7- and UGT1A9-overexpressing cell lines were sensitive to β-glucuronidase treatment (Fig. 3, C and E). The estimatedKm for the glucuronidation of NNAL by UGT2B7 was 9.3 mM, with aVmax/Km of 1.72. No detectable glucuronidation of NNAL was observed for homogenates of cell lines overexpressing UGT1A1, UGT1A4, UGT1A6, UGT2B4, and UGT2B15, even when increased amounts of cell homogenate protein (up to 8 mg) were used in the NNAL glucuronidation assays (Table 2).
To confirm the role of UGT2B7 in NNAL O-glucuronidation, assays were performed using baculosomes from Sf-9 cells infected with UGT2B7 cDNA-containing baculovirus. As was observed for homogenates of UGT2B7-overexpressing cells, NNAL-Gluc II was the major diastereomer formed for UGT2B7-overexpressing baculosomes (Fig. 3F). UGT2B7-expressing baculosomes metabolized both NNAL (Fig. 3F) as well as the UGT2B7 aglycone, androsterone (results not shown), at approximately one-third the rate observed for UGT2B7-overexpressing HK293 cell homogenates per milligram of baculosome or cell homogenate protein. The 14C-labeled products formed by homogenates from UGT2B7-overexpressing baculosome preparations were sensitive to β-glucuronidase treatment (Fig. 3G). No detectable activity against NNAL was observed in our assay system for baculosomes overexpressing UGT1A1, UGT1A3, UGT1A7, or UGT1A10 using up to 1.5 mg of baculosome protein (50 μg was recommended by the manufacturer). Detectable levels of activity were observed by HPLC for UGT1A7 and UGT1A10 baculosome preparations against 4-NP (results not shown), suggesting that the manufacturer-prepared baculosomes were in fact active in our assay system. No significant difference inO-glucuronidating activity of UGT1A7 or UGT1A10-overexpressing baculosome homogenates was observed against 4-NP in assays performed withdl-2-lysophosphatidylcholine palmital C16:0 (as described under Materials and Methods) or without detergent (as recommended by the manufacturer; results not shown).
Expression of NNAL-Glucuronidating UGTs in Human Liver and Lung.
To better evaluate the physiological importance of UGT1A9 and UGT2B7 in terms of the overall glucuronidation of NNAL in humans, RT-PCR experiments were performed to examine the levels of expression of the two UGT enzymes. For liver specimens, UGT expression was quantitated by comparison with the expression of β-actin within each RT-PCR. Semiquantitative RT-PCR demonstrated that UGT2B7 was expressed at similar levels for the two liver specimens analyzed (Fig.4, A and C). In contrast, a more than 10-fold difference in the expression of UGT1A9 was observed in the same specimens (Fig. 4, B and D). The level of expression of UGT2B7 was 28-fold that observed for UGT1A9 for liver specimen number 61297, but was only 2.5-fold that of UGT1A9 for liver specimen number 1049G. The amplification of UGT2B7, UGT1A9, and β-actin was linear at all cycles examined.

RT-PCR amplification of UGT1A9 and 2B7 transcripts in human liver tissue specimens.
RT-PCR was performed as described under Materials and Methods, with aliquots electrophoresed in 8% polyacrylamide. A, coamplification of human UGT2B7 and β-actin in human liver specimen 61297. B, coamplification of human UGT1A9 and β-actin in human liver specimen 61297. C, coamplification of human UGT2B7 and β-actin in human liver specimen 1049G. D, coamplification of human UGT1A9 and β-actin in human liver specimen 1049G. Numbers to the left of each panel indicate the liver specimen analyzed. The lane sequence was the same for all panels: lane 1, −RNA; lane 2, DNA marker (SK/HpaII); lane 3, RT-PCR (38-cycle PCR) using total RNA from UGT2B7-overexpressing HK293 (A) or UGT1A9-overexpressing V79 (B) cells; lanes 4 to 8, 20-μl aliquots of RT-PCRs at cycles 24 (lane 4), 26 (lane 5), 28 (lane 6), 30 (lane 7), and 32 (lane 8), using total RNA isolated from the indicated human liver tissue specimen.
