


Abstract
This article is a report on a symposium held at the March 1997 meeting of the American Society for Pharmacology and Experimental Therapeutics in San Diego. Current developments in the heterologous expression of cytochrome P450, NADPH-cytochrome P450 reductase, glutathione transferase, and UDP-glucuronosyltransferase enzymes are described. Systems include bacteria, insect cells, and transient and stable mammalian cells. Uses of the products are described for discernment of which enzymes are involved in metabolism of drugs, genotoxicity assays, mutagenesis (for structure-activity relationships), large scale production of enzyme products, antibody production, and production of proteins for biophysical studies.
During the past 15 years, there has been considerable progress in the characterization of the enzymes involved in drug metabolism. The work on purification, cDNA cloning, and heterologous expression has been extended from studies with experimental animal systems to humans. Today, it is possible to utilize recombinant human enzymes for a variety of purposes, and in the development of a new pharmaceutical drug candidate it is almost routine to determine which P450s1 and other enzymes contribute to their metabolism. This approach has been utilized in making rational in vitro predictions about bioavailability, toxicity, and drug-drug interactions.
Although much has been done in this area and many reagents are even commercially available, the systems are being improved and the methods of using the enzymes are still in a state of development. This article briefly summarizes four talks presented at a March 1997 American Society for Pharmacology and Experimental Therapeutics symposium on the subject, chaired by two of the authors (F.P.G. and B.B.). The individual sections are identified by contributing authors. Enzymes under consideration include P450, NADPH-P450 reductase, GSH transferase, and UGT.
Expression of Mammalian Microsomal Cytochromes P450 and Their Redox Partner, NADPH-P450 Reductase, in Escherichia coli (E.F.J., T.H.R., C.v.W., J.C., F.J.)
The principal advantages of the use of E. coli as a host for the expression of mammalian microsomal P450s are the relatively low costs to propagate cultures, the ease of producing large culture volumes, and the convenience of the vectors for protein engineering and mutagenesis. For these reasons, Johnson and colleagues have used E. coli as a host for the expression of a number of P450 2C enzymes and for the expression of NADPH-P450 reductase.
In general, modifications of the cDNA sequence are necessary to achieve adequate levels of expression in E. coli. Barnes et al. (1) demonstrated that two types of modifications enabled the expression of P450 17A in this host. First, the second codon was changed to a codon for Ala, which is found in many proteins that are highly expressed in E. coli. Second, silent substitutions were incorporated in the first eight codons to reduce the propensity of the nascent transcript to form stable secondary structures. These changes led to abundant expression of P450 17A in E. coliwith only a minor change in the sequence of the membrane anchor sequence found in the N terminus of the protein. As changes in the sequence of the membrane anchor sequence are unlikely to affect the catalytic properties of P450s, Richardson et al. (2) constructed a chimera of P450 2C3 that substituted the modified N-terminal coding sequence used for P450 17A for the native N terminus (fig. 1). The modified enzyme, P450 2C3mod, was readily expressed in E. coli, and the purified enzyme exhibited the catalytic properties of the unmodified enzyme isolated from rabbit liver.

Comparison of N-terminal sequences of modified forms of P450 2C3 with that of the native enzyme.
Modifications that altered the N-terminal amino acid sequence of P450 2C3 as shown were introduced to express the protein inE. coli as a membrane protein, 2C3mod, and as a peripheral membrane protein, 2C3d. The N terminus of the latter is similar to a truncated form of P450 7A, 7Δ2-24, that is expressed as a soluble enzyme in E. coli (3).
The polymerase chain reaction primer utilized in the construction of P450 2C3mod linked the N-terminal coding region used for the expression of P450 17A in E. coli to a primer designed to hybridize to templates for all of the rabbit and human 2C P450s. Due to a small number of mismatched nucleotides, the primer introduced additional alterations in the sequence of the membrane-spanning domains of each of seven rabbit (2, 4) and of the four known human P450 2C enzymes (4) that were subsequently expressed in E. coli. The levels of expression of the different P450 2C enzymes ranged from 100 to 1500 nmol/liter culture. In general, the cultures were maintained at temperatures ≤30°C during the induction of P450 expression to promote proper folding. In some cases, addition of the heme precursor δaminolevulinic acid increased the yield of P450, but in other cases, it did not. In each case tested, the enzymes exhibited their expected catalytic activities, further indicating that modifications to the membrane-spanning domain do not alter the catalytic activity of the enzyme.
