


Abstract
Capecitabine, a prodrug of 5-fluorouracil, is first metabolized to 5′-deoxy-5-fluorocytidine (5′-DFCR) by carboxylesterase (CES), which is mainly expressed in microsomes. Recently, we clarified that 5′-DFCR formation was catalyzed by the enzyme in cytosol as well as microsomes in human liver. In the present study, the cytosolic enzyme involved in 5′-DFCR formation from capecitabine was identified. This enzyme was purified in the cytosolic preparation by ammonium sulfate precipitation, Sephacryl S-300 gel filtration, Mono P chromatofocusing, and Superdex 200 gel filtration. The purified enzyme was identified by the amino acid sequence analysis to be CES1A1 or a CES1A1 precursor. Based on the result of the N-terminal amino acid sequence analysis, the purified enzyme has no putative signal peptide, indicating that it was CES1A1. The apparent Km and Vmax values of 5′-DFCR formation were 19.2 mM and 88.3 nmol/min/mg protein, respectively. The 5′-DFCR formation catalyzed by the purified enzyme was inhibited by both diisopropylfluorophosphate and bis(p-nitrophenyl)phosphate in a concentration-dependent manner. 7-Ethyl-10-hydroxycamptothecin (SN-38) formation from irinotecan also occurred in the purified enzyme, cytosol, and microsomes. In conclusion, the cytosolic enzyme involved in 5′-DFCR formation from capecitabine would be CES1A1. It is suggested that the cytosolic CES has significant hydrolysis activity and plays an important role as the microsomal CES in drug metabolism. It is worthy to investigate the metabolic enzyme in cytosol involved in the activation of ester-type prodrugs such as capecitabine.
Capecitabine (N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine) is a novel oral fluoropyrimidine carbamate aimed at preferential conversion to 5-fluorouracil (5-FU) within tumors. Capecitabine was designed for reduced adverse effects and improved selectivity toward tumors. It is considered to be bioactivated into 5-FU by three enzymes: capecitabine to 5′-deoxy-5-fluorocytidine (5′-DFCR) by carboxylesterase (CES), 5′-DFCR to 5′-deoxy-5-fluorouridine (5′-DFUR) by cytidine deaminase (CDA), and 5′-DFUR to 5-FU by thymidine phosphorylase (TP), respectively (Miwa et al., 1998). Although capecitabine is a prodrug, the hepatic metabolism is not fully understood, especially the involvement of CES.
CES (EC 3.1.1.1) is one of the serine esterases found in various tissues of numerous animal species (Munger et al., 1991; Satoh and Hosokawa, 1995). This enzyme hydrolyzes many different endogenous and xenobiotic compounds, indicating that CES plays an important role in drug metabolism. Two major isoforms, designated hCE-1 and hCE-2, were purified from human liver by Dean et al. (1991) and Pindel et al. (1997), respectively. On the other hand, a carboxylesterase isoform, which exhibited high homology to hCE-2, was purified from human liver and intestine by Schwer et al. (1997). Multiple CES isoforms have been reported in humans and several mammalian species (Hosokawa et al., 1990; Satoh and Hosokawa, 1995). However, the systematic nomenclature and classification of CES were inadequate, leading to considerable confusion. Based on the sequence homology, Satoh and Hosokawa (1998) proposed to classify CES isoforms into four families: CES1, CES2, CES3, and CES4. According to this classification, hCE-1 and hCE-2 are now labeled CES1A1 and CES2, respectively (Sanghani et al., 2003). In mammals, CES is mainly localized in the endoplasmic reticulum of many tissues (Satoh and Hosokawa, 1998; Zhu et al., 2000). In human brain, CES was reported to be expressed in the cytosolic fraction (Yamada et al., 1994; Dean et al., 1995). It was also suggested, based on Western blot analysis, that the CES2 protein is expressed in human liver cytosol, although there were no correlations between the cytosolic CES activities and CES2 protein concentration (Xu et al., 2002). At present, human liver S9 or microsomes are usually used as the enzyme sources for in vitro studies on CES. The role of the cytosolic CES in drug metabolism has received little notice and remains to be clarified.
In our laboratory, it was clarified that 5′-DFCR formation from capecitabine occurred in human liver cytosol with large interindividual variability and that the contribution of cytosol on 5′-DFCR formation was almost the same as that of microsomes (Tabata et al., 2004). It is necessary to elucidate the enzyme catalyzing 5′-DFCR formation in cytosol in humans, because the activation of capecitabine would be a crucial factor in terms of pharmacological and toxicological aspects.
The purpose of this study is to identify the cytosolic enzyme in human liver involved in 5′-DFCR formation and to investigate in detail the characteristics of this enzyme. In addition, the activation of irinotecan, which is a typical substrate of CES, was also measured with this purified enzyme, cytosol, and microsomes.
Materials and Methods
Chemicals. Capecitabine, 5′-DFUR, 5-FU, 5-chloro-2,4-dihydroxypyridine (CDHP), and 5-chloro-6-(2-iminopyrrolidin-1-yl)methyl-2,4-(1H,3H)-pyrimidinedione (TPI) were kindly provided by Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan). Diisopropylfluorophosphate (DFP) and camptothecin were purchased from Wako Pure Chemicals (Osaka, Japan). Bis(p-nitrophenyl)phosphate (BNPP) and tetrahydrouridine (THU) were obtained from Sigma-Aldrich (St. Louis, MO) and Calbiochem (San Diego, CA), respectively. Irinotecan hydrochloride trihydrate (CPT-11) and 7-ethyl-10-hydroxycamptothecin (SN-38) were kindly supplied by Yakult Honsha Co., Ltd. (Tokyo, Japan). Other chemicals used in this study were of the highest quality commercially available.
Enzyme Sources. For the purification of the cytosolic enzyme involved in 5′-DFCR formation from capecitabine, human liver cytosol was prepared as described previously (Yamazaki et al., 1999). Briefly, human liver samples from five Japanese individuals (K19, K20, K22, K24, and K25) were obtained at autopsy. The use of the human livers had been approved by the Institutional Committee of Dokkyo University School of Medicine. Liver tissues were rapidly frozen in liquid nitrogen immediately after excision and stored at -80°C. Liver tissues (approximately 8 g) were homogenized in 3 volumes of 0.1 M Tris-HCl buffer (pH 7.4) containing 1 mM EDTA and 0.1 M KCl, and the homogenate was centrifuged at 9,000g for 15 min. The supernatant was further centrifuged at 105,000g for 90 min to prepare the cytosol. A mixture of equal volumes of these five samples was used as the pooled human liver cytosol, which was dialyzed using a size 18 dialysis membrane (Wako Pure Chemicals) against 300 volumes of 20 mM potassium phosphate buffer (pH 7.4) containing 1 mM 2-mercaptoethanol according to the method described previously (Ishikawa et al., 1998a) and used in the following experiments.
For the measurement of SN-38 formation from CPT-11, pooled human liver cytosol and pooled human liver microsomes were purchased from BD Gentest (Woburn, MA). The pooled human liver cytosol from BD Gentest was also dialyzed using the same method described above.
Purification of the Cytosolic Enzyme Involved in 5′-DFCR Formation from Capecitabine. All procedures were carried out at 0 to 4°C. Pooled human liver cytosol was diluted 3-fold with 20 mM potassium phosphate buffer (pH 7.4) containing 1 mM 2-mercaptoethanol. Proteins precipitated with 40 to 60% saturated ammonium sulfate were dissolved in 10 mM Tris-HCl buffer (pH 7.4) containing 1 mM EDTA and 1 mM 2-mercaptoethanol and then dialyzed against the same buffer to remove the ammonium sulfate. The dialyzed fraction was applied to a Sephacryl S-300 gel filtration column (1.7 × 95 cm; Pfizer, Inc., Täby, Sweden) equilibrated with 10 mM Tris-HCl buffer (pH 7.4) containing 1 mM EDTA and 1 mM 2-mercaptoethanol. The proteins were eluted with the equilibration buffer at 0.4 ml/min. Each elute fraction (2.5 ml) was collected, and the protein concentration was monitored by the measurement of A280. The 5′-DFCR formation of each protein peak fraction was determined by the method described below. The active fractions (fractions 39-41) eluted from the Sephacryl S-300 column were pooled and dialyzed against 25 mM imidazole-HCl buffer (pH 7.4). The dialyzed fraction was loaded on a Mono P HR 5/20 chromatofocusing column (Pfizer, Inc.) equilibrated with 25 mM imidazole-HCl buffer (pH 7.4). The column was eluted at 0.8 ml/min with 9-fold diluted Polybuffer 74 adjusted to pH 4.0 by HCl. Each elute fraction (1.0 ml) was collected, and the 5′-DFCR formation was determined. The active fractions (fractions 20-22) eluted from the Mono P column were pooled and applied to a Superdex 200 HiLoad 16/60 gel filtration column (Pfizer, Inc.) equilibrated with 100 mM potassium phosphate buffer (pH 7.4). Proteins were eluted with the equilibration buffer at 0.5 ml/min. The 5′-DFCR formation was determined in each elute fraction (2.0 ml). The active fractions (fractions 37 and 38) from the Superdex 200 column were pooled.
Protein Concentration. The protein concentration of each active fraction was determined by the method of Bradford (1976).
Gel Electrophoresis. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the method described previously, with slight modifications (Emoto et al., 2000). Proteins were separated on 4.5% stacking gel and 7.5% separating gel. After electrophoresis, the gels were stained with 0.05% Coomassie Brilliant Blue or with a silver staining kit (Daiichi Pure Chemicals Co. Ltd., Tokyo, Japan). The molecular weight was determined using Precision Plus Protein Standards (Bio-Rad, Hercules, CA) as a standard.
In-Gel Digestion and Mass Spectrometric Identification of Proteins. In-gel digestion and mass spectrometric identification of proteins were performed using methods reported previously (Kristensen et al., 2000; Yamagata et al., 2002). The protein spot was excised from the dried silver-stained SDS-polyacrylamide gel using clean scalpels and rehydrated for 20 min in 100 mM NH4HCO3. The spot was then destained for 20 min in a solution of 15 mM potassium ferricyanide and 50 mM thiosulfate, rinsed twice with Milli-Q water (Millipore Corporation, Billerica, MA), and dehydrated again in acetonitrile until it turned opaque white. The spot was then dried in a vacuum centrifuge and subsequently rehydrated in 10 ml of digestion solution consisting of 50 mM NH4HCO3, 5 mM CaCl2, and 0.1 μg/ml of modified sequence-grade trypsin (Promega, Madison, WI). After incubation for 16 h at 37°C, the digestion was terminated by adding 10 ml of 5% trifluoroacetic acid (TFA). Peptides were extracted three times for 20 min with 50 ml of 5% TFA and 50% acetonitrile, and the extracts were pooled and dried in a vacuum centrifuge. The dried materials were resuspended with 10 ml of 0.1% TFA. To remove excess salts from the extracts, solid-phase extraction was performed using C18 ZipTip (Millipore Corporation) according to the manufacturer's instructions. Peptides were eluted from ZipTip with 3 ml of 50% acetonitrile and 0.1% TFA, and 1 ml of the eluents was spotted onto a target plate. The spot on the target plate was immediately mixed with 0.5 ml of a matrix solution containing 0.3 mg/ml α-cyano-hydroxycinnamic acid, 33% acetone, and 66% ethanol. Then they were completely air-dried at room temperature. The mass spectometry and tandem mass spectometry spectra were obtained using an Ultraflex TOF/TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). An external peptide mixture was used to calibrate the instrument. Identification of proteins was carried out using MASCOT software (Matrix Science Ltd., London, UK) with the National Center for Biotechnology Information database. These procedures were performed by Pro Phoenix Co., Ltd. (Hiroshima, Japan).
Amino Acid Sequence Analysis. Amino acid sequence analysis was performed using a Shimadzu PSQ-1 gas-phase sequenator (Shimadzu, Kyoto, Japan), and phenylthiohydantoin derivatives were identified with an online high-performance liquid chromatography (HPLC) system equipped with a TSK gel PTH pak (2 × 250 mm; Tosoh, Tokyo, Japan) by Pro Phoenix Co., Ltd.
N-terminal Amino Acid Sequence Analysis. The N-terminal amino acid sequence of the labeled protein and its peptide samples were determined with an HP G1005A protein sequencing system (Hewlett Packard, Palo Alto, CA) by the Advanced Protein Research Organization Life Science Institute, Inc. (Tokushima, Japan).
HPLC Analysis for 5′-DFCR Formation. A typical standard reaction mixture (total volume, 0.25 ml) consisted of the enzyme source, 30 mM Tris-HCl buffer (pH 7.4) containing 92.4 mM KCl and 1.2 mM EDTA, 2.5 mM nicotinamide adenine dinucleotide phosphate (reduced form), 100 μM CDHP, 500 μM THU, 0.1 μM TPI, and 100 μM capecitabine. CDHP, THU, and TPI are inhibitors of dihydropyrimidine dehydrogenase (Fukushima et al., 2000), CDA (Wentworth and Wolfenden, 1975), and TP (Tatsumi et al., 1987), respectively. Capecitabine was dissolved in dimethyl sulfoxide. CDHP, THU, and TPI were dissolved in water. The final concentration of the organic solvent in the reaction mixture was <1.0%. The reaction was initiated by the addition of capecitabine after a 2-min preincubation at 37°C. After incubation for 30 min at 37°C, the reaction was terminated by adding 1.5 ml of ethyl acetate and 10 μl of 0.5 M HCl. The 5-chlorouracil (0.2 nmol) was added as an internal standard. The reaction mixture was extracted twice with ethyl acetate. After centrifugation at 650g for 10 min, the organic phase was evaporated to dryness under a gentle N2 stream. The residue was dissolved in 100 μl of distilled water. The 5′-DFCR formation was determined by HPLC. HPLC analysis was performed using an LC-6A pump (Shimadzu), an SPD-6AV UV detector (Shimadzu), an SIL-9A autosampler (Shimadzu), an SLC-6B system controller (Shimadzu), a Chromatopak C-R7A plus integrator (Shimadzu), a noise-base clean Uni-3 (Union, Gunma, Japan), and a CO-965 column oven (Jasco, Tokyo, Japan) equipped with a C30 analytical column (Develosil C30-UG-5, 150 × 4.6 mm; Nomura Chemical Co., Ltd., Aichi, Japan). The flow rate was 1.0 ml/min, and the column temperature was 30°C. The mobile phases were solvent A (10 mM sodium phosphate buffer, pH 4.8) and solvent B (80% methanol). A typical condition for the elution was as follows: 97% A (0-9 min); 97 to 0% A (9-35 min); 0% A (35-40 min); and 0 to 97% A (40-42 min). A linear gradient was used for all solvent changes. The eluent was monitored at 265 nm (5′-DFCR and 5′-DFUR) and 315 nm (capecitabine). The retention time of 5′-DFCR was confirmed using the incubation product with the purified CES from porcine liver (Sigma-Aldrich). The 5′-DFCR formation was quantified using a standard curve of 5′-DFUR, because we could not obtain authentic 5′-DFCR. All data were analyzed using the mean of duplicate determinations.
Kinetic Analysis of 5′-DFCR Formation. Kinetic analysis of 5′-DFCR formation from capecitabine was performed at the range of 0.05 to 3 mM. The protein concentration of the purified enzyme in the reaction mixture was 4.4 μg/ml. Kinetic parameters were calculated from the fitted curves using a KaleidaGraph computer program (Synergy Software, Reading, PA) designed for nonlinear regression analysis.
Inhibition Studies on 5′-DFCR Formation. For the inhibition studies of DFP and BNPP, the protein concentration of the purified enzyme was 8.8 μg/ml. DFP and BNPP were used as inhibitors of CES (Brandt et al., 1980). DFP and BNPP were added to the reaction mixtures at 5 to 50 and 2 to 500 nM, respectively. DFP and BNPP were dissolved in water and methanol, respectively. The final concentration of the organic solvent in the reaction mixture was <1.0%.
HPLC Analysis for SN-38 Formation. SN-38 formation was determined according to the method described previously, with slight modifications (Gupta et al., 1994). A typical standard reaction mixture (total volume, 0.2 ml) consisted of the enzyme source, 100 mM potassium phosphate buffer (pH 7.4), and 5 μM CPT-11. SN-38 was dissolved in dimethyl sulfoxide. The final concentration of the organic solvent in the reaction mixture was <1.0%. The reaction was initiated by the addition of CPT-11 after a 2-min preincubation at 37°C. After incubation for 3 min at 37°C, the reaction was terminated by adding 0.2 ml of ice-cold acetonitrile and 10 μl of 1 M HCl. Camptothecin (50 pmol) was added as an internal standard. After centrifugation at 5,500g for 5 min, 50 μl of the supernatant was subjected to HPLC. SN-38 formation was determined by HPLC. HPLC analysis was performed using an LC-6A pump (Shimadzu), an AS-950 autosampler (Jasco), a Chromatopak C-R5A integrator (Shimadzu), and a CTO-6A column oven (Shimadzu) equipped with a C18 analytical column (Inertsil ODS-2, 150 × 4.6 mm; GL Sciences, Tokyo, Japan). The eluent was monitored using a Jasco FP-920II intelligent fluorescence detector (excitation, 380 nm; emission, 556 nm) with a noise-base clean Uni-3. The flow rate was 0.8 ml/min, and the column temperature was 35°C. The mobile phase consisted of a mixture of 100 mM KH2PO4 containing 3 mM sodium heptane sulfonate (pH 4.0)/acetonitrile (33:67). For the measurement of SN-38 formation, the purified enzyme, cytosol, and microsomes were used as the enzyme sources. All data were analyzed using the mean of duplicate determinations.
Kinetic Analysis of SN-38 Formation. Kinetic analysis of SN-38 formation from CPT-11 was performed at ranges of 0.5 to 300 μM. The protein concentrations of the purified enzyme, cytosol, and microsomes in the reaction mixture were 14 μg/ml, 1.0 mg/ml, and 0.2 mg/ml, respectively. Kinetic parameters were obtained from Eadie-Hofstee plots.
Inhibition Studies on SN-38 Formation. For the inhibition studies of DFP and BNPP on SN-38 formation, the protein concentrations of the purified enzyme, cytosol, and microsomes were 28 μg/ml, 1.0 mg/ml, and 0.2 mg/ml, respectively. DFP and BNPP were added to the reaction mixtures at 1 to 100 nM and 0.01 to 10 μM, respectively. DFP and BNPP were dissolved in water and methanol, respectively. The final concentration of the organic solvent in the reaction mixture was <1.0%.
Results
Purification of the Cytosolic Enzyme Involved in 5′-DFCR Formation. The recovery of protein and activity of 5′-DFCR formation in each step of the fractionation procedure are shown in Table 1. The pooled human liver cytosol was fractionated by the ammonium sulfate precipitation method. The 40 to 60% ammonium sulfate fraction was separated using a Sephacryl S-300 gel filtration column, since the 5′-DFCR formation in this fraction was higher than any other fractions in the preliminary study. By monitoring the eluent at an absorbance of 280 nm, four protein peaks were observed. Fractions from 39 to 41 exhibited most of the 5′-DFCR formation activity. After the pooled fraction (fractions 39-41) was dialyzed, it was loaded on a Mono P chromatofocusing column, which can separate according to the pI difference of biomolecules. Similarly, the active fraction (fractions 20-22) eluted from Mono P was further separated with a Superdex 200 gel filtration column. As shown in Fig. 1, three major protein peaks were detected by monitoring A280. The pooled fraction (fractions 37 and 38) exhibited the highest activity (399.4 pmol/min/mg protein; Table 1). In this pooled fraction, one major band was detected by SDS-PAGE and silver staining (Fig. 2) and regarded as a single purified enzyme. Thus, this approximately 60-kDa protein was identified as the cytosolic enzyme involved in the 5′-DFCR formation. Accordingly, 175.4-fold purification with a 4.6% yield was achieved.
