


Abstract
Liver microsomes, and more recently cryopreserved hepatocytes, are commonly used in the in vitro characterization of the metabolism of new xenobiotics. The flavin-containing monooxygenases (FMO) are a major nonP450 oxidase present in liver microsomes and hepatocytes. Since FMO is known to be thermally labile, and this enzyme may be involved in the metabolic clearance of some drugs, we sought to more completely characterize the metabolic competency of this enzyme in cryopreserved hepatocytes and in liver microsomes preincubated under various conditions using benzydamine as an in vitro and in vivo probe. The metabolism of benzydamine to its major metabolite, theN-oxide, is mediated by FMO3 in humans. We found that the in vitro microsomal t1/2 was 70% longer when incubations were prewarmed at 37°C in the absence of NADPH compared with prewarming in the presence of an NADPH-regenerating system, and N-oxide formation was inhibited >99%. Interestingly, the in vivo clearance predicted from these incubations and from human hepatocytes overpredicted the observed clearance of benzydamine in humans (>10.5 versus 2.4 ml/min/kg). In contrast, rat hepatocytes successfully predicted rat in vivo benzydamine clearance to within ∼30% (>68 versus 48 ml/min/kg). BenzydamineN-oxidation in liver microsomes from all common preclinical species demonstrated heat sensitivity. This information should be considered when extrapolating metabolism data of xenobiotics from these in vitro systems.
Drug metabolism studies have been continuously increased in relative importance in industry and have moved earlier in the drug discovery and development process (Ekins et al., 2000). Screening strategies early in lead optimization now typically incorporate high-throughput in vitro absorption, distribution, metabolism, and excretion (ADME1) measurements alongside potency determinations. This early screening has occurred due to the increased compound synthesis from combinatorial chemistry, the increase in knowledge and availability of techniques for studying a compound's ADME properties, and, most importantly, the degree of attrition from unacceptable pharmacokinetics of drug candidates in the latter, more expensive stages of drug development (Rodrigues, 1994; Eddershaw and Dickins, 1999; Eddershaw et al., 2000; Thompson, 2001). While the incorporation of ADME assays into a screening strategy allows the generation of large amounts of data on many compounds, a rational drug metabolism strategy that arises from a sound understanding of the relevant in vitro techniques is preferable over a “brute force” approach of data generation.
Liver microsomes, and more recently hepatocytes, from various species have been applied to the prediction of in vivo hepatic metabolic lability (Obach, 2001). The convenience of liver microsomes and hepatocytes exists in their ability to be cryopreserved until use. Since the cytochromes P450 (P450s) are responsible for the metabolism of the majority of xenobiotics, microsomes are often the in vitro system of choice. Alternatively, hepatocytes are being incorporated in place of or in addition to liver microsomes in ADME screens in drug discovery, as they are thought to include the full complement of oxidative and conjugative enzymes. However, information concerning the metabolic competency and/or in vitro–in vivo scaling of nonP450 enzymes from these in vitro systems is mostly lacking. Filling in this knowledge gap for nonP450 drug metabolism enzymes, such as UDP-glucuronosyltransferase (UGT), flavin-containing monooxygenase (FMO), and monoamine oxidase is required before in vitro drug metabolism techniques such as liver microsomes and hepatocytes can be used to their fullest capacities (Obach, 2001).
The FMOs are major mammalian nonP450 oxidative enzymes (Rettie et al., 1995), with the most abundant human liver isoform, FMO3, existing at levels similar to the major human liver P450 isoform, CYP3A4 (Haining et al., 1997). Human FMOs are characterized by heat lability in the absence of NADPH, and this property is often exploited to elucidate the enzyme involvement in a particular oxidation (Rettie et al., 1995). For example, preheating microsomes at 45 or 50°C for 1 to 3 min in the absence of NADPH is a common method for inhibiting FMO activity (Grothusen et al., 1996), and this inhibition is prevented in the presence of cofactor. Thus, prewarming of microsomes during a typical metabolic lability experiment in the absence of NADPH, which occurs when the incubation is initiated with cofactor, may have profound effects on the FMO activity and could potentially compromise the in vitro to in vivo scaling of FMO-mediated reactions. Additionally, given the known instability of FMOs, it remains to be seen whether enzyme activity survives hepatocyte cryopreservation or typical microsomal preincubation conditions.
