


Abstract
Tegaserod is a selective 5-HT4 receptor partial agonist with promotile activity in the gastrointestinal tract. This study was designed to describe the metabolic pathways of tegaserod in the human liver and small intestine in vitro, to identify the enzymes involved in tegaserod metabolism, and to investigate the effect of tegaserod on CYP-catalyzed reactions involving other compounds. Tegaserod was metabolized in human liver microsomes to O-desmethyl tegaserod at a low rate. This metabolite was also formed by cDNA expressed CYP2D6, and the reaction in human liver microsomes was inhibited by quinidine. In human liver slices, directN-glucuronidation of tegaserod at the guanidine nitrogens (M43.2, M43.8, and M45.3) was found, with M43.8 being the major metabolite. Human small intestine slices also metabolized tegaserod to the N-glucuronides, suggesting a contribution of the small intestine to the presystemic metabolism. 5-Methoxyindole-3-carboxylic acid (M29.0), the main metabolite in human plasma, was generated in vitro by a sequence of reactions starting with nonenzymatic acid-catalyzed hydrolysis, followed by enzymatic oxidation and conjugation with glucuronic acid. Tegaserod inhibited CYP2C8, CYP2C9, CYP2C19, CYP2E1, and CYP3A only to a small extent with IC50 values >30 μM. Tegaserod more effectively inhibited CYP1A2 and CYP2D6 with Ki values of 0.84 and 0.85 μM, respectively. However, these Kivalues are approximately 140-fold greater than the maximal tegaserod plasma concentrations following the clinically relevant 6-mg oral dose given to healthy volunteers. M29.0, the main circulating metabolite, did not demonstrate any inhibitory potential toward cytochrome P450 enzymes in vitro. Therefore, clinically relevant metabolic drug interactions with tegaserod seem unlikely.
Tegaserod is a novel selective 5-HT41receptor partial agonist (Buchheit et al., 1995a,b). Tegaserod exhibits a promotile effect throughout the gastrointestinal (GI) tract (Pfannkuche et al., 1995; Nguyen et al., 1997; Grider et al., 1998;Fioramonti et al., 1998); it modulates the sensitivity of rectal spinal afferents (Schikowski et al., 1999) and chloride/water flux (Stoner et al., 1999), indicating a role of 5-HT4 receptors in motor, sensory and secretory processes along the intestine. These properties make tegaserod a potential therapeutic agent for functional GI disorders, such as irritable bowel syndrome (IBS). Clinical studies have shown that tegaserod accelerates small bowel transit and improves overall GI symptoms in patients with constipation-predominant IBS (Lefkowitz et al., 1999; Prather et al., 2000; Müller-Lissner et al., 2001) and reduces esophageal acid exposure in gastro-esophageal reflux disease (Kahrilas et al., 2000).
Besides the GI tract, the 5-HT4 receptors are also found in other peripheral organs, including the heart (Bockaert et al., 1997). Some current prokinetic agents, notably cisapride, have been associated with a risk of cardiac arrhythmias (Bran et al., 1995; Wysowski and Bacsanyi, 1996; Carlsson et al., 1997); however, experimental evidence indicates that this is not related to 5-HT4 receptor activation (Tonini et al., 1999). The risk of proarrhythmia is further increased when cisapride is coadministered with drugs inhibiting cytochrome P450(CYP)3A4, such as ketoconazole, itraconazole, miconazole, troleandomycin, erythromycin, fluconazole, clarithromycin and ritonavir (Farrington, 1996; Bedford and Rowbotham, 1996; Gray, 1998). CYP3A is the main enzyme involved in cisapride clearance, whereas data on the enzymes responsible for tegaserod metabolism have not been published yet. Tegaserod does not prolong QT intervals and is not associated with major adverse reactions at therapeutic doses (Drici et al., 1999; Appel et al., 1997). Nevertheless, some patients with functional GI disorders may be taking multiple medications for comorbid illnesses. Therefore, an investigation of potential drug interactions of tegaserod with other drug classes is warranted. Before such interaction studies in humans were initiated, this study was undertaken to identify the metabolic pathways and the enzymes involved in the metabolism of tegaserod using human tissue slices and subcellular fractions, as well as individually expressed enzymes. Furthermore, the potential effect of tegaserod on the metabolism of potentially coadministered compounds was studied using CYP-isoenzyme-specific substrates.
Materials and Methods
Chemicals.