E. coli does not express an equivalent of the mammalian NADPH-P450 reductase. However, the catalytic properties of the expressed enzyme can be assessed following reconstitution with NADPH-P450 reductase using standard procedures. For this purpose, it is necessary to solubilize the P450 from the membrane fraction of the cell, centrifuge the solution to remove insoluble material, and subsequently remove the detergent before reconstitution with purified NADPH-P450 reductase. Richardson et al. (4) generally adsorb the detergent-solubilized enzyme to hydroxylapatite and then wash and elute the enzyme in detergent-free buffers.
Alternatively, a mammalian NADPH-P450 reductase can be co-expressed with the P450 in cellular membranes for direct use of the membranes in assays. For this purpose, the two coding regions can be linked in the same expression cassette to enable bicistronic expression. This approach has been described for P450s 2E1 (5), 17A (6), and 3A4 (7) (see also section by Guengerich and Parikh, vide infra). Johnson and colleagues have co-expressed P450 2C19 and human microsomal NADPH-P450 reductase by inserting the latter into the expression vector for P450 2C19mod following the stop codon for the P450. A short linker between the two coding sequences contains a ribosome binding site upstream of the initiation codon for the reductase. Modifications were also introduced to the reductase nucleotide sequence. These included substitution of the codon for Ala employed for P450 expression for the native second codon of human NADPH-P450 reductase and silent substitutions in codons 4, 6, and 8 to increase A/T content. Membranes isolated from the transformed E. coli contain a roughly 1:1 molar ratio of P450 2C19 and reductase. As shown in table 1, the membranes exhibit turnover numbers for the 4′-hydroxylation ofS-mephenytoin, the N-demethylation of diazepam, and the hydroxylation of R-warfarin by P450 2C19 that are very similar to the maximum rates obtained by reconstitution of the partially purified proteins at a 10:1 ratio of reductase to P450.
Catalytic activities of E. coli membranes containing co-expressed P450 2C19 and human microsomal NADPH-P450 reductase in comparison with a reconstituted system
Determination of the structure of a mammalian P450 would greatly aid in the prediction of drug metabolism and in the rational design of pharmaceutical agents. Expression of P450s in E. coliprovides an abundant source of protein for purification and subsequent crystallization procedures. P450 2C3mod can be highly purified from E. coli in large quantities (2). However, as seen for other microsomal P450s, P450 2C3mod is incorporated into E. colimembranes, and the purified enzyme forms large and poorly defined oligomers following its isolation (fig.2). Moreover, the crystallization of the protein may be hindered by residual, bound detergents that can not be completely removed from the purified proteins.
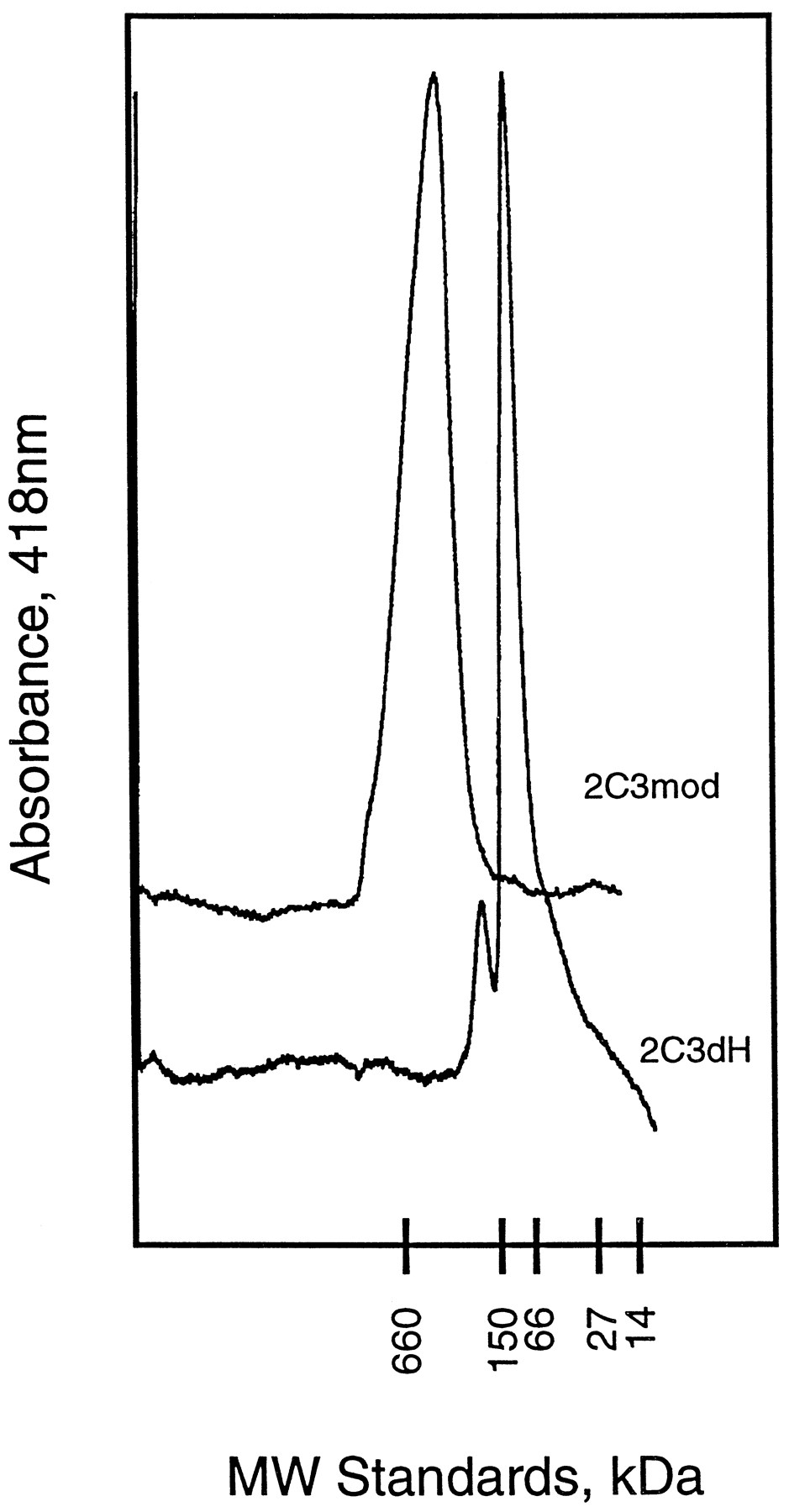
Superimposed chromatograms obtained for P450s 2C3mod and 2C3dH.
The purified proteins were each subjected to size exclusion chromatography on a Pharmacia Superdex 200 HR 10/30 column, and elution was monitored (A418). Theabscissa indicates the relative elution ofMr standards of the indicated sizes. The predicted molecular weights of the monomeric proteins are 55,736 and 54,408 for P450s 2C3mod and 2C3dH, respectively. However, the proteins elute as oligomers corresponding to octamers for 2C3mod and dimers for 2C3dH.