Methods and Materials
Materials.
Cryopreserved hepatocytes from Sprague-Dawley rats (two lots) or from three individual humans were purchased from In Vitro Technologies (Baltimore, MD). Williams E medium was purchased from Invitrogen (Carlsbad, CA). Benzydamine hydrochloride was from Sigma-Aldrich (St. Louis, MO). Benzydamine N-oxide maleate was a generous gift from Dr. Allan Rettie (University of Washington). Midazolam was obtained from Ultrafine Ltd. (Manchester, UK). Liver microsomes from mouse, Sprague-Dawley rat, beagle dog, cynomolgous monkey, and a mixture of humans were prepared as previously described (Kalvass et al., 2001). HPLC grade water and acetonitrile were purchased from J. T. Baker (Phillipsburg, NJ).
Hepatocyte Incubations.
Incubations for metabolite identification were performed in suspension in a 2.5-ml total volume. Vials of hepatocytes were thawed rapidly in a water bath set at 37°C, then diluted with cold Williams E medium (pH 7.4) containing 10% fetal bovine serum (Lave et al., 1997). Cells were isolated by centrifugation and pooled and resuspended in cold Williams E medium buffer at 2 million viable cells/ml. Membrane integrity of the cells, measured by trypan blue exclusion, was ≥0.76. Benzydamine, dissolved in dimethylsulfoxide (DMSO) at 10 mM, was diluted in the flask incubation to a final concentration of 10 μM (final DMSO concentration = 0.1%). An initial aliquot was immediately removed and frozen as a time 0 sample. Cells were incubated at 37°C in a 95% air/5% CO2 atmosphere in a shaking water bath for 4 h.
For hepatocyte clearance determination, cells were prepared as described above except resuspension was at 0.5 million/ml. Benzydamine, dissolved in DMSO at 1 mM, was diluted to a final concentration of 1 μM. Flasks were incubated as described above, except 0.4 ml of aliquots were removed at 0, 30, 60, 120, and 240 min and immediately frozen. For rat hepatocyte incubations, clearance was especially rapid, and timepoints needed to be modified to 0, 2, 5, 10, and 15 min.
Microsome Incubations.
For microsomal clearance determination, 1 μM benzydamine (final organic concentration = 0.1%) was incubated in a 1.5-ml incubation containing 1.0 mM MgCl2, 100 mM potassium phosphate buffer (pH 7.4), 0.5 mM NADPH, and microsomal protein was added to keep total P450 concentration at 0.5 μM. Incubations were performed two ways; either preincubations were performed without NADPH for 5 min, and reaction was initiated by addition of cofactor, or preincubations were with NADPH regenerating system (0.54 mM NADP, 6.2 mM dl-isocitrate, 0.5 U/ml isocitrate dehydrogenase, 11 mM MgCl2) and without benzydamine, and initiation was via substrate addition. Incubations were performed at 37°C either in a shaking water bath or in a heating block. Aliquots (100 μl) were removed at 0, 5, 10, 15, 20, and 30 min and placed in 200 μl of acetonitrile containing 2 μg/ml midazolam as internal standard. Samples were vortexed, centrifuged to pellet insoluble material, and 200 μl was transferred to a 96-well plate. For cross-species heat lability experiments, incubations were performed as described above, except mixtures were preincubated with or without NADPH at 45°C for 3 min, chilled on ice for 1 min to reduce the temperature to ∼37°C, and then the complete mixture (benzydamine concentration was 200 μM) was incubated at 37°C for 10 min.
Metabolite Identification.
Frozen hepatocyte incubations for metabolite identification were thawed on ice and diluted with 2 volumes of acetonitrile. Samples were placed on ice for 30 min, centrifuged to pellet insoluble material, and the supernatant was evaporated under a flow of nitrogen. The residue was reconstituted in 250 μl of 80% water/20% acetonitrile/0.1% acetic acid, centrifuged again to clarify the sample, and aliquots were analyzed for metabolite formation.
Metabolites were analyzed by reverse HPLC-tandem mass spectrometry (HPLC-MS/MS). A 50-μl aliquot was injected by a HTS PAL 96-well autoinjector (CTC Analytics, Zwingen Switzerland) onto a HP1100 HPLC system equipped with an Agilent Eclipse VDB-C8 5-μm column (4.6 × 150 mm; Agilent, Palo Alto, CA). Initial conditions were 95% solvent A (water containing 10 mM ammonium formate and 0.1% formic acid) and 5% solvent B (acetonitrile) at 1 ml/min. After 3 min, the %B was increased linearly for 20 min to 90% B, held there for 3 min, and then returned to the starting conditions. Detection was achieved on a PESciex model 2000 LC-MS/MS triple quadrupole mass spectrometer (PerkinElmerSciex Instruments, Boston, MA). Effluent was split so that 100 μl/min was flowing into the mass spectrometer, with the remainder into a ThermoSeparations Products SpectroMonitor 3200 UV detector set to 254 nm (ThermoSeparations Products, San Jose, CA). Ionization was conducted in the positive ion mode at a source temperature of 200°C and using nitrogen as the nebulizing gas. Ion spray voltage was 4.5 kV, and the orifice voltage was 30 eV. Initial Q1 scans were performed between m/z 100 to 700.
Potential metabolites were identified by comparing t = 0 samples to t = 4 h samples, and structural information was generated from collision-induced dissociation fragments generated from metabolite-protonated parent masses [M + H]+ via product ion scans. Nitrogen was used as a collision gas at a collision energy of −30 eV. Relative abundances of some metabolites were estimated from the intensities of the peaks in the UV chromatogram. Briefly, the fragmentation pattern of benzydamine (see Fig. 1; m/z310, 265, 174, 86, 58) was used to confirm the structure of theN-oxide (m/z 326, 265, 174, 102, 84) and to assign the probable sites of metabolism for the other proposed metabolites. In some cases, structures were assigned based on the observed fragmentation pattern and the structures of the known human metabolites (Chasseaud and Catanese, 1985; Schoenwald et al., 1987).
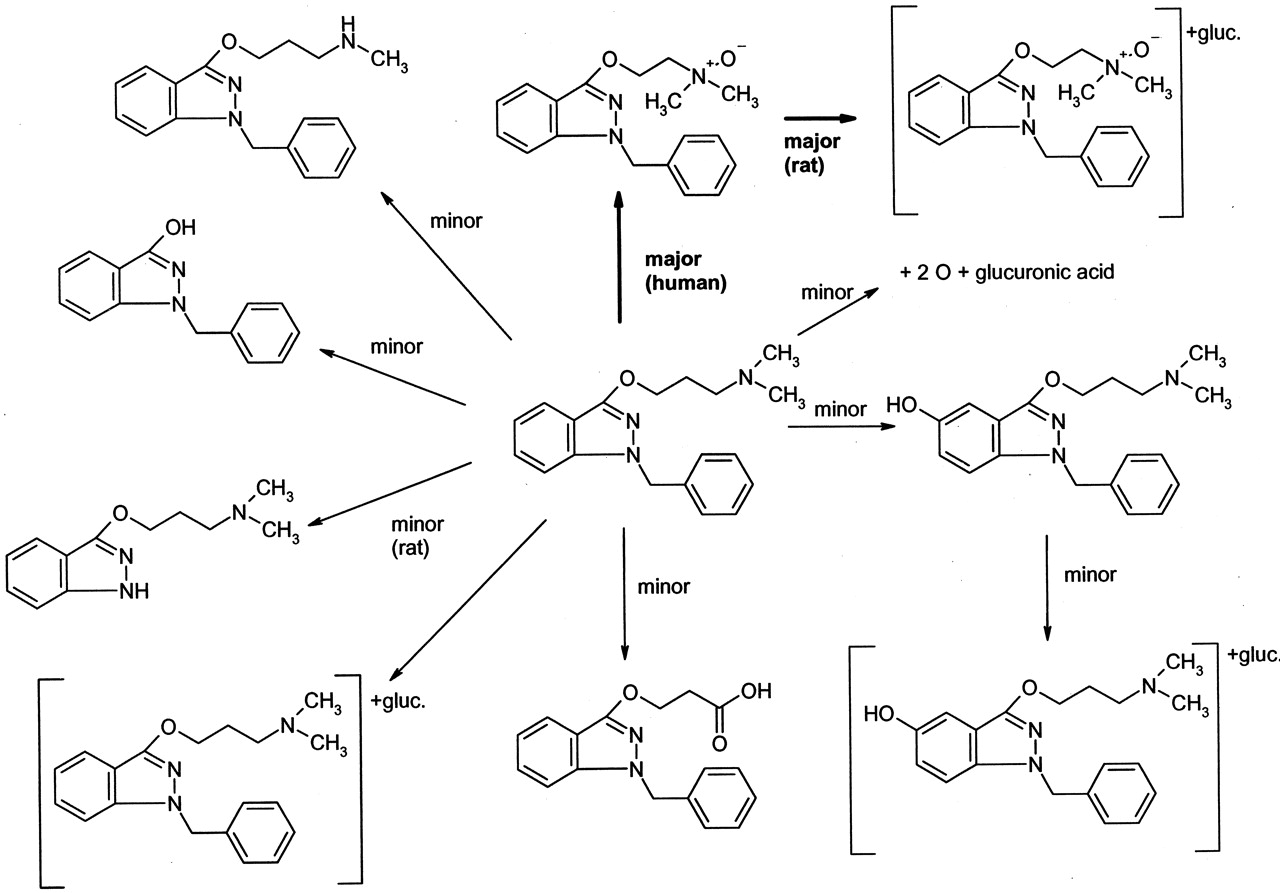
Biotransformation of benzydamine in rat and human hepatocytes.
Quantitative Incubations.
Frozen aliquots from hepatocyte incubations were thawed on ice, 100 μl was removed, transferred to a 96-well plate, and 200 μl of acetonitrile containing 1 μg/ml midazolam as internal standard was added. For both hepatocyte samples and microsome samples, plates were centrifuged, and 25 μl of the supernatant was injected for analysis by the HPLC-MS/MS system described above. Separation was achieved on a Phenomenex Primesphere 5 μm C18-HC 30 × 2-mm column (Phenomenex, Torrance, CA). Flow was 0.5 ml/min, with initial conditions of 90% solvent A and 10% solvent B. After injection, the %B was increased linearly from 10 to 95% over 0.5 min and held there for 1 min before rapidly returning to the initial conditions. Analytes were detected by employing collision induced dissociation using nitrogen as collision gas at a collision energy of −30 eV. Benzydamine and the internal standard midazolam were detected using multiple reaction monitoring, using the transitions 310→86 and 326→291, respectively. In some cases, a Micromass Ultima (Manchester, UK) HPLC-MS/MS was used in electrospray mode for this analysis, operating under similar chromatography conditions. Ionization conditions were capillary voltage 3.25 kV, cone voltage 27 V, collision energy −20 eV, and argon was used as collision gas. BenzydamineN-oxide was detected using the transition 326→102, and metabolite formation was quantitated by comparing peak area ratios (metabolite/internal standard) in incubations to ratios obtained from a standard curve containing known amounts of metabolite. The assay for benzydamine and its N-oxide was linear between 10 and 5000 ng/ml.
Rat Pharmacokinetics.
Four male Sprague-Dawley rats (jugular vein cannulated; 250–300g; Charles River Labs, Wilmington, MA) were dosed intravenously with a 10 mg/kg bolus dose of benzydamine dissolved in 70% water/20% cremophor EL/10% ethanol. All procedures were approved by the Pfizer Institutional Animal Care and Use Committee. Blood samples were collected into vacuum containers containing heparin, and plasma was prepared by centrifugation. Plasma was stored at −20°C prior to analysis. Plasma was thawed, and 100 μl was diluted with 2 volumes of acetonitrile containing internal standard as described above. The samples were centrifuged, and the supernatant was injected for HPLC-MS/MS analysis as described above. The values of the pharmacokinetic parameters were calculated using the noncompartmental method in WinNonLin v2.1 (Pharsight, Mountain View, CA).
Clearance Predictions.
For scaling up enzyme kinetics, theVmax/Kmratio was calculated to give Clint in milliliters per minute per milligram. For scaling recombinant FMO3 kinetics, Clint in ml/min/pmol, was multiplied by 80 pmol/mg microsomes (Haining et al., 1997) to convert CLint into milliliters per minute per milligram of human liver microsomes. This value was multiplied by 45 mg of microsomes/g liver and by 21 g of liver/kg body weight to yield CLint′ in milliliters per minute per milligram per kilogram (Obach, 2001). Intrinsic clearance was converted to a blood clearance (CLb) using the well stirred equation:
For hepatocytes and microsomes, the disappearance half-life method was used to predict clearance (Obach, 2001). Briefly, the in vitro disappearance half-life is incorporated into one of the following equations:
Results
Our goals for this work were to evaluate the loss in FMO activity that occurs during prewarming microsomes at 37°C in the absence of NADPH, to determine whether cryopreservation attenuates the activity of FMO in human and rat hepatocytes, to evaluate the in vitro to in vivo scaling of benzydamine metabolism in rats and humans, and to determine the relative heat lability of FMO activity in liver microsomes from preclinical species as determined by benzydamineN-oxidation. Benzydamine was chosen as the in vivo and in vitro probe since the enzymology of FMO-mediated metabolism to theN-oxide has been reported (Lang and Rettie, 2000; Stormer et al., 2000); its intravenous human pharmacokinetics are known; it is cleared predominantly (∼95%) by hepatic metabolism; the major metabolite is the FMO-generated N-oxide; and its plasma protein binding is minimal (<20%) (Chasseaud and Catanese, 1985;Schoenwald et al., 1987).
Figure 1 shows the qualitative results of metabolites formed after incubation of 10 μM benzydamine with rat and human hepatocytes. In addition to unmetabolized benzydamine at retention time of 12.6 min andm/z 310, the major peak in the total ion chromatogram had a retention time of 12.9 min andm/z 326. Product ion scans ofm/z 310 and 326 from human hepatocyte incubations and a mixture of benzydamine and benzydamine N-oxide synthetic standard showed by retention time and collision-induced dissociation spectrum that the peak at m/z 326 was the N-oxide (Fig. 2). Whereas other metabolites were apparently formed from P450 and UGT activity, it is clear that metabolites derived fromN-oxidation are a major component of the hepatic biotransformation of benzydamine, consistent with FMO activity being present in cryopreserved hepatocytes.

Characterization of benzydamine N-oxide.
Synthetic standards of benzydamine and benzydamine N-oxide, and metabolites from human hepatocyte incubation were generated and analyzed as described under Materials and Methods. Data shown for each sample are the TIC (total ion current) chromatograms from products of m/z 310 and 326, and the product ion spectra from m/z 326. Fragments from synthetic standard or biologically derived N-oxide are identical. TIC, total ion current.
It is well known that N-oxide metabolites can undergo in-source breakdown to the parent amine under some mass spectrometry conditions (Ramanathan et al., 2000; Tong et al., 2001). If this occurred with benzydamine N-oxide, it would complicate the determination of benzydamine lability and its quantitation in various matrices. Therefore, a requirement of our analytical methodology for quantitation is lack of analytical interference between the substrate and N-oxide metabolite. Figure3 shows chromatograms in multiple reaction monitoring (MRM) mode of benzydamine and itsN-oxide. The peak due to benzydamine, MRM transition 310→86, eluted at 1.56 min, and the peak due to itsN-oxide, MRM transition 326→102, eluted at 1.92 min. NoN-oxide was detected in the benzydamine channel, demonstrating that the N-oxide is stable under the liquid chromatography-mass spectrometry conditions used.

LC-tandem mass spectrometry quantitation of benzydamine and benzydamine N-oxide.
Synthetic standards were analyzed using MRM mode as described underMaterials and Methods. Shown are the TIC chromatogram and individual MRM traces for the transitions m/z310→86 and 326→102. No analytical interference between the amine and N-oxide was detected. TIC, total ion current
Table 1 shows the metabolic lability of benzydamine in human liver microsomes following prewarming of the incubation mixture for 4 min at 37°C in the presence or absence of NADPH. As expected, preincubation in the absence of cofactor resulted in a longer t1/2 and therefore lower predicted clearance. Interestingly, whereas benzydamine disappearancet1/2 was decreased 70% under this condition, benzydamine N-oxide formation rate was inhibited by >99% (3299 versus 7 pmol/min/mg).
Benzydamine metabolism by liver preparations
The predicted clearance as calculated from in vitro half-life under both preincubation protocols was dramatically higher than the in vivo clearance (Table 1). To determine whether this was a common occurrence, enzyme kinetics of benzydamine N-oxidation from human liver microsomes and recombinant FMO3 in previous reports (Haining et al., 1997; Lang and Rettie, 2000; Stormer et al., 2000) was scaled to predict in vivo clearance. Table 1 shows that in vitro FMO metabolism significantly overpredicted the in vivo clearance in all systems, as measured by benzydamine metabolism.
To evaluate whether this phenomenon also occurred in a whole cell system, the in vitro half-life was determined with 1 μM benzydamine in cryopreserved human hepatocytes that had been thawed and incubated in suspension. As shown in Table 1 and Fig.4A, the benzydamine half-life was determined to be 145 min, which results in a calculated CLb by the well stirred model of 14.4 ml/min/kg. This is similar to the observations in human liver microsomes and recombinant FMO3 and is significantly greater than the observed in vivo CL. Surprisingly, benzydamine half-life in rat hepatocytes was even shorter, determined to be 2 to 3 min (Table 1 and Fig. 4B). This results in a calculated CLb of >68 ml/min/kg, which agreed with the clearance scaled from enzyme kinetics taken from previous reports using rat liver microsomes (Kawaji et al., 1993).

Benzydamine lability in human and rat hepatocytes.
Incubations and analysis were performed as described underMaterials and Methods. Timepoints shown are the percentage of t = 0 expressed as log of the peak area ratio for human (A) and rat (B) samples. Humant1/2 = 144.9 min; Ratt1/2 = 2.3 min.
To determine whether the in vivo clearance was correctly predicted by this high in vitro clearance, the in vivo clearance of benzydamine in rat was determined by intravenous dosing at 10 mg/kg. The mean plasma concentration-time profile is shown in Fig.5. Mean pharmacokinetic parameters, estimated by fitting the plasma concentration profile to a noncompartmental model, were CL = 48 ml/min/kg, Vd(ss) = 29 l/kg, and t1/2 (terminal) = 12 h. Therefore, it appears that rat hepatocyte CL is consistent with the in vivo observations, predicting within ∼30% of the observed clearance.