Tegaserod and [14C]tegaserod (58.7 mCi/mmol) [3-(5-methoxy-1H-indol-3-yl[14C]methylene)-N-pentylcarbazimidamide hydrogen maleate] were prepared by the Preclinical Research Department and the Synthetic Tracer Laboratories of Novartis Pharma Ltd., respectively (Basel, Switzerland). The radiolabeled compound was determined to be 98% pure by means of HPLC. 5-Methoxyindole-3-carboxylic acid glucuronide (M29.0) was isolated from human urine (R. Dannecker, unpublished). [3H]Cyclosporine A (CSA) (10.1 Ci/mmol),S-[14C]mephenytoin (60 mCi/mmol), [14C]tolbutamide (54 mCi/mmol), and [14C]chlorzoxazone (59 mCi/mmol) were obtained from Amersham Pharmacia Biotech UK, Ltd. (Little Chalfont, UK). [14C]Phenacetin (12.3 mCi/mmol), dextromethorphan hydrobromide, chlorzoxazone, tolbutamide, and terfenadine were purchased from Sigma (St. Louis, MO). Dextrorphan tartrate monohydrate and fluoxetine hydrochloride were generous gifts from Hoffmann-La Roche (Basel, Switzerland) and from Eli Lilly (Indianapolis, IN), respectively. (±)Bufuralol hydrochloride, (±)hydroxybufuralol maleate, (±)4-hydroxymephenytoin, and 4-hydroxytolbutamide were obtained from Ultrafine Chemicals (Manchester, UK). [14C]Theophylline (52 mCi/mmol) was obtained from American Radiolabeled chemicals (St. Louis, MO), and [3H]paclitaxel (12.7 Ci/mmol) was purchased from Moravek Biochemicals (Brea, CA). [3H]Glyburide (51 Ci/mmol) and [14C]estradiol (54.1 mCi/mmol) were obtained from DuPont de Nemours (Brussels, Belgium). The radiochemical purity was 97% or greater for all chemicals as specified by the manufacturer. Tissue culture media components were obtained from Invitrogen (Carlsbad, CA). All other reagents were purchased from commercial sources and were of the highest purity grade available.
Biologicals.
Human liver microsomes HHM-0011 (895 pmol of CYP/mg of protein) and EIX 345-06 (550 pmol of CYP/mg of protein) were obtained from the International Institute for the Advancement of Medicine (Exton, PA) and from Human Biologics, Inc. (Phoenix, AZ), respectively. Human liver tissue, perfused with Belzer's University of Wisconsin solution and not suitable for transplantation, and human intestinal tissue was obtained through the Association of Human Tissue Users (Tucson, AZ). Microsomes or S9 fractions were prepared by differential centrifugation, as described previously (Ball et al., 1992). Microsomal protein was determined by the method of Bradford using bovine immunoglobulin as the standard (Bio-Rad, Glattbrugg, Switzerland). To determine the protein in the intestinal slices, the detergent 0.1% CHAPS was added to aid in the disruption of the mucosal tissue. CYP content was determined by the method of Omura and Sato (1964). The spectral CYP content (pmol of CYP/mg of microsomal protein) of these preparations were: HL43 (180), HL44 (230), GGM-002 (440), and M8 (290). Human livers HL569-1 and HL586-1, as well as human small intestine HI572-3, were used for tissue slice preparations.
Recombinant Chinese hamster ovary cells with stable expression of active CYP2D6 and CYP3A4 (108 and 49 pmol of CYP/mg of microsomal protein, respectively) were established as previously described (Fischer et al., 1998).
Organ Slices.
Tissue cores (8 mm in diameter) and slices (200 ± 25 μm in thickness) were prepared in cold Sacks preservation solution according to the method previously described using a Vitron tissue slicer (Vickers et al., 1995). The slices were placed onto roller culture inserts and maintained at 37°C in Waymouth's medium without phenol red and supplemented with 10 ml/l FungiBact solution, and 10% fetal calf serum. The viability of the human liver slices was assessed by determining the intracellular K+ content and the extracellular lactate dehydrogenase release in vehicle control, 0.1% dimethyl sulfoxide (DMSO), and tegaserod-exposed slice incubates. For human small intestine, the slice viability was assessed by the determination of the protein synthesis in vehicle control and treated slice incubates. The tegaserod concentrations used in this study had no effect on the viability of the slices (data not shown). Slice CYP metabolic functionality was confirmed with cyclosporine A (1 μM) biotransformation, which was 0.7 nmol/h/g of liver tissue.