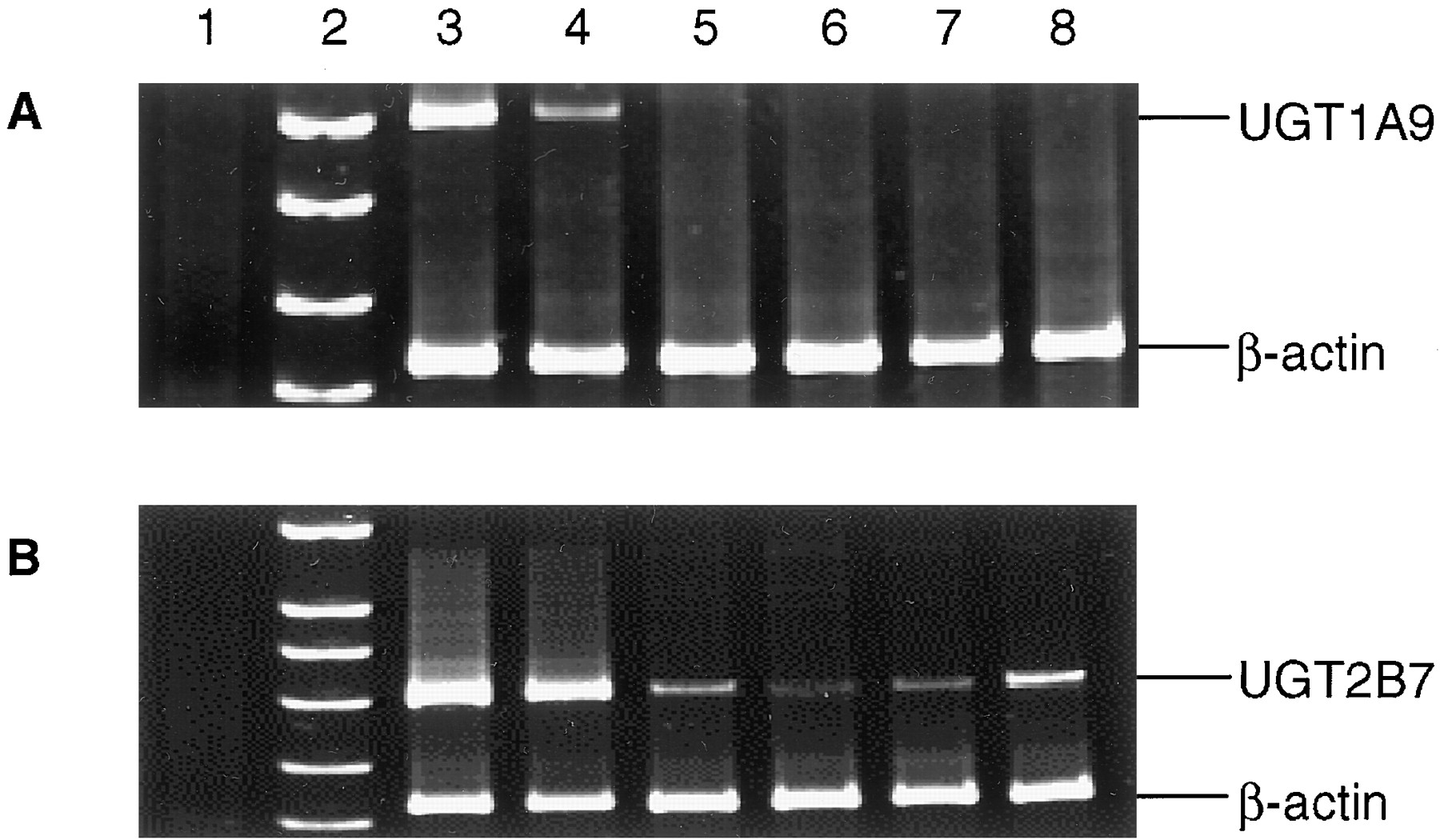
The levels of expression of UGT1A9 and UGT2B7 in four lung specimens after 38 cycles of PCR after reverse transcription of total RNA was determined by RT-PCR (Fig. 5). No UGT1A9 amplification was observed by RT-PCR in the four lung specimens analyzed in this study (A), nor was UGT1A9 amplified in the four lung specimens when the PCR cycles were increased from 38 to 50 or when β-actin primers were excluded from the PCR (results not shown). UGT1A9 mRNA was amplified in RT-PCRs using total RNA from 1) UGT1A9-overexpressing cells (A, lane 3), and 2) liver tissue (A, lane 4). UGT2B7 was expressed in the lung at much lower levels than that observed for human liver, but, in contrast to UGT1A9, UGT2B7 mRNA was detected in all four lung specimens examined (Fig. 5B).