Other membrane proteins have been crystallized following removal of membrane-spanning sequences (8, 9), and such modifications could facilitate the crystallization of microsomal P450s. The microsomal P450s are generally believed to be anchored to the endoplasmic reticulum by a single transmembrane segment found at the N terminus of the protein (10). However, removal of portions of the putative membrane-spanning domain from several microsomal P450s did not prevent incorporation of substantial amounts of the modified P450 into E. coli membranes, and the truncated enzymes could not be extracted from the membranes using Na2CO3 (11, 12). On the other hand, a soluble catalytic domain from P450 52A3 is liberated by proteolysis at a site introduced by site-directed mutagenesis near the N-terminal membrane anchor sequence (13). In addition, deletion of residues 2–24 of rat P450 7A, a cholesterol 7-hydroxylase, produced a soluble enzyme in E. coli that is catalytically active (3). These results suggest that the binding of the catalytic domain of microsomal P450s to membranes is peripheral in the absence of the N-terminal membrane-spanning domain and that suitable modifications of the N-terminal sequence could lead to expression of a soluble, catalytically active enzyme.
These latter observations led von Wachenfeldt et al. (14) to modify the N terminus of P450 2C3 with the aim of expressing a soluble form of the enzyme. The design of the construct removed the membrane-spanning domain by deleting amino acid residues 3–18 from the N terminus (fig. 1). This construct, P450 2C3d, also incorporated substitutions of an Ala for Asp2 to facilitate expression in E. coli and of serines for His24 and Gly25 to introduce a restriction site to simplify the generation of alternative constructs. The design of this construct is highly similar to the truncated form of P450 7A that was expressed as a soluble enzyme (13) in both the length of the truncation and the presence of charged residues in the modified N terminus (fig. 1). P450 2C3d is expressed at relatively high levels in E. coli (800–1200 nmol/liter culture medium).
In contrast to P450 2C3mod, which retains a membrane-spanning N-terminal sequence, the subcellular distribution of P450 2C3d in E. coli is dependent on the ionic strength of the buffer used for cell disruption. In low ionic strength buffers, 2C3d was mainly localized in the membrane fraction (70%), whereas in buffers containing 1 M NaCl or 0.5 M potassium phosphate, P450 2C3d is predominantly found in the soluble fraction (>90%), indicating that deletion of the hydrophobic segment from the N terminus converted the integral membrane protein to a peripheral one. P450 2C3d was further modified by the incorporation of four His residues at the C terminus (P450 2C3dH), enabling purification of the enzyme in the absence of detergents from high salt extracts using immobilized metal affinity chromatography. Subsequent purification employed ion exchange chromatography in the absence of detergents. However, incorporation of a detergent wash during the immobilized metal affinity chromatography seems to diminish the formation of aggregates during long term storage of highly concentrated, purified enzyme (>0.1 mM). The detergent used for this step can be selected from those that have been successfully employed for the crystallization of other proteins.