Biotransformation.
Microsomal incubations were performed at 37°C in 0.1 M phosphate buffer pH 7.4, containing 0.2 mM β-NADPH and β-NADPH-regenerating system (final concentrations of 1 mM NADP+, 5 mM isocitrate, 5 mM MgCl2, and 1 U of isocitrate dehydrogenase). The incubations with estradiol as substrate were performed in the presence of 1 mM ascorbic acid. The compounds were added in DMSO, methanol, polyethylene glycol 400 or water with final vehicle concentrations not to exceed 1%. Additional typical incubation conditions (i.e., substrate concentrations, incubation time, and microsomal protein content, respectively) were as follows: [14C]tegaserod, 10 μM, 4 h, 2 mg/ml; [3H]CSA, 1 μM, 15 min, 250 μg/ml; dextromethorphan, 5 μM, 30 min, 100 μg/ml; bufuralol, 5 μM, 60 min, 200 μg/ml; [14C]phenacetin, 20 μM, 30 min, 200 μg/ml; [14C]theophylline, 50 μM, 60 min, 2 mg/ml; [14C]chlorzoxazone, 40 μM, 20 min, 100 μg/ml; [14C]tolbutamide, 150 μM, 40 min, 200 μg/ml; [3H]paclitaxel, 10 μM, 20 min, 250 μg/ml;S-[14C]mephenytoin, 100 μM, 30 min, 1 mg/ml; [3H]glyburide, 5 μM, 40 min, 200 μg/ml; terfenadine, 20 μM, 30 min, 2 mg/ml; fluoxetine, 5, 10 and 50 μM, 60 min, 2 mg/ml and [14C]estradiol, 5 μM, 20 min, 500 μg/ml. Inhibitor concentrations were: tegaserod or M29.0 (1–200 μM), triacetyloleandomycin (10 μM), quinidine (10 μM), and ketoconazole (1 μM). The reactions were stopped with an equal volume of cold methanol, 70% perchloric acid or 10% trichloroacetic acid, with cooling on dry ice. Control incubations were performed in the absence of β-NADPH and the regenerating system or in the absence of microsomal protein. The incubation medium was separated from the denatured protein by centrifugation at 100,000g for 10 min using a Beckman TL-100 ultracentrifuge (Nyon, Switzerland). Aliquots of the supernatant were then applied directly onto the column for HPLC analysis.
Metabolism in precision-cut tissue slices was investigated following a preincubation period of 90 min in the absence of a substrate. Thereafter fresh media containing 1 or 5 μM [14C]tegaserod (0.06 and 0.3 μCi/ml medium) in DMSO (0.1%, final concentration) was added. At each time point, the slice and medium were transferred to separate vials. For mass balance of radioactivity the roller culture vial and insert were washed with methanol. All samples were stored at −80°C until analysis. Tegaserod was stable under the experimental conditions in the absence of the slice.
The human liver slices were disrupted in Eppendorf tubes containing phosphate-buffered saline (300 μl) by homogenization with a Teflon pestle, followed by brief sonication with a micro-ultrasonic cell disrupter (Kontes, Vineland, NJ) on ice. Aliquots (5 μl) of the slice homogenate, medium, and methanol wash fractions were taken for radioactivity determination to assess the extraction procedure. The medium was concentrated by evaporation to a volume of 200 μl, and the methanol wash evaporated to dryness. All fractions were pooled and the protein was pelleted at 100,000g for 10 min at 20°C. The pellet was re-extracted with methanol, and the resultant supernatant was evaporated to dryness and combined with the pooled aqueous supernatant. The human intestine samples were prepared similarly for HPLC analysis except that the disruption of the slices was performed directly in methanol. The extraction recovery for the liver and intestine slices was complete.
HPLC Analysis.
HPLC separations were performed on Kontron systems controlled by a 450-MT2 data system (Kontron Instruments, Zürich, Switzerland) with on-line detection using either a fluorescence detector F1000 (Merck, Darmstadt, Germany) or an LB 507A radioactivity monitor (Berthold AG, Wildbad, Germany). All compounds and their metabolites were characterized by their retention times and/or by LC-MS analysis.