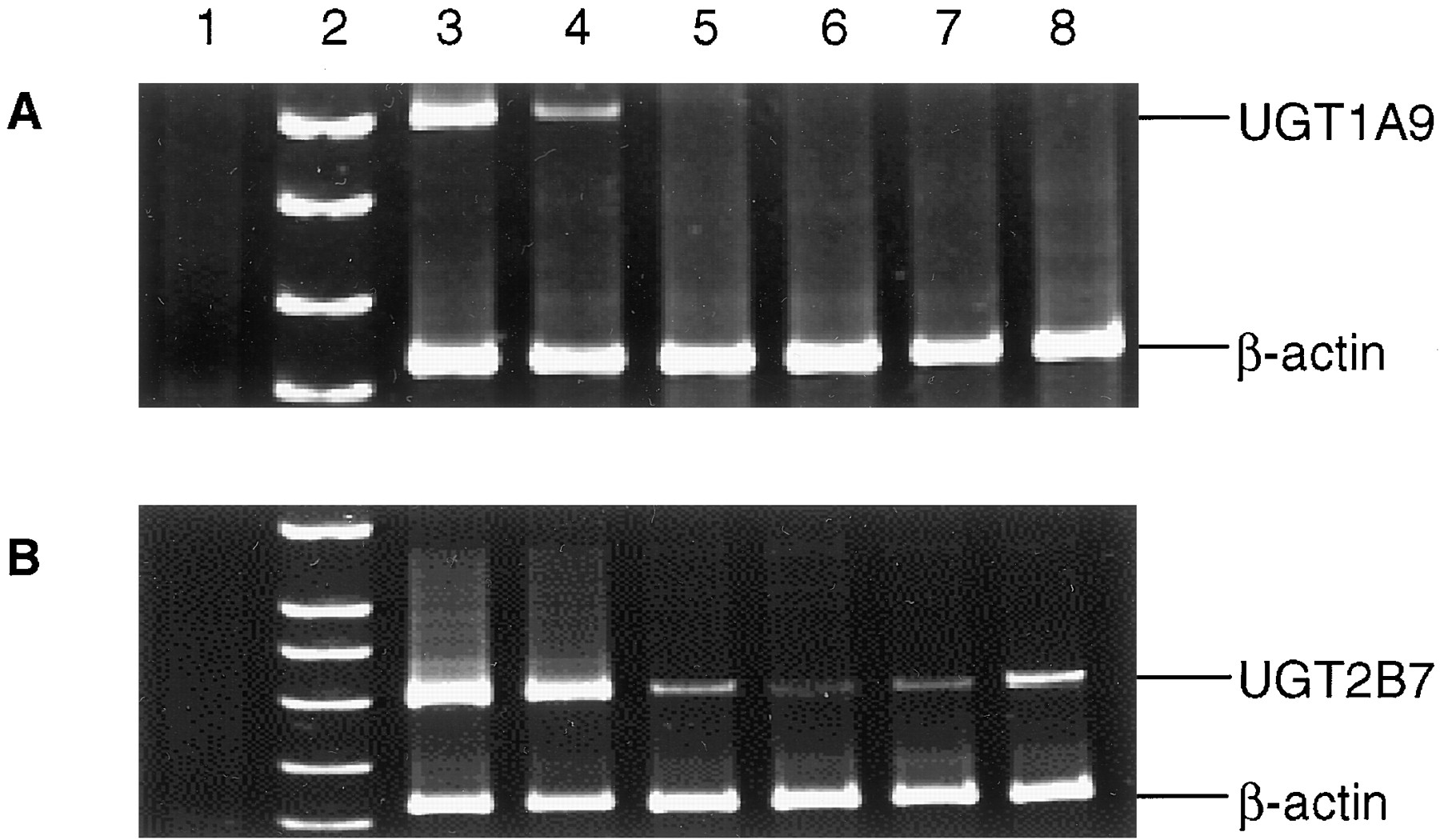
RT-PCR amplification of UGT1A9 and 2B7 in human lung tissue.