Benzydamine intravenous pharmacokinetics in rat.
Benzydamine (10 mg/kg) was dosed i.v. (n = 4), and plasma samples were generated and analyzed as described underMaterials and Methods. Estimated pharmacokinetic parameters were generated using WinNonLin (v2.1). CL = 48 ml/min/kg; Vd(ss) = 29 L/kg; t1/2(terminal) = 12 h.
Because liver microsomes from preclinical species, in addition to human liver microsomes, are often used in drug discovery for lability estimations, it is important to understand the cross species heat lability of mammalian FMO. Liver microsomes from mouse, rat, dog, monkey, and human were preheated at 45°C for 3 min in the presence and absence of NADPH, and the resulting benzydamine N-oxide formation activity was determined. As shown in Fig.6, all species formed benzydamineN-oxide at significant rates, and this formation was consistently heat-labile, with activity in heat-treated samples inhibited by >99%. This strongly suggests that the heat lability observed for human liver FMO is a consistent characteristic in other preclinical species, and heat lability is a useful and general method for differentiating between FMO and P450 in microsomal incubations. This is used routinely for human liver microsomes (Grothusen et al., 1996), and has been reported for rat, mouse, and cow (Kedderis and Rickert, 1985; Venkatesh et al., 1992; Blake et al., 1995; Ring et al., 1999; Santi et al., 2002). We extend this analysis here for other traditional preclinical species (i.e., dog and monkey).

Heat lability of benzydamine N-oxidation activity in liver microsomes from preclinical species.
Incubations were performed after preheating microsome samples at 45°C for 3 min in the presence (hatched bars) or absence (filled bars) of NADPH. Incubations were performed and samples were analyzed as described under Materials and Methods.
Discussion
Clearly, the degree of lability of hepatic FMO is not such that cryopreservation and thawing of human or rat hepatocytes is sufficient to significantly attenuate activity. The lability of benzydamineN-oxide formation in fresh hepatocyte monolayers from various species has been previously investigated (Ubeaud et al., 1999). It was reported that rat, hamster, rabbit, dog, and human FMO activity was relatively stable up to 72 h in culture, although rat, rabbit, and human showed modest activity losses whereas hamster and dog were very stable. To our knowledge, cryopreserved hepatocytes have not been previously examined for FMO stability.
However, the rate of benzydamine N-oxidation in microsomes is clearly dependent on the preincubation protocol. If reaction prewarming is with an NADPH-regenerating system or with NADPH present, and reaction is initiated with substrate, FMO activity should be fully expressed. However, if preincubation is performed with substrate present and reaction is initiated with cofactor, our data indicates that FMO activity is reduced by >99%. While numerous previous reports have documented FMO heat lability at 45 to 50°C (see above), to our knowledge this is the first report quantitating the tremendous loss in FMO activity during a typical incubation prewarming in the absence of cofactor. This observation has potentially significant implications, due largely to the reported FMO mechanism. The rate-limiting step in FMO-mediated reactions is substrate independent, so turnover number varies little across a diverse series of substrates and is typically high (Poulsen and Ziegler, 1995). If incubations are initiated with NADPH, and FMO is involved in the metabolism, a significant portion of the clearance could be overlooked. However, if hepatocyte clearance is incorporated into the screening strategy, either monolayer culture (Ubeaud et al., 1999) or cryopreserved (this work), FMO mediated clearance may be observed. The choice of in vitro system can clearly influence whether the result is an under-, over-, or a successful prediction of clearance.
An outcome of this work is the overprediction of FMO-mediated human benzydamine clearance (13–20 ml/min/kg in vitro compared with 2.4 ml/min/kg in vivo intravenous clearance, Table 1). Possible explanations for this overprediction are 1) enterohepatic recycling of benzydamine via gut flora reduction of N-oxide, 2) misquantitation of N-oxide as parent in the human clinical pharmacokinetics, or 3) enhanced FMO activity via an unknown mechanism during microsomal and hepatocyte preparation. 1) It is unknown if enterohepatic recirculation of benzydamine occurs in humans, although N-oxides and glucuronides are primary candidates for recirculation due to enzymes in gut flora with potent reductase and glucuronidase activity, respectively (Rowland and Tozer, 1995; Cashman, 1999). 2) The mass spectral source-induced and/or thermal deoxygenation of N-oxides is well documented and has recently been studied as a diagnostic tool for N-oxide structural assignment (Ramanathan et al., 2000; Tong et al., 2001). Mass spectral source-induced decomposition could easily result in misquantitation ofN-oxide metabolite as unchanged parent compound in pharmacokinetic studies and thus an artificially increased area under the curve for parent and reduced clearance value. Our results show that it is possible to avoid analytical interference between benzydamine andN-oxide and thus prevent misquantitation (Fig. 2), although it is unknown if this was a potential issue during the human clinical pharmacokinetic sample analyses.
It is known from previous work that various factors can modulate the in vitro–in vivo prediction of clearance. For example, microsomal “nonspecific” binding and plasma protein binding can lead to under- and overprediction, respectively (Kalvass et al., 2001; Obach, 2001). Since we observed an overprediction for human, and since plasma binding of benzydamine is considered minimal, we do not believe that this variable is responsible for our lack of successful clearance prediction for human. A possible reason for an erroneous prediction is the presence of excretion pathways that are significant components of total clearance. Obach (2001) uses nonrenal clearance as an estimate of hepatic clearance. As only ∼5% of a benzydamine dose is excreted in urine as parent, the in vivo clearance is predominantly hepatic. Additionally, benzydamine oral absorption is described as “rapid and complete” with a reported 97% oral bioavailability, suggesting that intestinal efflux is not a major clearance pathway and that total oral clearance is low (Chasseaud and Catanese, 1985; Schoenwald et al., 1987).
Another interesting observation in this work was the relatively moderate (70%) change in in vitrot1/2 with different preincubation protocols, especially in light of the dramatic inhibition ofN-oxide metabolite formation observed. As theN-oxide formation is known to be mediated by FMO, the 99% decrease in the rate of metabolite formation was not entirely surprising. However, as other enzymes (P450, UGT, monoamine oxidase) are clearly involved in benzydamine metabolism (Fig. 1), heat lability of FMO is only expected to partially decrease the in vitrot1/2. In fact, benzydamineN-oxide accounted for 46% of the urinary metabolites of benzydamine in man (or 40% of the total dose), although other minor metabolites in that study and in ours (Fig. 1) appeared to be due to sequential metabolism of the N-oxide. The ability to develop in vitro–in vivo correlations for this and other nonP450 enzymes largely depend on identifying a large number of drugs metabolized entirely via a single nonP450 pathway. Further work is necessary to extend this correlation.
Acknowledgments
The authors acknowledge Drs. M. Byron Kneller and Allan Rettie (University of Washington) for the generous gift of benzydamineN-oxide and Drs. Jennifer Liras and Cliff Fisher for many helpful discussions.
Footnotes
-
M.B.F. would like to dedicate this article to Michael R. Rowley (1969–2002).
- Abbreviations used are::
- ADME
- absorption, distribution, metabolism, and excretion
- P450s
- cytochromes P450
- UGT
- UDP-glucuronosyltransferase
- FMO
- flavin-containing monooxygenase
- HPLC
- high-performance liquid chromatography
- DMSO
- dimethylsulfoxide
- MS/MS
- tandem mass spectrometry
- MRM
- multiple reaction monitoring
- CLint
- microsomal intrinsic clearance
- CLint′
- organ intrinsic clearance
- CLb
- blood clearance
- Q
- blood flow
- fublood
- free fraction in blood
- Received March 5, 2002.
- Accepted June 24, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}