The catalytic properties of the purified, modified enzyme are similar to those of the native enzyme (14). P450 2C3dH is predominantly a dimer as indicated by size exclusion chromatography (fig. 2). Moreover, the detergents sodium cholate and CHAPS each dissociate P450 2C3dH dimers to monomers at concentrations that do not alter the aggregation state of P450 2C3mod (14). The latter is a larger oligomer (>8-mer) (fig. 2). The oligomerization of P450 2C3dH is similar to that of the proteolytic cleavage product of P450 52A3 that is also a dimer (13) and that of the truncated form of P450 7A that contained monomers and dimers in roughly equal proportions (3). Thus, appropriate modifications of the N terminus do not affect enzyme activity and introduce desirable characteristics that are likely to facilitate attempts to crystallize the catalytic domains of 2C3 as well as of other microsomal P450s.
Microsomal NADPH-P450 reductase is also anchored to the membrane by a single membrane-spanning domain in the N terminus of the protein. Trypsin cleavage of the membrane-bound enzyme liberates a cytosolic, flavin-containing domain. This soluble C-terminal fragment consists of a domain similar to flavodoxins that binds FMN and an FAD-containing domain associated with the NADPH binding site that is similar to flavodoxin reductases (15). The flavin-containing fragment liberated by trypsin is connected to the N-terminal membrane-spanning sequence by a highly conserved linker that includes a segment rich in aromatic residues and a highly charged segment (16) (fig.3). The soluble proteolytic fragment catalyzes the reduction of cytochrome c by NADPH but does not support P450-catalyzed reactions (17). However, fusion of this domain of the NADPH-P450 reductase directly to the C terminus of P450s leads to catalytically competent monooxygenases (18).

N-terminal fragment of human NADPH-P450 reductase corresponding to the portion of rabbit NADPH-P450 reductase removed by trypsin (16).
The N terminus of the reductase (hORd) was truncated to remove most of the hydrophobic segment that is likely to form a membrane-spanning region. The hORd construct retains a highly conserved cluster of aromatic residues flanked by a Thr and a segment containing predominantly charged amino acids. Nucleotide substitutions to facilitate expression in E. coli result in the substitution of Ala for Gly at position 2. In addition, introduction of a restriction site in the hORd cDNA resulted in the substitution of an arginine for the lysine preceding the trypsin cleavage site.
In contrast to the domain of the NADPH-P450 reductase released by trypsin cleavage, Johnson and colleagues found that an extended construct containing the linker region together with the flavin-containing domain functions as a redox partner for P450 2C3d. The construct, hORd, is expressed as a soluble monomer in E. coli with yields similar to that of the full-length reductase, and the hORd protein can be highly purified using affinity chromatography as employed for the full-length reductase. Reconstitution of the two soluble, truncated enzymes, P450 2C3d and hORd, yields roughly 25% of the catalytic activity observed for P450 2C3d with full-length hOR, which retains the membrane-spanning domain. The ability of hORd, relative to the inability of the trypsin-solubilized domain, to reconstitute P450 2C3d suggests that the linker region is likely to be a functionally important component of the structure of native microsomal NADPH-P450 reductases. In addition, these results indicate that both microsomal enzymes can be expressed in soluble forms that reconstitute the monooxygenase activity of the parental enzymes.
Use of Bacterial Expressions Systems with Human P450s and GSH Transferases (F.P.G., A.P.)
GSH transferases have been readily expressed in heterologous systems and even used to facilitate expression of other proteins as fusion constructs in bacteria. The expression of mammalian P450s was more difficult. In 1990–1991, several groups were successful in expressing P450s at high levels in E. coli after modifying the N-terminal (5′) sequence and applying several technological modifications (1, 3, 12). These basic approaches have now been applied to >30 P450s, as reviewed elsewhere (19, 20). In this laboratory, human P450s 1A1, 1A2, 2C9, 2D6, 2E1, 3A4, 3A5, and 3A7 have all been expressed in this manner. Most of these P450s can be purified by detergent solubilization and conventional ion exchange chromatography. With P450 2D6, a flavodoxin affinity method was used (21). Interestingly, when the classic MALLLAVFL… N-terminal sequence is used, the N-formylMet is retained, probably due to membrane sequestration (22).