RT-PCR was performed as described under Materials and Methods, with aliquots electrophoresed in 8% polyacrylamide. A, coamplification of UGT1A9 and β-actin. B, coamplification of UGT2B7 and β-actin. The lane sequence for A and B were as follows: lane 1, −RNA; lane 2, DNA marker (SK/HpaII); lane 3, RT-PCR (38-cycle PCR) of total RNA from UGT1A9-overexpressing V79 (A) or UGT2B7-overexpressing HK293 (B) cells; lanes 4 to 8, 20-μl aliquots of RT-PCRs (40-cycle PCR) of total RNA isolated from liver specimen 61297 (lane 4), lung specimen 17682 (lane 5), lung specimen 17702 (lane 6), lung specimen SEM (lane 7), and lung specimen 996041 (lane 8).
Discussion
In this study, we present results of experiments designed to elucidate the UGT isozymes that play a major role in the glucuronidation of NNAL in humans. This is the first study to directly measure the glucuronidation of this potent lung carcinogen by human UGTs. Of the UGTs screened in this study, UGT2B7 and UGT1A9 both catalyze the O-glucuronidation of NNAL in vitro. Both reactions are stereoselective, with UGT2B7 preferentially forming NNAL-Gluc II and UGT1A9 preferentially forming NNAL-Gluc I. Similar stereospecificity toward NNAL has been demonstrated with rat UGTs such as UGT2B1 (Ren et al., 1999). In addition, similar stereospecific activity has previously been shown for UGT2B7 with other aglycones such as (S)-oxazepam (Coffman et al., 1998), analogous to the preference we report here for (S)-NNAL.
Previous studies have established that there are differences in urinary NNAL glucuronide ratios in NNK-treated rats (Hecht et al., 1997; Murphy et al., 1997) versus snuff users (Murphy et al., 1994). Similar differences are observed in in vitro assays using microsomes isolated from either rat (Ren et al., 1999) or human (present study) liver samples. Recent studies have demonstrated that UGT2B1 is probably the major UGT isozyme involved in the glucuronidation of NNAL in the rat (Ren et al., 1999). However, the known human UGT isozyme with highest homology to UGT2B1, UGT2B15 (77% homology at the nucleotide level), exhibited no activity against NNAL in the present study. This was not totally unexpected because UGT2B15 and UGT2B1 lack significant overlap in substrate specificity (Mackenzie, 1987; Chen et al., 1993; Green et al., 1994; King et al., 1997). Family 1 UGTs do not appear to play a major role in the glucuronidation of NNAL in the rat. Surprisingly, at least one member of the human UGT1 family, UGT1A9, appears to contribute significantly to NNAL glucuronidation in humans. Together, the results presented here are consistent with the fact that there are significant differences in NNAL glucuronidation pathways in rats versus humans.