Purification of the enzyme involved in 5′-DFCR formation from capecitabine in human liver cytosol

Elution profile of human liver cytosolic protein and the activity of 5′-DFCR formation from capecitabine on Superdex 200. The active fractions (fractions 20-22) of 5′-DFCR formation eluted from Mono P HR 5/20 column were pooled and applied to a Superdex 200 HiLoad 16/60 column equilibrated with 100 mM potassium phosphate buffer (pH 7.4). Fractions 37 and 38 were used as the purified enzyme for further experiments in this study. The 5′-DFCR formation was determined in each fraction (2.0 ml) as described under Materials and Methods.

Protein profiles of human liver cytosol fractionated by ammonium sulfate and column chromatographies. The proteins were separated by SDS-PAGE. After electrophoresis, proteins were visualized by silver staining: lane 1, molecular weight standard; lane 2, human liver cytosol (5.55 μg of protein); lane 3, ammonium sulfate (40-60%) precipitation fraction (7.63 μg of protein); lane 4, Sephacryl S-300 fraction (5.47 μg of protein); lane 5, Mono P fraction (0.63 μg of protein); and lane 6, Superdex 200 fraction (0.41 μg of protein).
Identification of the Cytosolic Enzyme Involved in 5′-DFCR Formation. The final purified fraction from Superdex 200 was analyzed for the amino acid sequence. The obtained fragment sequences corresponded to those of the CES precursor (accession number p23141) in the National Center for Biotechnology Information database (Fig. 3; matched amino acid fragments to CES precursor sequence are represented in bold; 93-104, 172-199, 258-275, 303-313, and 499-523). To clarify the existence of the signal peptide (underlined in Fig. 3), we also investigated the 20 amino acid sequences from the N-terminal end by the Edman degradation method. The putative single peptide of CES could not be detected in this purified protein.

Amino acid sequence of CES1A1. Five peptide fragments of the purified enzyme identical to the CES1A1 sequence (National Center for Biotechnology Information accession number p23141) are represented in bold. The putative signal peptide is underlined.
Kinetic Analysis of 5′-DFCR Formation. The kinetic parameters of 5′-DFCR formation from capecitabine are shown in Table 2. Since the trace peak of 5′-DFCR was observed in the reaction mixture without incubation, the background levels were subtracted in the calculation of the enzymatic activities for 5′-DFCR formation at each substrate concentration. The 5′-DFCR formation was not saturated at the range of 0.05 to 3 mM capecitabine and increased linearly in a substrate concentration-dependent manner (Fig. 4). As shown in Table 2, the apparent Km and Vmax values of the purified enzyme were 19.2 mM and 88.3 nmol/min/mg protein, respectively. In our laboratory, kinetic analysis of 5′-DFCR formation was also performed in cytosol and microsomes (Tabata et al., 2004). The apparent Km and Vmax values of cytosol were 1.5 mM and 40.1 pmol/min/mg protein, respectively, and those of microsomes were 3.4 mM and 420.2 pmol/min/mg protein, respectively.
Apparent kinetic parameters of 5′ -DFCR formation from capecitabine in the purified enzyme, human liver cytosol, and microsomes
Capecitabine (0.05-3.0 mM) was incubated with the purified enzyme (4.4 μg/ml), cytosol (1.0 mg/ml), and microsomes (0.2 mg/ml) for 30 min. Kinetic parameters of cytosol and microsomes were obtained from our previous report (Tabata et al., 2004). Each data point represents the mean of duplicate determinations.

Kinetic analysis of 5′-DFCR formation from capecitabine by the purified enzyme. Capecitabine (0.05-3.0 mM) was incubated with the purified enzyme (4.4 μg/ml) for 30 min in the presence of THU (500 μM), TPI (0.1 μM), and CDHP (100 μM). Each data point represents the mean of duplicate determinations.
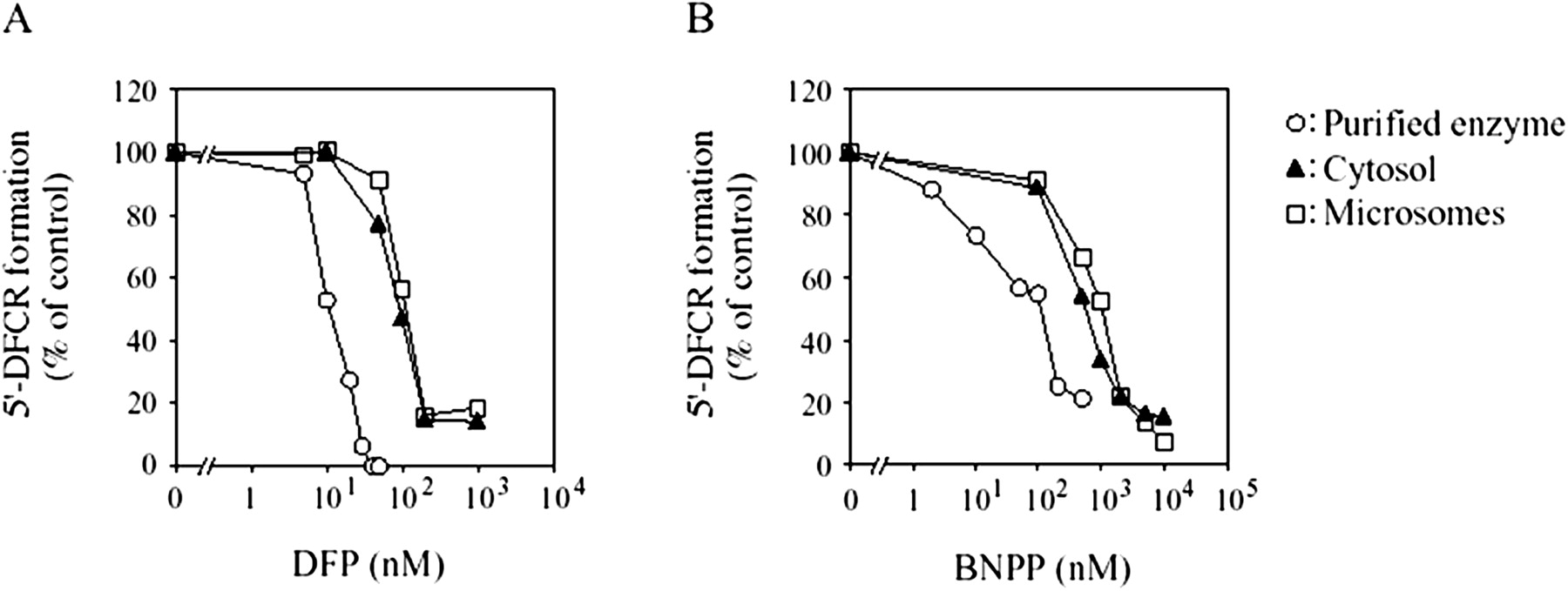
Inhibition Studies on 5′-DFCR Formation. The inhibitory effects of DFP and BNPP on 5′-DFCR formation from capecitabine catalyzed by the purified enzyme were investigated in the presence of CDA, TP, and dihydropyrimidine dehydrogenase inhibitors (Fig. 5). The 5′-DFCR formation was inhibited by DFP and BNPP in a concentration-dependent manner. The IC50 values of DFP and BNPP in the purified enzyme were 11.1 and 115.6 nM, respectively.

Effects of CES inhibitors on 5′-DFCR formation by the purified enzyme, cytosol, and microsomes. Capecitabine (100 μM) was incubated with the purified enzyme (8.8 μg/ml), cytosol (2.5 mg/ml), and microsomes (1.0 mg/ml) in the presence or absence of CES inhibitors. DFP (A) and BNPP (B) ranged from 5 to 50 nM and 2 to 500 nM, respectively. The incubation mixture contained 500 μM THU, 0.1 μM TPI, and 100 μM CDHP. The 5′-DFCR formation by the purified enzyme, cytosol, and microsomes without the CES inhibitors was 643.8, 1.9, and 6.2 pmol/min/mg protein, respectively. Each data point represents the mean of duplicate determinations. The inhibitory effects in cytosol and microsomes were derived from our previous report (Tabata et al., 2004). Each data point represents the mean of duplicate determinations.
Kinetic Analysis of SN-38 Formation. The kinetic parameters of SN-38 formation from CPT-11 are shown in Table 3. Since the trace peak of SN-38 was observed in the reaction mixture without incubation, the background levels were subtracted in the calculation of the enzymatic activities for SN-38 formation at each substrate concentration. In the present study, cytosol was used at a protein concentration 5 times higher than that of microsomes, because liver S9 contained approximately 5-fold more cytosolic protein than microsomal protein. In both cytosol and microsomes, Eadie-Hofstee plots for SN-38 formation were clearly biphasic over 0.5 to 300 μM of CPT-11. However, in the purified enzyme, SN-38 formation exhibited monophasic kinetics. As shown in Table 3, the calculated Km and Vmax values of the high-affinity enzyme were 10.8 μM and 0.09 pmol/min/mg protein in cytosol and 9.7 μM and 0.45 pmol/min/mg protein in microsomes, respectively. Those of the low-affinity enzyme were 66.1 μM and 0.28 pmol/min/mg protein in cytosol and 269.0 μM and 4.22 pmol/min/mg protein in microsomes, respectively. With the purified enzyme, the calculated Km and Vmax values were 108.2 μM and 32.71 pmol/min/mg protein, respectively.
Kinetic parameters of SN-38 formation from CPT-11 in the purified enzyme, human liver cytosol, and microsomes
CPT-11 (0.5-300 μM) was incubated with the purified enzyme (14 μg/ml), cytosol (1.0 mg/ml), and microsomes (0.2 mg/ml) for 3 min. The kinetic parameters were obtained from the Eadie-Hofstee plots. Each data point represents the mean of duplicate determinations.
Inhibition Studies on SN-38 Formation. The inhibitory effects of DFP and BNPP on SN-38 formation from CPT-11 catalyzed by the purified enzyme, cytosol, and microsomes were investigated (Fig. 6). SN-38 formation was inhibited by both DFP and BNPP in a concentration-dependent manner. The IC50 values of DFP were 24.3, 25.2, and 22.5 nM in the purified enzyme, cytosol, and microsomes, respectively. The IC50 values of BNPP were 106.6, 1730.3, and 1345.0 nM in the purified enzyme, cytosol, and microsomes, respectively.

Effects of CES inhibitors on SN-38 formation from CPT-11 by the purified enzyme, cytosol, and microsomes. CPT-11 (5 μM) was incubated with the purified enzyme (28 μg/ml), cytosol (1.0 mg/ml), and microsomes (0.2 mg/ml) in the presence or absence of the CES inhibitor. DFP (A) and BNPP (B) ranged from 1 to 100 nM and 10 nM to 10 μM, respectively. SN-38 formation by the purified enzyme, cytosol, and microsomes without the CES inhibitor was 2.72, 0.03, and 0.21 pmol/min/mg protein, respectively. Each data point represents the mean of duplicate determinations.
Discussion
Capecitabine is a new prodrug of 5-FU, which has improved selectivity toward tumors. The preferential activation of capecitabine in tumors may be caused by the tissue distribution of CES, CDA, and TP (Ishikawa et al., 1998b; Miwa et al., 1998). Generally, most drug-metabolizing enzymes exhibit high activity in the liver. However, the hepatic metabolism of capecitabine is not fully understood, especially 5′-DFCR formation from capecitabine catalyzed by CES.
In our laboratory, it was clarified that 5′-DFCR formation from capecitabine was catalyzed by both the cytosolic and microsomal enzymes in human liver (Tabata et al., 2004). Concerning 5′-DFCR formation in the liver, the contribution of cytosol appeared to be almost the same as that of microsomes, since liver S9 contains 5-fold more cytosolic protein than the microsomal protein, and large interindividual variability was observed in cytosol as in microsomes. Thus, 5′-DFCR formation, the activation of capecitabine, in cytosol would be important in terms of the pharmacokinetics. In the present study, the cytosolic enzyme involved in 5′-DFCR formation from capecitabine was identified and characterized by kinetic analyses.
To purify the cytosolic enzyme involved in 5′-DFCR formation, pooled human liver cytosol was fractionated by ammonium sulfate precipitation, Sephacryl S-300 gel filtration, Mono P chromatofocusing, and Superdex 200 gel filtration. Fractions 37 and 38 eluted from Superdex 200 exhibited the highest activity of 5′-DFCR formation, whereas fractions 30 and 31 also exhibited relatively high activity (Fig. 1). In the results of SDS-PAGE in each fraction from Superdex 200, fractions 30 and 31 also exhibited the same band as fractions 37 and 38 as a major band with the same molecular weight, suggesting that the enzyme was aggregated with other proteins (data not shown). In the preliminary study, ion-exchange chromatographies were not suitable for the purification procedure due to the protein aggregation (data not shown). The purified cytosolic enzyme was identified to be the CES precursor by amino acid sequence analysis. The amino acid sequence of this CES precursor is almost the same as that of CES1A1 except for the signal peptide (Fig. 3; MWLRAFILATLSASAAWG). Actually, the five peptide fragments of the purified enzyme completely matched CES1A1. The homology of amino acids between CES1A1 and CES2 is 48% (Pindel et al., 1997). The fragments of the purified enzyme in the present study showed 52% homology to the corresponding sequence of CES2, indicating that the purified enzyme is the CES1A1 precursor or CES1A1, not CES2. CES is mainly expressed in endoplasmic reticulum in human liver (Satoh and Hosokawa, 1998; Zhu et al., 2000). Generally, the signal peptide on the N-terminal end is suggested to play an important role in the localization of protein in mammalian cells (Kroetz et al., 1993). As compared with CES, the CES precursor has been reported to have 18 additional amino acids, which are the putative signal peptide on the N-terminal end (Munger et al., 1991; Mori et al., 1999). Since the signal peptide in the purified enzyme in this study could not be determined by amino acid sequence analysis, the N-terminal amino acid analysis was performed. It was clarified that the purified enzyme had no signal peptide. The purified enzyme in cytosol would be CES1A1, not a precursor. However, it is unknown whether the cytosolic CES initially had no signal peptide, the signal peptide had been cut off before it moved to microsomes, or the microsomal CES moved to cytosol. Further investigation is needed to clarify the mechanism of the localization of CES in human liver.
To characterize the purified enzyme, kinetic analysis was performed. As shown in Table 2, there was a large difference in the Km values between the purified enzyme and microsomes or cytosol. The Km and Vmax values of the purified enzyme were apparent values, since 5′-DFCR formation was not saturated at the range of 0.05 to 3 mM capecitabine (Fig. 4). Until now, similar phenomena have been reported. In the case of aldehyde dehydrogenase (Manthey and Sladek, 1988; Manthey et al., 1990), Km values of mouse liver cytosol were 22 and 390 μM using aldophosphamide and acetaldehyde as the substrates, respectively. Using the different fractions of purified enzymes, Km values ranged from 16 to 1,200 μM and 53 to 30,500 μM, respectively. In the case of N-acetyltransferase (Yerokun et al., 1989; Trinidad et al., 1989), Km values of the purified enzyme and cytosol from hamster liver and hamster bladder were extremely different using acetyl coenzyme A lithium salt, 2-aminofluorene, p-aminobenzoic acid, 4-aminobiphenyl, and isoniazid as the substrates. The protein binding of capecitabine did not differ among these three enzymes sources (data not shown). Capecitabine could hardly be bound to the other proteins in each enzyme source at the concentration used in this study (data not shown). Therefore, it was suggested that other factors might affect the difference of Km values of the purified enzyme and cytosol, but the reason is still unclear. Further investigations will be needed to clarify the difference of Km values.
As shown in Fig. 5, 5′-DFCR formation catalyzed by the purified enzyme as well as by cytosol and microsomes was inhibited by both DFP and BNPP in a concentration-dependent manner. Therefore, the purified enzyme was also identified to be CES by the inhibition study. It has been reported that capecitabine is metabolized to 5′-DFCR in the liver but not in the small intestine (Shimma et al., 2000), indicating that 5′-DFCR formation is mainly catalyzed by an isoform of the CES1 family, not the CES2 family, because of their tissue localization (Hosokawa et al., 1995). These results also supported that the purified enzyme would be CES1A1.
Recently, based on Western blot analysis, the existence of CES in human liver cytosol has been suggested (Xu et al., 2002). In this report, there was no relationship between the protein concentration and enzymatic activity in the cytosol fraction. Humerickhouse et al. (2000) purified two CES isoforms from human liver, CES1A1 (hCE-1) and CES2 (hCE-2), but they did not mention the subcellular localization. In this study, we first clarified the existence of the cytosolic CES with a kinetic analysis of 5′-DFCR formation. Generally, it has been considered that CES is mainly expressed in microsomes in human liver. According to the Vmax/Km values in Table 2, the cytosolic CES appeared to play an important role in 5′-DFCR formation as the microsomal CES.
For further characterization of the purified enzyme, the metabolism of CPT-11, which is a prodrug well known as a typical substrate of CES, was investigated. It has been reported that two CES isoforms are involved in SN-38 formation from CPT-11, and the CES2 family mainly catalyzed this pathway (Humerickhouse et al., 2000). SN-38 formation from CPT-11 exhibited biphasic kinetics in microsomes (Slatter et al., 1997). As shown in Table 3, SN-38 formation was shown to be catalyzed by two enzymes in both cytosol and microsomes from BD Gentest. This result in microsomes was consistent with that of the previous report (Slatter et al., 1997). The Km values of cytosol corresponded with those of previous reports (Hennebelle et al., 2000; Humerickhouse et al., 2000). It is suggested that SN-38 formation in human liver is catalyzed by four enzymes, assuming that enzymes in cytosol and microsomes are different from each other. With both high- and low-affinity isoforms, the intrinsic clearance of microsomes was approximately 5-fold higher than that of cytosol. Taking the protein level into consideration, the contributions of the cytosolic enzyme and microsomal enzyme are the same, indicating that the cytosolic CES would be as important as the microsomal CES. Since the protein binding of CPT-11 was not investigated, it would be difficult to compare the Km and Vmax values of each enzyme source. The purified enzyme in the present study exhibited a monophasic pattern. It is surmised that the purified enzyme would be a low-affinity isoform in SN-38 formation, because this enzyme, which catalyzes 5′-DFCR formation from capecitabine, would be CES1A1.
In conclusion, we clarified that the cytosolic enzyme involved in 5′-DFCR formation from capecitabine would be CES1A1. Until now, only the microsomal CES had been well characterized in terms of the catalysis of endogenous and exogenous compounds. In this study, it is suggested that the cytosolic CES has significant hydrolysis activity and plays an important role as the microsomal CES. The therapeutic and adverse effects of the prodrug could be influenced by the metabolic activation process. Therefore, it is worthy to investigate the metabolic enzyme involved in the activation of prodrugs such as capecitabine and CPT-11. We hope that this study will contribute to the appropriate use of ester-type prodrugs such as capecitabine.
Acknowledgments
We thank Brent Bell for reviewing the manuscript.
Footnotes
-
This study was supported by a grant-in-aid for Encouragement of Young Scientists of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
-
doi:10.1124/dmd.104.000554.
-
ABBREVIATIONS: 5-FU, 5-fluorouracil; 5′-DFCR, 5′-deoxy-5-fluorocytidine; CES, carboxylesterase; 5′-DFUR, 5′-deoxy-5-fluorouridine (doxifluridine); CDA, cytidine deaminase; TP, thymidine phosphorylase; CDHP, 5-chloro-2,4-dihydroxypyridine; TPI, 5-chloro-6-(2-iminopyrrolidin-1-yl)methyl-2,4-(1H,3H)-pyrimidinedione; DFP, diisopropylfluorophosphate; BNPP, bis(p-nitrophenyl)phosphate; THU, tetrahydrouridine; CPT-11, irinotecan hydrochloride trihydrate; SN-38, 7-ethyl-10-hydroxycamptothecin; PAGE, polyacrylamide gel electrophoresis; TFA, trifluoroacetic acid; HPLC, high-performance liquid chromatography.
- Received May 8, 2004.
- Accepted July 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}