[14C]Tegaserod and its metabolites were analyzed at 40°C using two columns (20 × 4.6 mm and 150 × 4.6 mm) in series (5 μm particle size, LC-18, Supelco Inc., Bellefonte, PA). The mobile phase consisted of 10 mM ammonium acetate (pH 5.4; A) and acetonitrile (B), with a total flow rate of 1 ml/min. The proportion of B was 0% up to 10 min and then increased linearly to reach 10% at 30 min, 90% at 71 min and 100% at 90 min. Detection was by on-line radioactivity monitor with a scintillator flow of 3 ml/min.
Terfenadine and its metabolites were analyzed on Supelcosil LC-CN columns (5-μm particle size, Supelco) [i.e., a precolumn (20 × 4.6 mm) and an analytical column (250 × 4.6 mm)]. The mobile phases were 12 mM ammonium acetate buffer (solvent A) and acetonitrile (solvent B). The proportion of solvent B was 0% up to 3 min and was increased linearly to reach 50% at 5 min, 90% at 30 min, and 100% at 35 min. The samples were eluted at 35°C with a flow rate of 1 ml/min. Fluorescence detection was used with excitation and emission wavelengths of 230 and 280 nm, respectively.
[14C]Estradiol and fluoxetine and their metabolites were analyzed at room temperature on Supelcosil LC 18-DB columns (20 × 4.6 mm and 150 × 4.6 mm; 5-μm particle size). For estradiol, the mobile phases consisted of water containing 0.1% acetic acid (solvent A) and acetonitrile (solvent B). The proportion of solvent B was 20% during the first 5 min and was increased linearly to reach 22% at 20 min, 35% at 65 min, and 100% at 70 min. The total flow was 1 ml/min. On-line radioactivity detection was performed with a scintillator flow of 3 ml/min. For fluoxetine, the mobile phases consisted of 50 mM ammonium acetate buffer (solvent A) and acetonitrile (solvent B). The proportion of solvent B was 0% up to 2 min and was increased linearly to reach 40% at 27 min and maintained until 47 min, then increased to reach 100% at 60 min. The flow rate was 0.3 or 1 ml/min. A fluorescence detector with excitation and emission wavelengths set to 235 and 310 nm, respectively, was used.
[3H]CSA (Kronbach et al., 1988), dextromethorphan (Fischer et al., 1994), [14C]phenacetin, [14C]chlorzoxazone, [14C]tolbutamide, bufuralol, [14C]theophylline, [3H]paclitaxel,S-[14C]mephenytoin, [3H]glyburide (Fischer et al., 1998), and their metabolites were analyzed as previously described.
Liquid Chromatography-Mass Spectrometry (LC-MS).
Metabolites of tegaserod in human liver slice incubates were characterized by LC-MS using a TSQ 700 triple-stage quadruple instrument (Finnigan MAT, San Jose, CA), equipped with an electrospray LC-MS interface. The samples were prepared and metabolites were separated chromatographically essentially as described above. After the column, the total flow was split into two parts. Approximately 0.75 ml/min was passed into a radioactivity monitor. The remaining 0.25 ml/min was combined with 0.1 ml/min of acetonitrile and then directed into the electrospray interface. The latter was operated with methanol as sheath liquid (0.1 ml/min) and nitrogen as sheath gas (45 psi) and as auxiliary gas (5 flowmeter units). The spray voltage was 4.5 kV and the transfer capillary was heated to 240°C. Single-stage mass spectra were taken at unit mass resolution by using the first quadrupole as mass analyzer. Fragmentation in the ion source region was induced by applying an up-front collision offset voltage of 30 V between the skimmer and the transfer-octapole.
LC-MS and/or liquid chromatography-tandem mass spectrometry was also used to confirm the assignment of known metabolites of terfenadine and fluoxetine. The HPLC conditions in these runs were as described above for the respective compounds. The mass spectrometric instrumentation and the operating conditions of the electrospray LC-MS interface were similar to those used for characterizing the tegaserod metabolites.
Data Analysis.
IC50 values were determined graphically by plotting the percentage of the control activity against the inhibitor concentration. Michaelis-Menten parametersKm, Vmax, and standard errors were determined by nonlinear curve fitting using Fig.P (BIOSOFT, Cambridge, UK) with the following equation: V= Vmax × [S]/(Km + [S]).
Ki values were calculated using Enzpack 3 (BIOSOFT, Cambridge, UK) with the following equation (Segel, 1993):Ki = [I]/[(Km,i/Km,u)(Vmax,u/Vmax,i) − 1], where Km,i andKm,u are the Michaelis-Menten constants andVmax,i andVmax,u are the maximal velocities in the presence and absence of inhibitor, respectively.
