


Abstract
Fexofenadine hydrochloride (FEX), a second generation H1-receptor antagonist, is mainly eliminated from the liver into bile in unchanged form. Recent studies have shown that FEX can be accepted by human MDR1 (P-glycoprotein), OATP1A2 [organic anion-transporting polypeptide (OATP)-A, and OATP2B1 (OATP-B)] expression systems. However, other transporters responsible for the hepatic uptake of FEX have not yet been identified. In the present study, we evaluated the contribution of OATP family transporters, namely OATP1B1 (OATP2/OATP-C), OATP1B3 (OATP8), and OATP2B1 (OATP-B), to FEX uptake using transporter-expressing HEK293 (human embryonic kidney) cells. The uptake of FEX in OATP1B3-expressing cells was significantly greater than that in vector-transfected cells. On the other hand, OATP1B1- or OATP2B1-mediated uptake of FEX was not statistically significant. OATP1B3-mediated transport could be explained by a one-saturable component with a Michaelis constant (Km) of 108 ± 11 μM. The inhibitory effect of FEX on the uptake of estrone-3-sulfate (E1S), cholecystokinin octapeptide (CCK-8), and 17β-estradiol-17β-d-glucuronide (E217βG) was also examined. Both OATP1B1- and OATP1B3-mediated E217βG uptake was inhibited by FEX. The Ki values were 148 ± 61 and 205 ± 72 μM for OATP1B1 and OATP1B3, respectively. FEX also inhibited OATP1B3-mediated CCK-8 uptake and OATP1B1-mediated E1S uptake with a Ki value of 83.3 ± 15.3 and 257 ± 84 μM, respectively, suggesting that FEX could not be used as a specific inhibitor for OATP1B1 and OATP1B3, although FEX was preferentially accepted by OATP1B3. In conclusion, this is, to our knowledge, the first demonstration that OATP1B3 is thought to be a major transporter involved in hepatic uptake of FEX in humans.
Several members of different uptake transporter families are thought to be involved in the hepatic uptake of substances in human liver. Since the uptake of substances from blood into hepatocytes is the first step in the hepatocellular elimination, it is increasingly recognized that uptake transporters in the basolateral membrane play an important role in substrate disposition. Organic anion-transporting polypeptides (OATPs) form a superfamily of the sodium-independent transport system that mediates the transmembrane transport of a wide range of amphiphilic organic compounds including bile salts, organic dyes, steroid conjugates, thyroid hormones, anionic oligopeptides, and many drugs, such as pravastatin (Hagenbuch and Meier, 2004).
Fexofenadine hydrochloride (FEX) (Aventis Pharmaceuticals, Inc., Kansas City, MO), a selective histamine H1-receptor antagonist, is clinically effective in the treatment of seasonal allergic rhinitis and chronic idiopathic urticaria, for which it is considered as first-line therapy (Markham and Wagstaff, 1998; Simpson and Jarvis, 2000). FEX is the active metabolite of terfenadine (Seldane) and FEX showed no significant effect on the prolongation of the corrected QT interval (QTc) in contrast to terfenadine (Pratt et al., 1999). After oral administration of [14C]FEX (60 mg), most of the total dose was recovered in the urine (12%) and feces (80%), with the majority of the dose (>85%) recovered as the unchanged form (Lippert et al., 1996). This shows that metabolism is an insignificant elimination route and that FEX is poorly absorbed and/or is mainly eliminated from the liver into bile in its unchanged form.
Hepatic metabolism is of minimal importance in the elimination of FEX. On the other hand, coadministration of erythromycin (500 mg three times a day) or ketoconazole (400 mg once daily) with FEX resulted in substantial increase in steady-state plasma concentration of FEX and its plasma AUC by 109 and 164%, respectively (product information, Aventis Pharmaceuticals, Inc.). However, a regional perfusion study showed that ketoconazole did not have a significant effect on the in vivo intestinal absorption of FEX when coadministered or given as a pretreatment (Tannergren et al., 2003).