The purified P450s can be used to reconstitute catalytic activities when mixed with NADPH-P450 reductase (with both recombinant proteins purified from E. coli). In general, the reaction rates measured in these systems are similar to those in human liver microsomes when expressed on an nmol P450 basis (vide supra). Comparisons have been made among a number of recombinant systems involving P450 3A4, including baculovirus microsomes, liver microsomes, and the reconstituted systems.2 The kcat andKm parameters vary among these systems in a manner that is not always predictable, and the use of these parameters in modeling experiments must be done with caution. It is of interest that the reconstituted P450 3A4 system can be frozen, stored, and thawed many times without loss of activity, even in the absence of glycerol.
Bacteria are normally devoid of NADPH-P450 reductase, and three different approaches have been considered for the purpose of developing bacteria with in vivo oxidation capability. The first involves overexpression of flavodoxin, a bacterial protein that can bind and transfer electrons to mammalian P450s, albeit slowly (1, 23,24). The second approach is to express fusion proteins containing a P450 domain covalently linked to a modified NADPH-P450 reductase domain, as originally developed by Ohkawa and co-workers (25) in yeast and then Estabrook and colleagues in bacteria (18). Those with P450s 1A1, 1A2, and 3A4 have been prepared and purified in this laboratory (26-28). The third approach involves bicistronic expression. Atac/tac promoter is used to produce a single RNA containing both the P450 and reductase messages, each with an independent site for ribosome binding. The messages are separated by a short oligonucleotide gap (∼30 base pairs). Such systems yield P450 and NADPH-P450 reductase proteins in ∼1:1 ratio. Human P450s 1A1, 1A2, 2C9, 2D6, 2E1, 3A4, 3A5, and 3A7 have been expressed with NADPH-P450 reductase in such systems (29). The expected marker catalytic activities are observed in both E. coli membranes and whole cells (fig. 4).

A, phenacetin O-deethylation by bicistronic E. coli constructs containing human P450 1A1 or 1A2 plus human NADPH-P450 reductase. Assays were done with 25–100 pmol of P450 using whole cells or membranes. B, chlorzoxazone 6-hydroxylation by bicistronic E. coli (cells or membranes) containing human P450 2E1 and NADPH-P450 reductase. ctrl indicates cells transformed with the empty (null) pCW expression vector.
An expression vector developed for E. coli was used to express rat GSH transferase 5-5 in S. typhimurium TA1535, one of the base pair mutation-sensitive systems used in the Ames test (30, 31). The expression level was relatively high, and the modified bacteria showed mutations when exposed to several alkyl dihalides (fig.5A). Similar results were obtained when human GSH transferase T1-1, an apparent ortholog of the rat 5-5 enzyme, was expressed in S. typhimurium TA1535 (34). In these situations, the enzyme reaction products react with bacterial DNA, and some of the adducts result in a forward mutation in thehis operon, which allows growth in the absence of histidine [it has also been possible to directly express a human P450, 1A2, inS. typhimurium TA1538 and activate promutagens to reactive species (35), apparently utilizing electron transfer from flavodoxin].
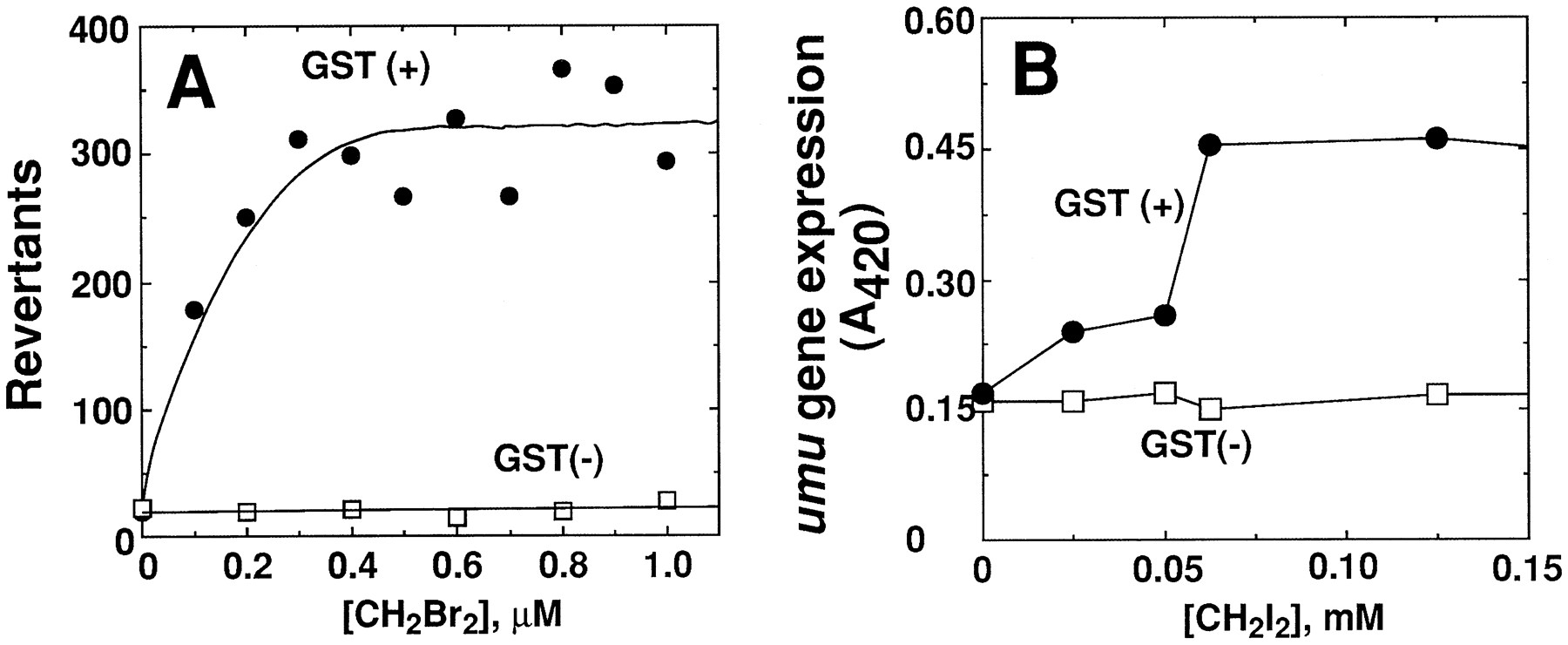
Another approach involves the detection of bacterial DNA damage by reactive chemicals with the SOS system, in which a cascade of events results in the activation of a chimeric plasmid containing aumu gene promoter and a lacZ reporter (which produces β-galactosidase) (36). Rat GSH transferase 5-5 has been inserted into such a system, and the system is sensitive to some dialkylhalides (32, 33) (fig. 5B). In principle, this system should also prove useful for human GSH transferases and P450s.