As reported in previous studies (Guengerich et al., 1997; Strassburg et al., 1997), both UGT1A9 and UGT2B7 are expressed in normal human liver specimens. Because NNAL-Gluc II is the major O-glucuronidated diastereomer formed in vitro with human liver microsomes (present study) and is the major form observed in the urine of snuff users in vivo (Murphy et al., 1997), UGT2B7 probably plays a major role in the glucuronidation of this NNAL diastereomer in humans. The nonhepatic UGTs, UGT1A7 and UGT1A10, did not catalyzeO-glucuronidation of NNAL in the present study. This is consistent with the possibility that the majority of the NNAL-Gluc II observed in the urine of smokers is derived from UGT2B7-induced conjugation in the liver. The data presented here further suggest that UGT1A9 is likely to play an important role in the formation of the less abundant NNAL-Gluc I diastereomer observed in the urine of snuff users (Murphy et al., 1997), a diastereomer not efficiently formed (relative to NNAL-Gluc II) by UGT2B7. The preferential formation of NNAL-Gluc I by UGT1A9-overexpressing cell homogenates is similar to that observed for the rat UGT2B1 (Ren et al., 1999) and is similar to that observed in the urine of NNK-treated rats (Hecht et al., 1997, Murphy et al., 1997; Ren et al. 1999).
A greater than 2-fold higher estimatedVmax/Km was observed for UGT2B7 toward NNAL (1.72) than was observed for UGT1A9 (0.79). In addition, in the two liver specimens examined, UGT2B7 was expressed at higher levels than UGT1A9. Both sets of data are consistent with the NNAL-Gluc diastereomer patterns observed in the urine of snuff users and in in vitro assays using human liver microsomes (present study), where NNAL-Gluc II was the predominant diastereomer. These data suggest that UGT1A9 and UGT2B7 play important roles in the in vivo formation of NNAL-Gluc I and NNAL-Gluc II, respectively, particularly in the liver. Although the data presented here indicate that UGT2B7 may be the major UGT enzyme involved in NNALO-glucuronidation, recent preliminary studies suggest that UGT1A9 also metabolizes NNAL by N-glucuronidation (Nguyen et al., 2000). Therefore, although the ratio of N- versusO-glucuronidation of NNAL has not yet been studied in human tissue or bodily fluids (e.g., urine), it is likely that both UGT1A9 and UGT2B7 play major roles in the overall glucuronidation of NNAL in humans.
Previous studies examining the expression of UGT2B7 in human lung tissue were inconclusive (King et al., 1999). The present studies demonstrate that the expression of UGT2B7 mRNA is much lower in human lung than liver; however, expression of UGT2B7 mRNA was observed in all lung specimens examined. In contrast, no UGT1A9 mRNA was detected in the same lung specimens, a pattern similar to that reported previously for human lung RNA purchased from CLONTECH Laboratories (Palo Alto, CA) (Albert et al., 1999). These data suggest that UGT2B7 may play an important role in the detoxification of NNAL (and therefore, NNK) in human lung tissue. No NNAL-glucuronidating activity was observed in any of the human lung microsomes tested. However, the same microsomal specimens exhibited significantly reduced glucuronidation activity (260-fold) against a prototypical UGT substrate, 4-NP, compared with that observed for liver microsomes. Therefore, NNAL-Gluc formation by lung microsomes may be below the levels of detection for the methods used in the present study. In addition, the tissue specimens obtained for the above studies were procured from either tissue network banks or pathology tissue procurement facilities. Hence the investigators in this study did not have full control of how tissues were isolated immediately after surgery or postmortem. Hence, it is therefore also possible that microsomal enzyme fractions from these tissues were not of optimal quality for enzyme analysis.