FEX was shown to be a substrate of P-glycoprotein and OATP1A2 (OATP-A), and its disposition was altered in mdr1a (-/-) mice (Cvetkovic et al., 1999). In addition, rifampin increased the oral clearance of FEX, suggesting an up-regulation of P-glycoprotein and, possibly, other transport processes (Hamman et al., 2001). Currently, several OATP family transporters such as OATP1B1 (OATP2/OATP-C), OATP1B3 (OATP8), and OATP2B1 (OATP-B) have been identified on the basal membrane of human liver (Konig et al., 2000a,b; Kullak-Ublick et al., 2001). OATP1A2- and OATP2B1-mediated uptake of FEX had been determined previously (Cvetkovic et al., 1999; Nozawa et al., 2004); however, other transporters responsible for the hepatic uptake of FEX have not yet been investigated, and their clinical relevance has not yet been determined.
In this study, we especially focused on the involvement of OATP1B1 and OATP1B3 in the hepatic uptake of FEX because they are exclusively expressed in liver and exhibit similar broad substrate specificities, which suggests that they play a crucial role in the hepatic uptake of several anionic endogenous compounds and drugs (Ismair et al., 2003; Hagenbuch and Meier, 2004).
The substrate specificity of OATP1B3 commonly overlaps that of OATP1B1 (Ismair et al., 2001; Kullak-Ublick et al., 2001). However, there are some differences as far as their substrate recognition and transcriptional regulation are concerned (Hagenbuch and Meier, 2003; Kullak-Ublick et al., 2004). Therefore, it is important to estimate the relative contribution of OATP1B1 and OATP1B3 to the hepatic uptake in humans separately. Recently, two kinds of methodologies were established to identify a quantitative contribution of OATP1B1 and OATP1B3 to the overall hepatic uptake. One is to compare the uptake clearance of transporter-selective compounds in transporter-expressing HEK293 cells and human cryopreserved hepatocytes, and the other is to compare the relative expression level of each transporter in expression system and hepatocytes estimated by Western blot analysis. (Hirano et al., 2004).
In the present study, we evaluated the contribution of OATP1B1 and OATP1B3 to FEX uptake using transporter-expressing HEK293 cells. We also examined the inhibitory effect of FEX on the uptake of a number of reference compounds, which are estrone-3-sulfate (E1S), a selective substrate of OATP1B1; CCK-8, a selective substrate of OATP1B3; and 17β-estradiol-17β-d-glucuronide (E217βG), a substrate of both transporters.
Materials and Methods
Materials. FEX was a gift from Aventis Pharmaceuticals, Inc. (Kansas City, MO). [3H]E1S (57.3 Ci/mmol) and [3H]E217βG (45.0 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). [3H]CCK-8 (68.0 Ci/mmol) was purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Unlabeled E1S, CCK-8, and E217βG were purchased from Sigma (St. Louis, MO). All other chemicals were of analytical grade and commercially available.
Construction of Stably Transfected HEK293 Cells Expressing Human OATP2B1. The human OATP2B1 gene was isolated by polymerase chain reaction using ATCC IMAGE clone (I.D. 5752976) purchased from Summit Pharmaceuticals International Corp. (Tokyo, Japan). To obtain the full-length cDNA of the OATP2B1 gene, pCMV-SPORT6 vector containing OATP2B1 cDNA was digested with EcoRI and NotI. Then, cDNA fragment was ligated into EcoRI and NotI sites of the pcDNA3.1 (+) (Invitrogen, Carlsbad, CA). OATP2B1-expressing HEK293 cells were constructed by the transfection of expression vector into cells using FuGENE6 (Roche Diagnostics, Indianapolis, IN), according to the manufacturer's instruction and selection by 800 μg/ml of the antibiotic G418 sulfate (Promega, Madison, WI) for 3 weeks.
Cell Culture. Transporter-expressing or vector-transfected HEK293 cells were grown in Dulbecco's modified Eagle's medium low glucose (Invitrogen) supplemented with 10% fetal bovine serum (Cansera International Inc., Toronto, ON, Canada), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% CO2 and 95% humidity. Cells were then seeded in 12-well plates (coated with 50 mg/l poly(l-lysine) and 50 mg/l poly(l-ornithine); Sigma) at a density of 1.5 × 105 cells/well. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before transport assay to induce the expression of transporters.