Expression and Characterization of UGTs (C.P.S., M.P.M., R.H.T.)
The UGTs are a family of proteins that plays a key role in the inactivation and elimination of many endogenous and exogenous agents, such as the catabolic heme byproduct bilirubin, drugs, dietary constituents, and potential carcinogenic compounds. Based upon sequence alignments of predicted amino acid sequence from cloned RNAs, the UGTs are classified into two families that have been designated UGT1 and UGT2 (37). The human UGT1A gene locus has been mapped to human chromosome 2 (38), where it has been predicted that at least nine functional proteins may be encoded. Transcription and RNA processing proceeds by a process of exon sharing, where each transferase is transcribed by a unique first exon, encoding approximately 285 amino acids, and a carboxyl-terminal portion of 245 amino acids, which is encoded by exons 2–5 and is identical for all of the UGT1s. In contrast, the human UGT2B proteins are encoded by individual structural genes on chromosome 4 (39).
The biophysical and biochemical similarity of this large protein family has confounded studies utilizing protein preparations from human tissues and characterizations based on substrate specificity analyses. However, cloning of individual UGT cDNAs and recombinant protein expression in eukaryotic cell systems have enabled investigation of the specific substrate specificities of the different UGT proteins. Although significant information has been obtained through transient and stable expression in mammalian tissue culture cells (40, 41), this laboratory has pursued the utilization of baculovirus and insectSpodoptera frugiperda (Sf9) cells for UGT expression (42). Although the expression of UGTs by transient transfection into COS-1 cells is effective, the insect-derived UGT proteins showed nearly 100-fold greater protein accumulation, with recombinant protein yields of up to 5–10% of total cellular protein. The lower amounts of recombinant protein expression in COS-1 cell extracts are reflected in 10–20-fold lower Vmax values when compared with baculovirus-generated extracts, but theKm values for specific substrates are independent of the expression system. This would suggest that posttranslational N-glycosylation and integration into the endoplasmic reticulum of SF9 cells have little impact on functional properties of the UGTs. These results indicate that recombinant eukaryotic expression of UGTs in baculovirus serves as an efficient means to examine the substrate specificity of these proteins.
The utilization of recombinant UGT1A proteins in baculovirus has recently been implemented as a diagnostic tool in human B-cell autoimmunity (43, 44). Chronic hepatitis D virus infection and type 2 autoimmune hepatitis have been found to be associated with LKM-3 autoantibodies directed toward UGT1A proteins. Baculovirus-generated recombinant human UGT1A1, human UGT1A6, and rabbit UGT1A6 have been utilized in immunoblot and enzyme-linked immunosorbent assays for the detection and characterization of autoantibodies. The human autoimmune response targeting UGT1A proteins recognizes different epitopes in idiopathic autoimmune hepatitis vs. virus-associated autoimmunity. This work provides evidence for a differential immunological response in these clinically distinct disease entities. In addition, the detection of anti-UGT1A autoantibodies in hepatitis D virus infection may indicate a more severe course of the disease and may represent a significant diagnostic tool.