Different levels of UGT1A9 expression were observed for the two liver samples examined in the present study. Some variation was also observed for UGT2B7 expression between lung specimens, although accurate quantitation of UGT2B7 in these specimens was made difficult because of relatively low overall levels of UGT2B7 expression. There are several mechanisms by which such differences in expression could occur, including genetic differences and/or induction by exogenous agents. Such variability in UGT expression could potentially be important in explaining the significant interindividual variability in the ratio of NNAL-Gluc/NNAL observed in the urine of smokers (Carmella et al., 1995). Previous studies have suggested that isothiocyanates may decrease NNK-induced carcinogenicity by increasing the glucuronidation of NNAL (Hecht and Hoffmann, 1989; Hecht, 1998). The possibility exists that such activity is manifested via induction of specific UGT enzymes such as UGT2B7. Another possibility is that tobacco smoke carcinogens may induce expression of specific UGT enzymes. Because UGT expression levels were determined in only a few specimens, a larger quantitative study examining NNAL glucuronidation phenotype from a large number of subjects for whom complete exposure/demographic questionnaire data are collected will be necessary to better evaluate UGT expression in people.
Previous investigators have suggested that the NNAL glucuronidation pathway is an important determinant of individual susceptibility to NNK- and tobacco-related cancer (Richie et al., 1997). They have hypothesized that smokers with lower NNAL-Gluc/NNAL ratios may be more susceptible to the carcinogenic effects of NNK and NNAL because these individuals would be less able to detoxify these agents, thereby increasing their carcinogenic potential. This hypothesis, however, depends on the value of the urinary NNAL glucuronide ratio as a marker for lung cancer risk. Urinary ratios are likely to best reflect the metabolic capabilities of metabolizing organs such as the liver and may not necessarily be indicators of metabolic pathways in other tissues such as the lung where metabolizing enzyme gene expression levels may differ. In this study, UGT2B7 but not UGT1A9 mRNA was detected by RT-PCR in total RNA samples isolated from normal human lung specimens. Therefore, although differences in expression or activity of either UGT1A9 and/or UGT2B7 may play a role in the differential urinary NNAL-Gluc/NNAL ratios observed in smokers, it is possible that potential differences in UGT-linked susceptibility to NNK-/NNAL-induced lung cancer may in part be due to differences in the expression or activity of UGT2B7. Further studies will be required to establish the relative importance of UGT2B7 as a detoxification mechanism for NNAL in the lung.
In addition to the NNAL-glucuronidating activity exhibited by UGT1A9 and UGT2B7, these isozymes have been reported to play a major role in the glucuronidation and potential detoxification of other major carcinogens. UGT1A9 has been reported to glucuronidate the colon carcinogen, 2-hydroxyamino-1-methyl-6-phenylimidaza[4,5-b] pyridine (Nowell et al., 1999) whereas UGT2B7 has been reported to glucuronidate metabolites of the lung carcinogen benzo[a]pyrene, including benzo[a]pyrene-trans 4,5-dihydrodiols and benzo[a]pyrene-7,8-dihydrodiols (MacKenzie et al., 1993). Thus, UGT1A9 and UGT2B7 play an important role in the detoxification of several tobacco carcinogens and may serve as important targets for chemoprevention of cancer in the future.
Footnotes
-
Send reprint requests to: Philip Lazarus, Ph.D., Divisions of Cancer Control and Molecular Oncology, H. Lee Moffitt Cancer Center, University of South Florida, MRC-2E, 12902 Magnolia Dr., Tampa, FL. E-mail: plazarus{at}moffitt.usf.edu
-
This research was supported by National Institutes of Health Grants CA52216 (to P.L.) and DE12206 (to P.L.). This research was presented at the American Association for Cancer Research Meeting, 2000, in San Francisco, CA. [Ren et al. (2000) Proc Am Assoc Cancer Res 41:5336.]
- Abbreviations used are::
- NNK
- 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNAL
- 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol
- UGT
- UDP-glucuronosyltransferase
- NNAL-Gluc
- β-O-[4-(methylnitrosamino)-1-(3-pyridyl)-1-but-1-yl]-d-glucosiduronic acid
- UDPGA
- UDP-glucuronic acid
- 4-NP
- 4-nitrophenol
- TLC
- thin layer chromatography
- RT
- reverse transcriptase
- PCR
- polymerase chain reaction
- Received June 15, 2000.
- Accepted August 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}