Transport Study Using Transporter Expression Systems. The transport study was carried out as described previously (Hirano et al., 2004). After cells had been washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 15 min, uptake was initiated by adding Krebs-Henseleit buffer containing radiolabeled and unlabeled substrates. The Krebs-Henseleit buffer consisted of 118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM d-glucose, and 1.5 mM CaCl2 adjusted to pH 7.4. For inhibition studies, inhibitor was added in the incubation buffer. At designated times, the incubation buffer was removed and uptake was terminated by adding ice-cold Krebs-Henseleit buffer. Then, cells were washed twice with 1 ml of ice-cold Krebs-Henseleit buffer and lysed with 500 μlof0.2 N NaOH overnight at 4°C. Then, 250 μl of 0.4 N HCl was added to the cell lysate and aliquots (500 μl) were transferred to scintillation vials. The radioactivity associated with the cells and incubation buffer was measured in a liquid scintillation counter (LS6500; Beckman Coulter, Fullerton, CA) after adding 2 ml of scintillation fluid (Clear-sol-I; Nacalai Tesque, Kyoto, Japan) to the scintillation vials. The cellular protein amount was determined by the Lowry method using 50 μl of lysate with bovine serum albumin as a standard.
When using FEX as a substrate, aliquots (150 μl) of lysates were transferred to microtubes, mixed with 30 μl of 1 N HCl and 300 μl of 2 ng/ml midazolam (internal standard of LC/MS) in methanol, and deproteinized by centrifugation for 10 min at 13,000 rpm. Then, 50 μl of the supernatants were used for LC/MS analysis.
LC/MS Analysis. FEX concentrations were determined by high-performance liquid chromatography with electrospray mass spectrometry (LC/MS) using the modified protocol described in the previous report (Hofmann et al., 2002). The LC/MS system was operated by MassLynx and QuanLynx software (Waters, Milford, MA). The contents of the mobile phase for high-performance liquid chromatography were 0.05% formic acid in water (A) and methanol (B). Chromatographic separation was achieved on a Capcell Pak C18 MG analytical column (4.6 mm i.d. × 75 mm; particle size 3 μm; Shiseido, Tokyo, Japan) kept at 30°C, using a linear gradient from 55% B to 70% B over 5 min, and a flow rate of 0.8 ml/min. Electrospray parameters were as follows: capillary voltage, 3.10 kV; cone voltage, 50.00 kV; extractor voltage, 5.00 V; source temperature, 100°C; cone temperature, 20°C; desolvation temperature, 350°C; cone gas flow, 50 l/h; and desolvation gas flow, 300 l/h. The mass spectrometer (ZQ 2000 MS detection; Waters) was operated in the selected ion monitoring mode using the respective positive ions, m/z 502.30 for FEX and m/z 326.10 for midazolam (internal standard). The retention time of FEX was approximately 3.3 min. Standard curves were linear over the range of 2.5∼100 nM. The coefficient of variation (CV) of the interassay variability (n = 14; quality controls containing 10 nM FEX) ranged between 3.6 and 11.8%. The CV of the intra-assay variability (n = 9) ranged between 4.7 and 9.8%.
Data Analysis. Kinetic analysis was carried out as described previously (Hirano et al., 2004). Ligand uptake was expressed as the uptake volume (μl/mg protein), given as the amount of radioactivity associated with the cells (dpm/mg protein) divided by its concentration in the incubation medium (dpm/μl). In the case of FEX, ligand uptake (μl/mg protein) was given as the amount of FEX associated with the cells (pmol/mg protein) divided by its concentration in the incubation medium (pmol/μl). Specific uptake was obtained by subtracting the uptake into vector-transfected cells from that into cDNA-transfected cells. Kinetic parameters were obtained using the following equation: 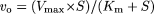 where vo is the initial uptake rate of substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), and Vmax is the maximum uptake rate (pmol/min/mg protein). To obtain kinetic parameters, the data were fitted to eq. 1 by a nonlinear least-squares method using the MULTI program (Yamaoka et al., 1981).
where vo is the initial uptake rate of substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), and Vmax is the maximum uptake rate (pmol/min/mg protein). To obtain kinetic parameters, the data were fitted to eq. 1 by a nonlinear least-squares method using the MULTI program (Yamaoka et al., 1981).
The inhibition constant (Ki) of FEX for the uptake of radiolabeled compounds was obtained by fitting the following equation to the data as described previously (Chu et al., 1997): 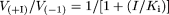 where V(+I) and V(-I) represent the transport velocity in the presence and absence of inhibitor, respectively, and I is the inhibitor concentration. This equation was derived based on two assumptions: the mode of inhibition was competitive or noncompetitive and the concentration of substrates (0.1 μM) used was much lower than their Km values.
where V(+I) and V(-I) represent the transport velocity in the presence and absence of inhibitor, respectively, and I is the inhibitor concentration. This equation was derived based on two assumptions: the mode of inhibition was competitive or noncompetitive and the concentration of substrates (0.1 μM) used was much lower than their Km values.

Time-dependent uptake of FEX in OATP1B1, OATP1B3, and OATP2B1-expressing HEK293 cells. Uptake of FEX (10 μM) was measured for 1, 2, 5, and 10 min using HEK293 cells expressing OATP1B1 (♦), OATP1B3 (•), OATP2B1 (▪), or vector-transfected cells (control; ○). Data are shown as the mean ± S.E. of three to eight independent experiments, and each experiment was performed in triplicate. The asterisks represent a statistically significant difference from uptake in the control cell shown by a paired Student's t test (*, p < 0.05; **, p < 0.005).
Statistical Analysis. Statistical differences were determined using a paired Student's t test and differences were considered significant at P < 0.05.
Results
Uptake of FEX by Transporter-Expressing Cells. To evaluate whether FEX was a substrate for OATP1B1, OATP1B3, or OATP2B1, the uptake of FEX (10 μM) was investigated using OATP1B1-, OATP1B3-, and OATP2B1-expressing cells and vector-transfected HEK293 cells (control). The hepatic uptake clearances of probe substrates E217βG by OATP1B1, CCK-8 by OATP1B3, and E1S by OATP2B1 were 14.9 ± 0.3, 5.52 ± 0.28, and 10.8 ± 1.0 μl/min/mg protein, respectively (data not shown). The uptake of FEX in OATP1B3-expressing cells was significantly greater than that in vector-transfected cells (P < 0.005) (Fig. 1). Its uptake clearance, which was calculated by the difference in the slope of uptake amount at 1 min and 10 min in expression system and vector-control cells, was 0.409 ± 0.056 μl/min/mg protein. On the other hand, OATP1B1- and OATP2B1-mediated uptake of FEX was only slightly observed, although not statistically significant. The uptake clearance by OATP1B1 and OATP2B1 was 0.0622 ± 0.0586 and 0.0414 ± 0.0614 μl/min/mg protein, respectively (Fig. 1).
The concentration-dependent uptake of FEX was studied using OATP1B3-expressing cells and vector-transfected HEK293 cells. Kinetic analysis revealed that the OATP1B3-mediated uptake of FEX was saturable with a Km of 108 ± 11 μM and a Vmax of 56.7 ± 3.2 pmol/min/mg protein (Fig. 2).
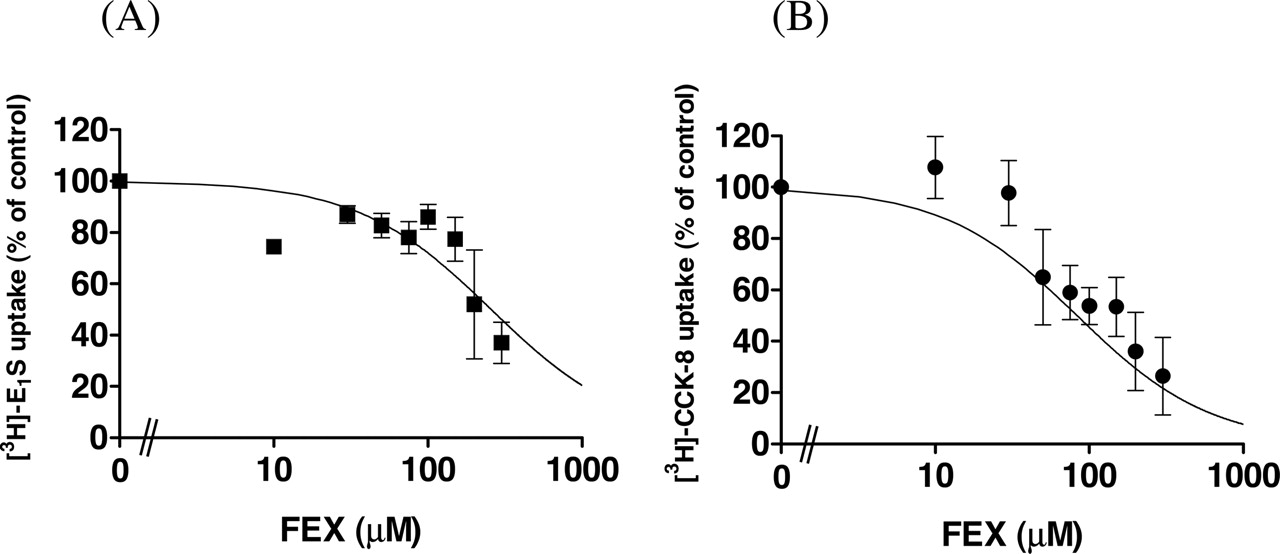
Effect of FEX on the Uptake of [3H]E1S, [3H]CCK-8, and [3H]E217βG by Transporter-Expressing Cells. The effect of FEX on the uptake of [3H]E1S and [3H]CCK-8 by transporter-expressing cells is shown in Fig. 3. OATP1B3-mediated CCK-8 uptake was inhibited by FEX (Fig. 3B), whereas OATP1B1-mediated E1S uptake was weakly inhibited by FEX (Fig. 3A). The Ki values were 257 ± 84 and 83.3 ± 15.3 μM for OATP1B1-mediated E1S uptake and OATP1B3-mediated CCK-8 uptake, respectively.
The effect of FEX on the uptake of [3H]E217βG by transporter-expressing cells is shown in Fig. 4. In this case, the Ki values were 148 ± 61 μM for OATP1B1 and 205 ± 72 μM for OATP1B3, respectively.
Discussion
In the present study, we have shown that FEX is efficiently transported by OATP1B3 in human liver, whereas OATP1B1 did not transport FEX significantly. Hirano et al. (2004) have recently established a method for determining the contribution of OATP1B1 and OATP1B3 to the overall hepatic uptake of compounds.

Concentration-dependence of the OATP1B3-mediated uptake of FEX. Uptake of FEX was measured for 5 min using HEK293 cells expressing OATP1B3 (•) or vector-transfected cells (control; ○). OATP1B3-mediated uptake (▴) was calculated by subtracting the uptake in control cells from that in OATP1B3 cells. Kinetic parameters were obtained by fitting the data for OATP1B3-mediated uptake to the Michaelis-Menten equation (eq. 1) by nonlinear least-squares analysis. The inset is an Eadie-Hofstee plot of the same data. Data are shown as the mean ± S.E. of three to four independent experiments, and each experiment was performed in triplicate.
Following one of those approaches, we estimated the relative expression level of OATP1B1 and OATP1B3 in transporter-expression systems and human hepatocytes by using transporter-selective ligands. We found previously that E1S can be used as an OATP1B1-selective ligand, whereas CCK-8 is an OATP1B3-selective ligand. As shown in the results, we measured the uptake clearance of E217βG in OATP1B1-expressing cells and CCK-8 in OATP1B3-expressing cells. As for OATP1B1, we can estimate the uptake clearance of E1S in our expression systems [124 μl/min/mg protein (= 132*14.9/15.8)] by comparing the uptake of E217βG in our cells (14.9 μl/min/mg protein) with that previously reported (15.8 μl/min/mg protein) (Hirano et al., 2004). Then, following the method as described previously (Hirano et al., 2004), the ratio of the uptake clearance of reference compounds in each batch of human hepatocytes to that in the OATP1B1- and OATP1B3-expression system (Ract, OATP1B1 and Ract, OATP1B3) was found to be 0.887 and 1.43 for lot OCF, 1.08 and 0.634 for lot 094, and 0.465 and 0.366 for lot ETR, respectively. Therefore, we can estimate the predicted uptake clearance of FEX mediated by a specific transporter by multiplying Ract by the uptake clearance of FEX in each expression system. If the contribution of OATP1B1 to the hepatic uptake of FEX is assumed to be equal to that of OATP1B3, the following equation should be satisfied: 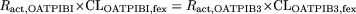 where CLOATP1B1, fex and CLOATP1B3, fex represent the uptake clearance of FEX in our OATP1B1- and OATP1B3-expressing cells. Assuming that OATP1B1 is more important than OATP1B3 in the hepatic uptake of FEX, CLOATP1B1, fex should be 1.61-, 0.587-, and 0.787-fold larger than CLOATP1B3, fex in lot OCF, 094, and ETR, respectively. However, in Fig. 1, OATP1B1-mediated uptake was less than half the OATP1B3-mediated uptake. This finding suggests that the contribution of OATP1B3 is at least more than 50% of the overall hepatic uptake of FEX.
where CLOATP1B1, fex and CLOATP1B3, fex represent the uptake clearance of FEX in our OATP1B1- and OATP1B3-expressing cells. Assuming that OATP1B1 is more important than OATP1B3 in the hepatic uptake of FEX, CLOATP1B1, fex should be 1.61-, 0.587-, and 0.787-fold larger than CLOATP1B3, fex in lot OCF, 094, and ETR, respectively. However, in Fig. 1, OATP1B1-mediated uptake was less than half the OATP1B3-mediated uptake. This finding suggests that the contribution of OATP1B3 is at least more than 50% of the overall hepatic uptake of FEX.
We showed that OATP2B1 does not transport FEX significantly, although slight enhancement of its uptake by OATP2B1 could be observed (Fig. 1). This is not consistent with a previous result demonstrating the uptake of FEX in OATP2B1-expressing cells at both pH 5.0 and 7.4 (Nozawa et al., 2004). This discrepancy might be due to the low level of OATP2B1 in our expression system, compared with their cells, because the uptake clearance of E1S at pH 7.4 in our cells (10.8 ± 1.0 μl/min/mg protein) was smaller than that reported previously (Nozawa et al., 2004).

Inhibitory effect of FEX on the OATP1B1-mediated uptake of E1S (A) and OATP1B3-mediated uptake of CCK-8 (B). OATP1B1-mediated uptake of [3H]E1S (A) or OATP1B3-mediated [3H]CCK-8 (B) was calculated by subtracting the uptake in control cells from that in transporter-expressing cells. The concentration of E1S or CCK-8 was set at 0.1 μM. The inhibition constant (Ki) of FEX for the transporter-mediated uptake was obtained by fitting to eq. 2 using nonlinear least-squares analysis, and the solid line represents the fitted line. Data are shown as the mean ± S.E. of three independent experiments, and each experiment was performed in triplicate.

Inhibitory effect of FEX on the uptake of OATP1B1 (A)- or OATP1B3 (B)-mediated uptake of [3H]E217βG. OATP1B1 (A)- or OATP1B3 (B)-mediated uptake of E217βG was calculated by subtracting that in control cells from that in OATP1B1- or OATP1B3-expressing cells (♦, OATP1B1; ▴, OATP1B3). The concentration of E217βG was set at 0.1 μM. The inhibition constant (Ki) of FEX for the OATP1B1- or OATP1B3-mediated uptake of E217βG was obtained by fitting to eq. 2 using nonlinear least-squares analysis, and the solid line represents the fitted line. Data are shown as the mean ± S.E. of three independent experiments, and each experiment was performed in triplicate.
Kobayashi et al. (2003) have demonstrated that OATP2B1 is localized on the apical membrane of human intestine, and Nozawa et al. (2003) showed pH-dependent uptake of some organic anions in OATP2B1-expressing cells. In addition, Satoh et al. (2005) have indicated that citrus juice can inhibit the OATP2B1-mediated uptake of anionic compounds, which is consistent with the clinical report showing that fruit juices decreased the bioavailability of FEX in humans (Dresser et al., 2002). Therefore, OATP2B1 might be involved in the intestinal absorption of FEX. On the other hand, we found that the ratio of expression level of OATP2B1 in human hepatocytes per 106 cells to that in our transporter expression system per milligram of cellular protein was about 5 times lower than that of OATP1B1 and OATP1B3 (M. Hirano, K. Maeda, Y. Shitara, and Y. Sugiyama, unpublished observation), suggesting that OATP2B1 does not play a major role in the hepatic uptake of FEX, even though it is a substrate for ATP2B1.
We also examined the effect of FEX on the OATP1B1- or OATP1B3-mediated uptake of probe substrates (Figs. 3 and 4). FEX inhibited both OATP1B1- and OATP1B3-mediated uptake of the three compounds we tested. The substrate-dependence of the Ki values was not clearly observed. These inhibitory effects are not likely to be clinically significant because the Ki values were much higher than the reported Cmax of FEX (459 nM, product information). The inhibitory effect of FEX for OATP1B1 was slightly weaker than that for OATP1B3. The results suggest that FEX was not a specific inhibitor for OATP1B1 or OATP1B3, although it is thought to be a selective substrate for OATP1B3 in human liver.
OATP1B3 shares 80% amino acid identity with OATP1B1, and the spectrum of substrates of OATP1B3 is similar to that of OATP1B1 (Kullak-Ublick et al., 2001). Both OATP1B1 and OATP1B3 have broad substrate specificities, including bile salts (glycocholate, taurocholate), hormones and their conjugates [dehydroepiandrosterone sulfate, E217βG, thyroid hormones (T3 and T4), leukotriene C4), peptides [cyclo(l-Leu-d-Trp-d-Asp-l-Pro-d-Val) (BQ-123), [d-Pen2,5]-enkephalin (DPDPE)], drugs (methotrexate, rifampicin), other organic anions (monoglucuronosyl bilirubin, bromosulfophthalein), and natural toxins (microcystin, phalloidin) (Hagenbuch and Meier, 2003). To characterize the contribution of OATP1B3, it is necessary to identify the specific substrate(s) or inhibitor(s) of OATP1B3. Previously, only a few unique substrates for OATP1B3 have been reported, such as CCK-8 (intestinal peptide) (Ismair et al., 2001), deltorphin II (opioid peptide), and digoxin (cardiac glycoside) (Kullak-Ublick et al., 2001) in transporter-expressing Xenopus laevis oocytes. However, considering the direct evaluation of the function of OATP1B3 in humans by measuring the hepatic clearance of probe drugs, they cannot be easily used as a clinically applicable probe drug in humans because of the narrow safety margin. In this study, we demonstrated for the first time that FEX may also be a relatively selective substrate for OATP1B3. Because severe side effects of FEX were not reported, FEX may be a good probe drug for evaluating the function of OATP1B3 in a clinical situation.
Some reports have indicated that genetic polymorphisms of OATP1B1 affect the pharmacokinetics of pravastatin in vivo (Nishizato et al., 2003;. Mwinyi et al., 2004; Niemi et al., 2004). Therefore, pravastatin can be used as a probe drug for evaluating the function of OATP1B1. Currently, one rare polymorphism in OATP1B3 has been reported to affect substrate specificities using in vitro analysis (Letschert et al., 2004). However, the impact of the functional change in OATP1B3 by genetic polymorphisms, diseases, and drug-drug interactions on the pharmacokinetics of drugs in humans remains to be clarified, and FEX might be one of the candidate drugs to estimate the OATP1B3 function in vivo.
While this article (manuscript) was under review, Niemi et al. (2005) published the interesting results of a clinical study suggesting that polymorphism in OATP1B1 (T521C), which affected the pharmacokinetics of pravastatin (Nishizato et al., 2003; Mwinyi et al., 2004; Niemi et al., 2004), increased the plasma AUC of fexofenadine. This result appears to conflict with our findings. However, the genetic polymorphism in OATP1B3 was not evaluated in the study by Niemi et al. (2004). The relative importance of OATP1B1 and OATP1B3 in the hepatic uptake of FEX will be determined in larger clinical trials, since the frequency of the polymorphism in OATP1B3 was very low. In addition, specific inhibitors for OATP1B1 and OATP1B3 that can be clinically used will give us a chance to validate our conclusion.
In conclusion, FEX can inhibit both OATP1B1- and OATP1B3-mediated transport, and FEX can be recognized by OATP1B3 preferentially compared with OATP1B1 or OATP2B1. This is the first demonstration that OATP1B3 is thought to be a major transporter involved in the hepatic uptake of FEX.
Acknowledgments
We are grateful to Aventis Pharmaceuticals, Inc. for kindly providing fexofenadine hydrochloride. We thank Masaru Hirano for valuable discussions, Tian Ying and Miyuki Kambara for great assistance with the construction of OATP2B1-expressing cells, and Ayumi Sakai, Keiko Ohsone, and Mika Munemasa for excellent technical assistance.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.004622.
-
ABBREVIATIONS: OATP, organic anion-transporting polypeptide; HEK, human embryonic kidney; AUC, area under the plasma concentration curve; FEX, fexofenadine hydrochloride; E1S, estrone-3-sulfate; CCK-8, cholecystokinin octapeptide; E217βG, 17β-estradiol-17β-d-glucuronide; LC/MS, liquid chromatography/mass spectrometry.
- Received March 7, 2005.
- Accepted July 12, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}