Hepatic glucuronidation has been extensively studied and characterized. However, the precise extent of UGT1A expression in hepatic and extrahepatic tissues has not been established. Physiological considerations would suggest that organ systems with an extensive exterior surface such as the gastrointestinal and respiratory systems might have specific glucuronidation requirements as a metabolic line of defense against dietary and environmental compounds. The implementation of a quantitative duplex reverse transcription polymerase chain reaction (“D-RT-PCR”) assay has provided the first evidence for tissue-specific regulation of the UGT1A locus in hepatocellularvs. biliary tissue (45). When the entire locus was analyzed for expression in hepatic and biliary tissue, biliary tissue not only displayed lower overall levels of UGT1A mRNA expression but also did not express UGT1A9, a protein found in liver epithelium. In contrast, biliary epithelium expresses UGT1A10, a transcript that has not yet been identified in human tissues (table2). Both tissues express UGT1A1, UGT1A3, UGT1A4, and UGT1A6. Cloning and sequence analysis of the novel biliary-derived UGT1A10 cDNA indicates that this protein lacks the 5′ signal sequence portion present in other characterized UGT sequences, which may be significant in light of its extrahepatic expression. The identification of extrahepatic members of the UGT1A subfamily of proteins, their expression in eukaryotic expression systems, and subsequent characterization have helped to understand the complex physiology of human glucuronidation.
Identification of UGT1 RNA transcripts in human liver and biliary epithelial tissues 2-a
Heterologous Expression of Human UDP-Glucuronosyltransferases in Mammalian Cells and E. coli (B.B., M.P., S.F-G.)
Human UGTs catalyze the conjugation of a wide variety of endobiotics and xenobiotics, generally resulting in a less toxic, more polar product, thereby facilitating transport excretion and detoxication, as pointed out above (46). This family of enzymes has proved extremely difficult to purify even from some rare high quality tissue. Thus, characterization of these enzymes was not possible without the advent of cDNA cloning and expression technologies (47). Subsequently, it has become apparent that these expression systems allow examination of the specificity of xenobiotic glucuronidation, indicate tissue specific metabolism in vivo, and provide specific antigens for antibody production and protein for structure-function analysis by NMR or even crystallization work. Expression of human UGTs in bacteria should provide the pharmaceutical firms with a reagent for drug metabolism studies directly relevant to man.
Twenty human UGT cDNAs have been cloned and sequenced (37). An updated nomenclature based on protein sequence similarity has appeared in 19973. Biochemical studies of UGT proteins and computer-aided analysis of protein sequences have suggested a similar architecture for all known UGTs, where the mature protein has the N-terminal signal sequence removed, leaving the majority of the protein inside the lumen of the endoplasmic reticulum attached to the membrane by a C-terminal transmembrane sequence and a multi-lysine stop transfer signal (48).
Heterologous expression of human UGTs in COS-7 monkey kidney cells was achieved almost 10 years ago (49), and subsequently several enzymes have been stably expressed in V79 and HEK293 cells (47). These enzymes have been extensively assayed using more than 100 substrates (47). Unfortunately, they have not been consistently assayed under optimal conditions with the same range of substrates to determineKm and Vmaxvalues (with Km probably being the most important determinant of specificity in expression systems), where levels of UGT expression and hence Vmaxwill be variable (47). Nonetheless, this substantial body of work does provide indications of probe substrates for seven human UGTs as shown in tables 2 and 3. This knowledge and these reagents provide a basis for investigation of the glucuronidation of novel drugs and their potential in drug-drug or drug endobiotic interactions. This work can be done in whole cells in tissue culture where drug(s) are added to the culture medium and glucuronides are assayed from the medium (50). Incubations can be continued for 48 hr with poorly glucuronidated drugs such as non-steroidal antiinflammatory drugs. Further, the potential toxicity of carboxylic acid drugs through acyl migration may be indicated by protein binding within tissue culture cells (51).