Metabolic rates were extrapolated to a human subject using an adult body weight of 70 kg using a liver weight of 1.69 kg and a yield of 52.5 mg of microsomal protein from 1 g of human liver (Iwatsubo et al., 1997). The intestinal weight was estimated to 600 g (Rietsch and Belocq, private communication).
Results
Biotransformation Pathways.
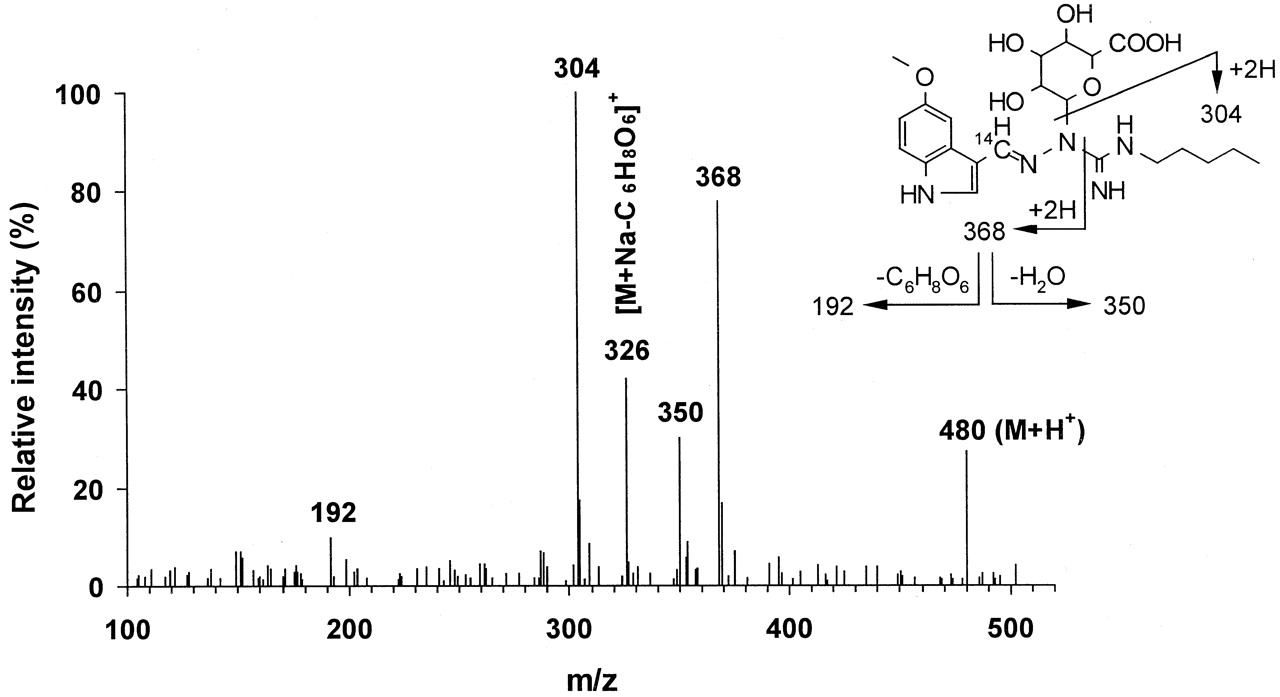
Human liver microsomes, human liver slices and human small intestine slices were incubated with [14C]tegaserod (1–10 μM) and the metabolite profiles of tegaserod were defined by HPLC (Fig. 1). O-Desmethyl tegaserod (M52.8) was the main metabolite in all four human liver microsomal preparations. This metabolite was identified based on its identical retention time by HPLC with synthetic reference material (Fig. 2). The O-demethylation pathway was inhibited in the presence of quinidine (10 μM) but not with ketoconazole (1 μM) nor following preincubations with triacetyloleandomycin (10 μM) (data not shown).O-Desmethyl tegaserod was also formed in microsomes of recombinant Chinese hamster ovary cells expressing active human CYP2D6 but not in those expressing CYP3A4. O-Desmethyl tegaserod was neither formed in human liver slice incubates nor human small intestinal slices. The major metabolites obtained from human liver slices and small intestine slices were characterized by LC-MS (Table1). M43.2, M43.8, and M45.3 were identified as N-glucuronides of tegaserod (Fig. 2). M43.2 and M43.8 coeluted in the radiochromatograms but were partially separated in the LC-MS runs. Due to the high degree of labeling (>90%14C), ions containing the label appeared in the spectra at two mass units higher than expected for the corresponding unlabeled metabolites. M43.2 was found to be glucuronidated on the nitrogen atom carrying the pentyl substituent, as indicated by the fragment at m/z 264. The high relative intensity of this fragment ion, compared with the analogous fragment of the parent drug (m/z 88), indicates a preferred cleavage between the guanidino carbon atom and the nitrogen atom carrying the glucuronyl group. The site of glucuronidation in M43.8 was not directly revealed by the mass spectrum. However, the high relative intensity of the fragment ion at m/z 368 suggests that glucuronidation had taken place on the nitrogen atom of the guanidino group next to the indole ring (Fig.3), in analogy to the fragmentation behavior of M43.2. Metabolite M45.3 is proposed to be glucuronidated on the unsubstituted nitrogen atom of the guanidino group, as suggested by the fragment ion at m/z 287. The corresponding loss of glucuronic acid imine (or equivalent) would not be possible from other N-glucuronides, except after complex rearrangements. All three metabolites underwent loss of anhydroglucuronic acid (176 mass units) from M + H+, as expected of glucuronides, forming the protonated aglycone at m/z 304. The major metabolite formed by both liver and small intestine slices was tegaserod-glucuronide M43.8. While cytochrome P450-mediated reactions were dominant in human liver microsomal preparations, direct conjugation was the dominant metabolic pathway in tissue slices.

Typical radiochromatograms of tegaserod incubates with human tissue preparations.
Tegaserod metabolic profiles following incubations of [14C]tegaserod with human liver microsomes (HL44, 2 mg of protein/ml; 10 μM tegaserod; 4 h), a human liver slice (HL586-1, 5 μM tegaserod, 12 h) and a human small intestine slice (HI572-3, 1 μM tegaserod, 24 h).

Proposed metabolic pathways of tegaserod in human in vitro systems.
Mass spectrometric data on tegaserod and its metabolites from LC-MS analyses of human liver slice incubates, electrospray ionization in positive ion mode, up-front collision offset 30 V

Electrospray ionization mass spectrum of one of the tegaserod N-glucuronides (M43.8) and proposed interpretation.
Electrospray ionization in positive ion mode, up-front collision offset 30V, background subtracted.
Product formation rates in liver slices were approximately linear with concentration. The 4-h rates were 3.2 nmol/h/g of tissue for the 1 μM and 14.1 nmol/h/g of tissue for the 5 μM substrate concentrations (Table 2). Rates of metabolism were similar for both human liver slice preparations and decreased in prolonged incubations. However, a distinction between the role of substrate depletion versus enzyme degradation was not established in this study. In human liver microsomes at a higher 10 μM substrate concentration the rate was 0.8 nmol/h/g of liver tissue when extrapolated to the intact liver, as described under Materials and Methods.
Metabolite formation from tergaserod in incubations with human tissue preparations
Human small intestine slices metabolized tegaserod at a somewhat slower rate compared with the liver [i.e., over a 24-h period 1.1 or 4.9 nmol/h/g of tissue were metabolized at 1 or 5 μM tegaserod, respectively (Table 2)]. It is estimated that the capacity of the liver (1690 g) to metabolize tegaserod is ∼3.5- to 5-fold the capacity of the small intestine (600 g).
Stability at Gastric pH.
The predominant metabolite found in human plasma after oral dosing of tegaserod is M29.0 (R. Dannecker, unpublished results). This metabolite was not detected in either human liver or intestinal preparations. The metabolic pathway, most likely associated with low gastric pH, involves the hydrolytic cleavage of the imine bond of tegaserod. Incubations of 50 μM tegaserod at 37°C and pH 2 for 1 h resulted in about 60% of tegaserod degradation. The primary degradation product (not characterized; most likely 5-methoxyindole-3-carboxaldehyde), was converted by human liver S9 fractions to 5-methoxyindole-3-carboxylic acid, as identified by cochromatography with synthetic reference material. The aldehyde oxidase inhibitor isovanillin inhibited this reaction, indicating that the acid degradation product is the aldehyde. Finally, 5-methoxyindole-3-carboxylic acid formed the glucuronic acid conjugate in the presence of human liver microsomes and uridine 5′-diphosphoglucuronic acid cofactor. Therefore, when tegaserod is administered orally, its hydrolysis in the stomach appears to be a significant presystemic degradation process.
Effects of Tegaserod on Metabolism of Other Drugs.
The effect of tegaserod and its main metabolite in human plasma, M29.0, on the metabolism of 13 different compounds was studied at “inhibitor” concentrations up to 200 μM (Table3). The substrates were selected because they are agents used in common disease states and therefore could be coadministered in comorbid GI conditions. In addition, the selected agents are representative substrates of major CYP isoenzymes. M29.0 had no effect on the investigated metabolic reactions of any of the substrates tested. Tegaserod itself also had little or no effect on most of these probe reactions. The IC50 values for paclitaxel 6-α-hydroxylation (CYP2C8) (Rahman et al., 1994), tolbutamide 4-hydroxylation and glyburide cyclohexyl hydroxylation (CYP2C9) (Relling et al., 1990; Fischer et al., 1998),S-mephenytoin 4-hydroxylation (CYP2C19) (Goldstein et al., 1994), chlorzoxazone 6-hydroxylation (CYP2E1) (Peter et al., 1990), CSA metabolism (Kronbach et al., 1988), p-hydroxyphenyl C3′ paclitaxel formation (Cresteil et al., 1994), norfluoxetine formation (Stevens and Wrighton, 1993), terfenadine metabolism (Yun et al., 1993) and glyburide phenylethyl hydroxylation (CYP3A) (Fischer et al., 1998) were all >30 μM. The decrease in IC50 values for norfluoxetine formation at lower substrate concentrations can be explained: at clinical doses norfluoxetine formation correlated with the phenotype for debrisoquine metabolizer status (Hamelin et al., 1996) suggesting CYP2D6 is involved at lower concentrations. Tegaserod inhibited more specific CYP2D6-catalyzed reactions, such as dextromethorphan O-demethylation (IC50, <1 μM) and bufuralol 1′-hydroxylation (IC50, ∼1 μM) (Kronbach, 1991). The inhibition of CYP2D6-mediated reactions exhibited a strong competitive component, as shown for bufuralol 1′-hydroxylation (Ki = 0.85 ± 0.47 μM; Fig.4). The Vmaxfor bufuralol 1′-hydroxylation did not change in the presence of tegaserod although its apparent Km was increased. Tegaserod does not inhibit CYP3A mediated reactions. Hence, the observed inhibition of estradiol 2/4-hydroxylation by tegaserod must be due to inhibition of CYP1A2, since estradiol is metabolized by CYP1A2 and CYP3A (Aoyama et al., 1990). Typical CYP1A2 reactions, such as phenacetin O-deethylation (IC50, ∼4 μM) and theophylline metabolism (IC50, ∼8 μM) (Tassaneeyakul et al., 1993), were strongly inhibited by tegaserod. Inhibition of CYP1A2 was predominantly noncompetitive, the apparent Km for phenacetinO-deethylation was unchanged in the presence of tegaserod while the Vmax was decreased (Ki = 0.84 ± 0.47 μM; Fig.5).
Effect of tegaserod on the metabolism of characteristic CYP substrates and potentially coadministered compounds in human liver microsomes

Inhibition of bufuralol 1′-hydroxylation by tegaserod.
Bufuralol (2–40 μM) was incubated with human liver microsomes (M8) in the absence or presence of 0 to 5 μM tegaserod.

Inhibition of phenacetinO-deethylation by tegaserod.
Phenacetin (5–60 μM) was incubated with human liver microsomes (M8) in the absence or presence of 0 to 5 μM tegaserod.
Discussion
Tegaserod metabolism was initially characterized in human liver microsomes in the presence of NADPH and found to occur predominantly byO-demethylation. In human liver slices, however, products of direct glucuronidation at the guanidine nitrogens were identified by LC-MS while O-desmethyl tegaserod was not detected. This difference can be explained by a ∼40-fold difference in the rate of glucuronidation versus O-demethylation. This estimate assumes equal substrate concentrations at the active site in the two in vitro systems. If the rate comparison is made using CSA metabolism as a reference activity, it can be estimated that tegaserodO-demethylation is more than 3 orders of magnitude slower compared with direct glucuronidation in the intact tissue: the mean 20-h rate of tegaserod (1 μM) glucuronidation in human liver slices was 2-fold greater than the 24-h rate for the metabolism of CSA (1 μM) whereas tegaserod O-demethylation was about 1000-fold slower compared with CSA metabolism using the same human liver microsomes (Fischer et al., 1994). Glucuronide conjugation was found in both liver and small intestine although the metabolic capacity of the liver is estimated to be 3-fold greater than the capacity of the intestine. This suggests a limited contribution of theN-glucuronidation in the small intestine to the presystemic metabolism of tegaserod. The data are consistent with those from a study with oral administration of radiolabeled tegaserod in humans where direct conjugation with glucuronic acid was a major pathway andO-desmethyl tegaserod formation was not significant (R. Dannecker, personal communication).
In this radiolabeled human study, M29.0 was the most abundant tegaserod metabolite in plasma, formed by an initial hydrolysis followed by oxidation, probably by aldehyde oxidase, and glucuronidation. The absence of products from this pathway in both liver and intestinal tissue incubates suggests that the initial hydrolysis occurs exclusively in the stomach under the influence of gastric acid. These findings were confirmed in an additional study in healthy subjects. When tegaserod (12 mg/day) was given with either 6 μg/kg pentagastrin (to attain a gastric pH <2) or a combination of 40 mg of oral omeprazole and 30 ml 0.4 M NaHCO3 (to attain a gastric pH >3.5), plasma tegaserod concentrations were significantly reduced and M29.0 concentrations were increased after reducing stomach pH (Zhou et al., 2000a). Taken together, the data indicate that CYP mediated metabolism plays only an insignificant role in the elimination of tegaserod and inhibitors of CYP-mediated metabolism, such as the azole antifungals, antibiotics and antiviral agents, are not expected to affect tegaserod metabolism as they do the metabolism of cisapride. Most compounds do not change the gastric pH and can therefore also not be expected to alter the acid hydrolysis of tegaserod. The likelihood of tegaserod pharmacokinetics being affected by inhibitors of glucuronyl transferase is also small because these compounds do not typically reach high enough concentrations to effectively inhibit glucuronic acid conjugation in vivo (Hansten and Horn, 2000). If tegaserod concentrations should be increased however, the consequences should be minimal, because the safety profile of tegaserod is favorable and unlike the 5-HT3antagonist/5-HT4 agonist, cisapride, tegaserod does not prolong QTC intervals at therapeutic doses (Drici et al., 1999).
Both tegaserod and its major metabolite in human plasma, M29.0, were also investigated for their potential to inhibit the CYP-mediated metabolism of other compounds. Tegaserod most effectively inhibited CYP1A2 and CYP2D6 with Ki values of 0.84 and 0.85 μM, respectively. However, theseKi values are approximately 140-fold greater than the maximal tegaserod plasma concentrations following a single 6-mg oral dose (Zhou et al., 2001). Thus tegaserod coadministration does not affect the kinetics of the CYP1A2 substrate theophylline (Zhou et al., 2001), and the CYP2D6 substrate dextromethorphan (Kalbag et al., 2000). These substrates are sensitive for inhibition of CYP1A2 and CYP2D6, respectively, and thus it should be possible to extrapolate the lack of inhibition to other CYP enzymes. The narrow therapeutic index drug warfarin is metabolized by CYP2C9 and coadministration of tegaserod does not affect its pharmacokinetics (Ledford et al., 2000). Furthermore the efficacy of contraceptive prodrugs such as mestranol, which depend on activation via CYP2C9, should not be altered in the presence of tegaserod. Absence of CYP3A inhibition by tegaserod has been confirmed when tegaserod was coadministered with a triphasic oral contraceptive in healthy females. Tegaserod coadministration does not increase the risk of oral contraceptive failure (Zhou et al., 2000b). Most HMG-CoA reductase inhibitors are also metabolized by CYP3A and inhibition of their metabolism has been implicated in rhabdomyolysis (Sachse et al., 1998;Gruer et al., 1999). Tegaserod does not inhibit CYP3A and is therefore not expected to affect the metabolism of these commonly prescribed compounds.
Acknowledgments
We thank R. Nufer, U. Glänzel, B. Keller, and J. Molpeceres for technical assistance.
Footnotes
- Abbreviations used are::
- 5-HT4
- 5-hydroxytryptamine
- GI
- gastrointestinal
- IBS
- irritable bowel syndrome
- CYP
- cytochrome P450
- CSA
- cyclosporine A
- DMSO
- dimethyl sulfoxide
- LC-MS
- liquid chromatography-mass spectrometry
- Received April 17, 2001.
- Accepted June 14, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}