Probe substrates for human UGTs
Multiple UGTs may catalyze the glucuronidation of a single substrate in expressed systems assayed in vitro. For example, 1-naphthol glucuronidation may be catalyzed by several UGTs, although kinetic analysis has suggested that the primary UGT responsible for this reaction is UGT1A6. However, it is important to assess the contribution of each enzyme to substrate metabolism by human tissue microsomes. No specific inhibitors of UGTs are known; therefore, specific antibodies are required to quantitate each UGT in human tissue microsomes. Fortunately, these specific polyclonal antibodies can be prepared from heterologously expressed and purified UGT antigens. Expression of specific peptide fragments overcomes the problems encountered due to the high sequence similarity of UGT family members (52, 53). The specific inhibitory antibodies obtained can be combined to titrate tissue microsomal preparations to elucidate catalytic selectivity. This type of analysis has shown that UGT1A6 is only responsible for approximately 50% of 1-naphthol glucuronidation in liver and kidney microsomes (52, 53), whereas UGT2B4 is the only UGT catalyzing 6-O-glucuronidation of hyodeoxycholic acid (54). These studies have to be completed with all the major UGTs.
Heterologous expression of UGTs provides the opportunity to study structure and function of UGTs. Comparative sequence inspection allows intuitive identification of potential key functional residues in the enzymes. Work with site-specific inhibitors such as diethylpyrocarbonate (reacts with His) and 2,3-butanedione (modifies Arg) helps to confirm suggested functional residues. Then the human UGT can be modified by site-directed mutagenesis and expressed in cell cultures. Kinetic analysis of expressed mutants will provide information about active site structure. Work done to date has suggested key residues involved in function architecture but not catalysis (53).
Expression of larger amounts of His-tagged protein in bacteria or baculovirus (see Strassburg, Manns, and Tukey, vide supra) facilitates protein purification for NMR or crystallization studies, although crystallization of a membrane-bound UGT will be a formidable task. Expression of human UGTs in E. coli provides the simplest reagent for drug metabolism studies without the need for tissue culture facilities. The successful expression of the human liver UGT isoforms UGT1A6 and 2B4 in E. coli for the production of specific anti-UGT antibodies and for the expression of functional proteins has been described (52-54). The human liver UGT1A6 was expressed as a membrane-bound enzyme in a pET-derived expression system (55). Modification of the two first codons of the UGT cDNA to decrease secondary structure of the expressed mRNA and replacement of the mammalian signal peptide of UGT1A6 by the bacterial OmpA or PelB leader sequence strongly increased the level of expression. These results were confirmed by recent work performed in this laboratory allowing the functional expression of PelB-UGT1A6 constructed in the vector pCW inE. coli JM109.4Bacterial growth at 27°C produced the highest level of functionally expressed UGT1A6 exhibiting activity toward 4-methylumbelliferone (1.2 mmol/min/mg protein). The His-tagged product was partially purified on a Ni-agarose column for use in antibody production.4 This preliminary work has provided the impetus for further development of these systems as an expression system for drug metabolism and structural studies.
Footnotes
-
Send reprint requests to: Prof. F. Peter Guengerich, Department of Biochemistry and Center in Molecular Toxicology, Vanderbilt University School of Medicine, Nashville, TN 37232-0146.
-
↵4 M. Pritchard, unpublished results.
-
This work was supported in part by United States Public Health Service Grants CA44353, ES000267 (F.P.G.), GM07347 (A.P.), GM31001 (E.F.J.), GM 49135 (R.H.T.), and The Wellcome Trust and BBSRC (B.B.).
-
↵3 P.I. Mackenzie, I.S. Owens, B. Burchell, K. W. Bock, A. Bairoch, A. Belanger, S. Fournelgigleux, M. Green, D. W. Hum, T. Lyanagi, D. Lancet, P. Louisot, J. Magdalou, J. R. Chowdhury, J. K. Ritter, H. Schachter, T. R. Tephly, K. F. Tipton, and D. W. Nebert: The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence.Pharmacogenetics 7, 255–269 (1997).
-
↵2 P. M. Shaw, N. A. Hosea, D. V. Thompson, J. M. Lenius, and F. P. Guengerich: Reconstitution premixes for assays using recombinant human cytochrome P450, NADPH-cytochrome P450 reductase and cytochromeb5. Arch. Biochem. Biophy. (in press).
- Abbreviations used are::
- P450
- cytochrome P450
- GSH
- glutathione
- UGT
- UDP-glucuronosyltransferase
- Received May 27, 1997.
- Accepted July 17, 1997.
- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Expression of Mammalian Microsomal Cytochromes P450 and Their Redox Partner, NADPH-P450 Reductase, in Escherichia coli (E.F.J., T.H.R., C.v.W., J.C., F.J.)
- Use of Bacterial Expressions Systems with Human P450s and GSH Transferases (F.P.G., A.P.)
- Expression and Characterization of UGTs (C.P.S., M.P.M., R.H.T.)
- Heterologous Expression of Human UDP-Glucuronosyltransferases in Mammalian Cells and E. coli (B.B., M.P., S.F-G.)
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters