


Abstract
The aim of this study was to characterize rat small intestinal and colon tissue slices as a tool to study intestinal metabolism and to investigate gradients of drug metabolism along the intestinal tract as well as drug-induced inhibition and induction of biotransformation. Tissue morphology and the intestinal mucus layer remained intact in small intestinal and colon slices during 3 h of incubation, while alkaline phosphatase was retained and the rate of metabolism of three model compounds (7-hydroxycoumarin, 7-ethoxycoumarin, and testosterone) appeared constant. Phase I and phase II metabolic gradients, decreasing from stomach toward colon were shown to be clearly different for the model compounds used. Furthermore, the observed slice activities were similar or even higher compared with the literature data concerning metabolism of in vitro intestinal systems. Preincubation with β-naphthoflavone for 24 h induced the O-deethylation of 7-ethoxycoumarin from nearly undetectable to 140 pmol/min/mg protein in small intestine (fresh slices, 43 pmol/min/mg protein) and to 100 pmol/min/mg protein in colon slices (fresh slices, undetectable). Ketoconazole inhibited metabolism of testosterone by 40% and that of 7-ethoxycoumarin by 100%. In conclusion, we showed that the intestinal slice model is an excellent model to study drug metabolism in the intestine in vitro, since we found that the viability parameters remain constant and the measured enzyme activities are relevant, sensitive to inhibitors, and inducible. Therefore, it is a promising tool to study intestinal drug metabolism in human intestine in vitro in the future.
Although the liver has long been considered as the main drug-metabolizing organ, the ability of the small intestine to metabolize drugs is increasingly recognized. Several reports have now been published showing significant contribution of intestinal tissue to the metabolism of drugs (Krishna and Klotz, 1994; Doherty and Charman, 2002; Kaminsky and Zhang, 2003). On the other hand, no exhaustive studies of drug metabolism in the colon have been published (Ding and Kaminsky, 2003).
In vivo assessment of intestinal metabolism is difficult due to the contribution of other organs and especially the liver to drug metabolism. Therefore, in vitro studies are necessary. Among the in vitro systems used for studying metabolism in the intestine are subcellular fractions like microsomes (Emoto et al., 2000a; Takemoto et al., 2003) and S9 fractions (Sohlenius-Sternbeck and Orzechowski, 2004), which are commonly used due to their easy handling and applicability to human tissue. However, these systems lack membrane transporters and several enzymatic systems. Moreover, results are highly dependent on the method of their isolation (Plumb et al., 1987).
Intact tissue systems, such as the everted sac preparation (Emoto et al., 2000b) and the Ussing chamber (Lennernas et al., 1997), are commonly used intestinal systems in which the in vivo tissue structure is retained. However, the viability of these systems is open to question and, in fact, rapidly deteriorates in a few hours of experimentation.
The Caco-2 cell line is also an often applied in vitro method for absorption studies (Pelkonen et al., 2001). Although Caco-2 cells during culture express many small intestinal epithelial functions, drug metabolism cannot be accurately predicted by these cells (Pelkonen et al., 2001; Davis and Riley, 2004).
Many factors, such as drug-drug and drug-diet interactions, have been reported to influence drug metabolism and absorption in intestinal tissue by inhibition and induction (Pelkonen et al., 2001), thereby changing the bioavailability and causing an imbalance between toxification and detoxification (Lin and Lu, 1998). To date, only Caco-2 cells are available to study drug-induced induction of biotransformation in intestinal cells in vitro (Galijatovic et al., 2000). However, these cell cultures clearly differ from the complex structure and metabolic function of normal intestinal tissue. Pharmaceutical research, therefore, would greatly benefit from an in vitro system with retained intestinal drug-metabolizing function that allows better prediction of drug metabolism and interactions in the small intestine of animals and humans.
Recently, a new in vitro model was developed in our laboratory to study drug metabolism in intact tissue of rat small intestine and colon using tissue slices (de Kanter et al., 2005). Such tissue slices show high metabolic activity toward a variety of drugs (De Kanter et al., 2004). The technique has several advantages compared with the above-mentioned methods: 1) it is highly efficient, considering the large amount of slices that can be prepared even from small tissue samples; 2) in the slices, enzyme systems, cofactors, and transporters are present in their physiological context; and 3) slices are relatively easy to process. These features make the intestinal slice system suitable to study metabolic properties, metabolic clearance, and interactions between diet components and drugs, with respect to metabolism and transporters in animals and humans.
In the present study, we characterized the viability of the intestinal slices in more detail by evaluating the morphology, with special regard to the maintenance of the mucus layer during 3 h of incubation, which may be important to maintain the physiological microclimate for the epithelial cells. Furthermore, the viability of the epithelial cells, which are responsible for drug metabolism, in particular, was evaluated by assessing alkaline phosphatase (AP) retention; this enzyme is abundantly present in the brush border of the small intestinal epithelial cells (van Goor et al., 1989). In addition, the rate of drug metabolism reactions was assessed during the first 3 h of incubation using testosterone (TT) and 7-ethoxycoumarin (7EC) as substrates to evaluate phase I metabolism and 7-hydroxycoumarin (7HC) as a substrate to evaluate phase II reactions. Subsequently, the metabolic rate of the three model compounds along the intestinal tract was studied. To investigate to what extent the precision-cut slice model is suitable to study drug interactions, inhibition of 7EC and TT metabolism by ketoconazole (KT) was studied. Furthermore, drug-mediated induction of 7EC metabolism was investigated in slices of small intestine and colon after 24 h of incubation with β-naphthoflavone (βNF).
Materials and Methods
Chemicals.para-Nitrophenylphosphate, para-nitrophenol, 6β-, 14α-, 15α-, and 11β-hydroxytestosterone (TOH), testosterone, androstenedione, 7HC, 7HC-glucuronide (7HC-GLUC), low gelling temperature agarose (type VII-A), DMSO, eosin Y solution, and KT were purchased from Sigma-Aldrich (St. Louis, MO). Gentamicin, Williams medium E with Glutamax-I, and amphotericin B (Fungizone)-solution were obtained from Invitrogen (Paisley, UK). Alcian Blue, hematoxylin solution according to Mayer, 7EC, βNF and dextran from leuconostoc SSP (dextran-70) were obtained from Fluka Chemie (Buchs, Switzerland). 2-amino-2-methyl-1,3-propanediol (ammediol) was purchased from Aldrich (Steinheim, Germany). 2α-, 16α-, 16β-, 19-, and 11α-TOH were obtained from Steraloids (Newport RI). DePeX mounting medium was purchased from BDH Laboratory (Poole, Dorset, UK). The TissueTek 4583 was obtained from Sakura Finetek Europe BV (Zoeterwoude, The Netherlands). Acetic acid, sodium azide, sodium chloride, calcium chloride, and formaldehyde solution (37%) were obtained from Merck (Darmstadt, Germany). HEPES was obtained from MP Biomedicals (Eschwege, Germany). 7HC-sulfate (7HC-SULF) was a kind gift from P. Mutch (GlaxoSmithKline, Uxbridge, Middlesex, UK). All reagents and materials were of the highest purity that is commercially available.
Animals. Male Wistar (HsdCpb:WU) rats weighing ca. 350 g were purchased from Harlan (Horst, The Netherlands). Rats were housed in a temperature- and humidity-controlled room on a 12-h light/dark cycle with food [Harlan Chow no. 2018 or standard RMH Chow (Hope Farms, Woerden, The Netherlands)] and tap water ad libitum. Pilot experiments showed no significant differences in metabolic rates in intestine from rats fed with the two chows with regard to metabolism of the model substrates (unpublished observation). The animal ethical committee of the University of Groningen approved the use of animals for these experiments.
Preparation of Precision-Cut Slices. Under isoflurane/N2O/O2 anesthesia, the small intestine and colon were excised from the rat and put in ice-cold, oxygenated Krebs-Henseleit buffer (containing 10 mM HEPES and 25 mM d-glucose, pH 7.4). Segments of 3 cm were excised at distinct distances from the stomach and subsequently flushed with ice-cold Krebs-Henseleit buffer. One side of the segment was tightly closed, and the segment was filled with 3% (w/v) agarose solution in 0.9% NaCl (37°C) and then cooled in ice-cold Krebs-Henseleit buffer, allowing the agarose solution to gel. Subsequently, the filled segment was embedded in 37°C agarose solution using a precooled (0°C) tissue embedding unit (Alabama Research and Development, Munford, AL). After the agarose solution had gelled, precision-cut slices (thickness about 300 μm and weight about 2 mg) were cut using a Krumdieck tissue slicer as described before (de Kanter et al., 2005).
Incubation of Precision-Cut Slices. The slices were incubated individually in a six-well culture plate (Greiner Bio-One GmbH, Frickenhausen, Austria) in 3.2 ml of Williams Medium E (with Glutamax-I), supplemented with d-glucose (final concentration 25 mM), gentamicin (final concentration 50 μg/ml), and amphotericin B (final concentration 2.5 μg/ml). The culture plates were placed in a prewarmed cabinet (37°C) in plastic boxes. Slices were incubated under humidified carbogen (95% O2 and 5% CO2) and shaken back and forth 90 times per minute.
Viability Testing.AP Retention. AP is only present in the brush border of epithelial cells in the intestine. The activity retained in slices compared with the total activity is assumed to be indicative of the viability of the epithelial cells. The precision-cut slices were incubated in triplicate for 0, 1, and 3 h. After incubation, the slices were taken out of the wells and placed in 1 ml of 0.05 M ammediol buffer (pH 9.8, at 4°C), and medium and slices were stored separately at 4°C until further analysis. Pilot experiments showed that the AP enzyme activity was stable during the storage period up to 48 h. Just before analysis, slices and medium samples were homogenized by 15 s of sonication using a microsonicator (Sonics & Materials, Danbury, CT). A total of 5 μl of slice homogenate or 15 μl of medium homogenates were added to 190 μl of ammediol buffer (0.05 M, pH 9.8) containing MgCl2 (final concentration, 2 mM) in a 96-well plate (Costar, Cambridge, MA), which was stored on ice. Subsequently, 10 μl of para-nitrophenylphosphate (final concentration 1.25 mM) was added and the plates were incubated for 40 min at 37°C. The reaction was stopped by addition of 20 μl of ice-cold 1 N NaOH. The amount of p-nitrophenol formed was measured using a plate reader at 405 nm. AP retention in slices was calculated as the ratio of the amount of AP activity present in the slices and the total AP activity present in slice and medium together (× 100%). Experiments were performed in five rats in triplicate.
Histomorphology. Precision-cut slices were incubated for 0 and 3 h. After incubation, slices were embedded in TissueTek, snap-frozen in isopentane (–80°C), and stored at –80°C until further usage. Cryostat sections of 5 μm were cut, fixed in formalin-Macrodex (0.9% NaCl, 5.4% dextran-70, 4% formaldehyde, and 1% CaCl2 solution at pH 7.4) for 10 min at 4°C, and rinsed in demi water. Subsequently, the sections were stained with 1% Alcian Blue solution in 3% acetic acid (8 min) and counterstained with hematoxylin (15 min), washed with tap water, incubated with eosin Y solution (2 min), dehydrated, and embedded in DePeX mounting medium.
Stability of Drug Metabolism Rate. Slices were prepared from small intestine (25–40 cm from the stomach) and colon and subsequently incubated in triplicate for 15, 30, 60, and 180 min with TT (final concentration 250 μM), 7EC (final concentration 500 μM), or 7HC (final concentration 500 μM) by addition of 32 μlofa100× stock solution in methanol to the 3.2 ml of medium (n = 4–6). As controls, slices were incubated under standard incubation conditions without substrate, and substrates were incubated without slices. After TT incubation, slice and medium were collected together and stored at –20°C until further use. After thawing (at 4°C), the slices were homogenized for 15 s using a sonicator (Sonics & Materials). Subsequently, 10 μl (1 mg/ml) of 11β-TOH was added as an internal standard, followed by addition of 6 ml of methanol to precipitate the proteins. The total mixture was vortexed and centrifuged using a Beckman-Coulter CS-6KR centrifuge (Beckman Coulter, Fullerton, CA) (10 min, 4°C, at 800g). The supernatant was evaporated under N2, and the residue was dissolved in 500 μl of 50% methanol and stored at –20°C until further analysis. Just before analysis, samples were centrifuged using an Eppendorf centrifuge 5415R during 5 min (4°C, 16,000g) and then analyzed by HPLC as described earlier (van't Klooster et al., 1993), using testosterone and known metabolites as a reference.
Since it was previously shown that 7EC and 7HC and their metabolites are not significantly retained in the tissue, analysis was performed on medium samples only (de Kanter et al., 2005). During 7EC and 7HC incubations, medium samples (200 μl) were harvested after 15, 30, 60, and 180 min and stored at –20°C until further use. After thawing, sodium azide (final concentration 1 mg/ml) was added to inhibit bacterial growth during the analysis and centrifuged using an Eppendorf centrifuge 5415R for 5 min at 4°C, at 16,000g. 7EC, 7HC, 7HC-GLUC, and 7HC-SULF were used as reference. Analysis of 7EC, 7HC, and their metabolites was performed using an HPLC method as described before (Walsh et al., 1995). Total 7EC metabolism, which occurs via phase I metabolism and subsequent conjugation with either glucuronide or sulfate, was calculated from the total 7HC, 7HC-GLUC, and 7HC-SULF formation. All experiments were performed in four to seven rats in triplicate.
Assessment of Metabolic Rate Along the Length of the Intestine and Colon. Precision-cut slices were prepared from small intestine at 5, 15, 25, 40, 60, or 80 cm and the last 3 cm (ileum) from the stomach, and from three colon segments, equal in length (about 5 cm). The slices were incubated in triplicate with 7EC (final concentration 500 μM), 7HC (final concentration 500 μM), or TT (final concentration 250 μM) for 3 h and analyzed as described above. Per experiment, slices to assess the metabolic activity along the small intestine and colon of both 7EC and 7HT as TT were obtained from the same animal. Each experiment was performed in seven rats using three slices from each incubation procedure.
Inhibition Studies. Precision-cut slices were prepared from small intestine (at 25–40 cm from the stomach) and preincubated for 1 h with or without the inhibitor ketoconazole (50 μM). Subsequently, TT (final concentration 250 μM) or 7EC (final concentration 500 μM) was added and slice incubation was prolonged for 3 h under standard conditions. Samples were harvested and analyzed as described above. Each experiment was performed in three rats using three slices from each incubation procedure.
Induction Studies. Precision-cut slices were prepared from small intestine (at 25–40 cm from the stomach) and colon and incubated with βNF (final concentration 50 μM or 100 μM, added as a 100× concentrated stock solution in DMSO) for 24 h under standard incubation conditions, as described under Incubation of Precision-Cut Slices. Control slices were incubated in medium with DMSO but without βNF. After 24 h of incubation, slices were transferred to fresh medium and incubated for 3 h with 7EC (final concentration 500 μM) to assess metabolic activity. Samples were analyzed as described above. Each experiment was performed in three rats in triplicate.
Protein Determination. After incubation with 7EC or 7HC, slices were stored at –20°C until further use. After thawing, 20 μl of 5 N NaOH was added to the slice followed by 40 min of incubation at 37°C to dissolve the tissue. Water (980 μl) was added to dilute the NaOH concentration to 0.1 M, after which the mixture was homogenized by 5 s of sonication. Samples were diluted and the protein content was determined using Bio-Rad protein assay dye reagent (Bio-Rad, Munich, Germany) using bovine serum albumin as standard. After testosterone incubation, the protein was not determined. For these slices, the average protein contents of comparable incubations with 7EC and 7HC within the same experiment were used.
Liver Slices. Data on liver slice metabolism were taken from earlier studies (De Kanter et al., 2002).
Statistics. Statistical significance was determined using Student's t test.
Results
Viability of Intestinal and Colon Slices. Three different parameters were evaluated after 3 h of incubation to assess the viability of small intestinal and colon slices.
Alkaline Phosphatase Retention. AP retention was measured to assess the viability of epithelial cells of small intestinal slices. In absolute amounts, 100% AP retention was 18.0 ± 1.3 U/slice (mean ± S.D.). Only a slight decrease to 86 ± 2% (mean ± S.E.M.) in AP retention was observed after 1 h of incubation. The AP retention after 3 h of incubation (77 ± 4%) did not significantly differ from that after 1 h of incubation (p = 0.08). AP activity was below the detection level in colon.
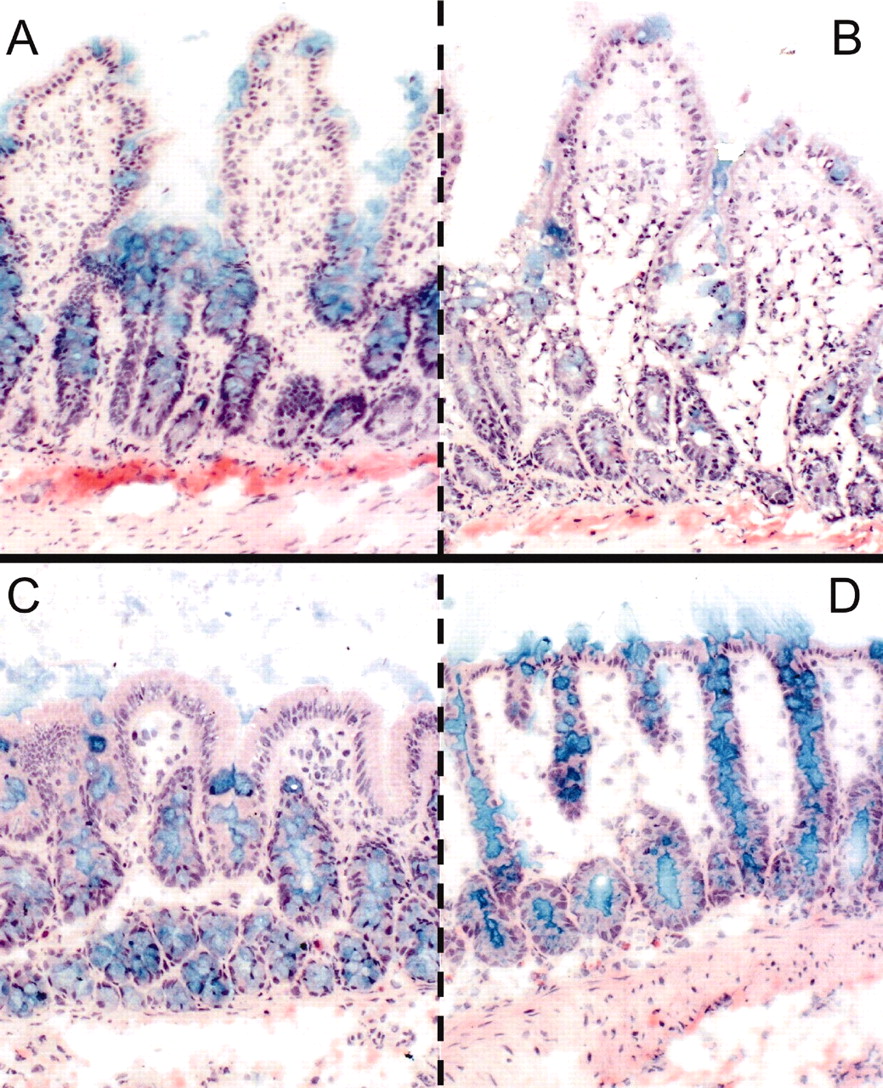
Histomorphology. Viability of colon and small intestinal slices was assessed by histomorphological evaluation. Alcian Blue staining indicated that goblet cells still contained mucus, and the mucus layer was partly retained after preparation and after 3 h of incubation. In addition, epithelial cells showed intact morphology, but villi of small intestine appeared somewhat flattened/broadened during incubation (Fig. 1).
Stability of Drug Metabolism Rate.Figure 2 shows the stability of metabolic rates of 7EC, TT (phase I substrates), or 7HC (phase II substrate) during the first 3 h of incubation in slices prepared from small intestine and colon. A time-dependent linear formation rate of metabolites was observed during 3 h of incubation for all compounds tested in both small intestine (R2 > 0.99) and colon (R2 > 0.98). 7EC metabolism in colon was low and, in some experiments, below the detection level. After incubation with testosterone, only androstenedione formation was detected both in small intestine and colon.
Assessment of Metabolic Rate Along the Length of the Intestine. The metabolic activity of model compounds was assessed in slices from small intestine and colon excised at distinct distances from the stomach. Activities showed a decreasing gradient along the small intestine toward colon, with the slope of the gradient depending on the model compound used.
O-Deethylation (mediated by CYP1A and 1B) of 7EC was constant and highest in the first 40 cm of the proximal site of the small intestine (about 40 pmol/min/mg protein or 4.4 nmol/min/g wet weight since the intestinal protein content appeared to be 109 mg/g wet weight) (Fig. 3A), after which the metabolic rates gradually and significantly decreased (5, 15, and 25 cm toward 60, 80 cm, and ileum p < 0.05) to 8 pmol/min/mg protein (or 0.9 nmol/min/g wet weight) in ileum (Fig. 3A). Although 7EC O-deethylation in colon slices was below the detection level in some experiments and in others, significantly lower compared with the proximal part of the small intestine (p < 0.001), the average metabolic rate in colon (about 2 pmol/min/mg protein or 0.2 nmol/min/g wet weight) was nonsignificantly lower than that in the distal part of the small intestine. The increase in metabolic rate in colon in the distal direction was not significant. 7EC O-deethylation was significantly (p < 0.01) lower in small intestine (4–23%) and colon (1%) compared with liver slices [207 pmol/min/mg protein (De Kanter et al., 2002) or 22 nmol/min/g wet weight].
To compare the activity of the metabolically active cells in liver and intestine (hepatocytes and epithelial cells), metabolic activity was recalculated per milligram of hepatocyte or epithelial protein. Due to lack of data, activity is expressed per epithelial protein and not per enterocyte protein, despite the fact that enterocytes are the metabolically active epithelial cells. Thus, taking into account that small intestine contains 18.1 mg of epithelial protein per g wet weight (Koster and Noordhoek, 1983) and our slices contained 109 mg of protein per g wet weight, the specific activity per milligram of epithelial protein is 109/18.1 = 6 times the activity per milligram of small intestinal protein. Due to lack of data, the same factor is used for colon. Likewise, with liver, about 80% of liver protein is hepatocyte protein. Assuming that roughly 50% of the hepatocytes in the slice contribute to the metabolic activity (unpublished research), the activity per milligram of hepatocytes is 2 × 100/80 = 2.5.
The metabolic activities of 7EC O-deethylation in epithelial cells of small intestine and colon were, respectively, 4 to 26 and 1.3 nmol/min/g epithelial cell, which is 7 to 47%, respectively, or 2.4% of the hepatocyte activity (55 nmol/min/g hepatocyte).
Androstenedione was the only metabolite that could be detected after incubation of slices with TT and appeared to be formed at the highest rate in the first 40 cm of the proximal site of the small intestine (about 550 pmol/min/mg total protein or 60 nmol/min/g total wet weight) (Fig. 3B) and decreased significantly (p < 0.01) in the distal direction to 61 pmol/min/mg protein (or 7 nmol/min/g wet weight). In colon, the metabolic rate of androstenedione formation was significantly lower (232–105 pmol/min/mg protein or 25–11 nmol/min/g wet weight) compared with the proximal part of the small intestine (p < 0.01), but significantly higher compared with the distal part of the small intestine (p < 0.02), and also tended to decline in the distal direction. The activity found in the proximal part of the small intestine was comparable to that of liver [666 pmol/min/mg total protein (De Kanter et al., 2002) or 72 nmol/min/g total wet weight]. On the other hand, colon (16–35%) and the distal part of the small intestine (11%) showed significantly lower specific activity compared with liver. P450-mediated reactions, such as 6β-, 15α-, 19-, 16α-, 16β-, and 2α-TOH formation, were not detectable in both small intestine and colon. These metabolites were readily detected using liver slices at rates up to 563 pmol/min/mg protein, resulting in a total metabolic rate in liver for testosterone of 1230 pmol/min/mg protein (or 131 nmol/min/g wet weight). Converted to the amount of active cells, the metabolic rates toward testosterone per epithelial cell in small intestine (proximal, 360; and distal, 42 nmol/min/g epithelial cell) were comparable or higher compared with liver (328 nmol/min/g hepatocytes).

Morphological evaluation of slice preparations of the small intestine after 0 h (A) and 3 h (B) of incubation and colon slices after 0 (C) and 3 h (D) of incubation. Staining: Alcian Blue counterstained with hematoxylin and eosin (100×).
Phase II metabolism was assessed by measuring 7HC biotransformation. The glucuronidation rate was high in the proximal part of the small intestine (663 pmol/min/mg protein or 72 nmol/min/g wet weight) (Fig. 3C) and decreased significantly only after 60 cm (p < 0.05) in the distal direction to 326 pmol/min/mg protein (or 36 nmol/min/g wet weight). Subsequently, in the proximal part of the colon, the metabolic rate increased again to even higher rates (800 pmol/min/mg protein or 87 nmol/min/g wet weight) compared with the distal part of the small intestine and decreased significantly (p < 0.05) to 490 pmol/min/mg protein (or 53 nmol/min/g wet weight) distally. Glucuronidation rates in both small intestine and colon slices were comparable to rates measured in liver slices [700 pmol/min/mg protein (De Kanter et al., 2002) or 74 nmol/min/g wet weight], being 49 to 97% and 72 to 118%, respectively. Converted to the specific activity of epithelial cells in small intestine (214–435 nmol/min/g epithelial cell) and colon (322–525 nmol/min/g epithelial cell), the activity was 1 to 3 times higher compared with hepatocytes (186 nmol/min/g hepatocyte).
Sulfation of 7HC (Fig. 3D) showed a different pattern along the small intestine than for other metabolic pathways: the metabolic rate initially increased nonsignificantly from 43 pmol/min/mg protein (4.7 nmol/min/g wet weight) to 58 pmol/min/mg protein (or 6 nmol/min/g wet weight), with the highest metabolic rate measured at 15 cm from the stomach. The metabolic rates decreased significantly from 40 cm onward to a final rate of 21 pmol/min/mg protein (2 nmol/min/g wet weight) in ileum (p < 0.05). In colon, the proximal metabolic rate was significantly higher than in the distal part of the small intestine (p < 0.05). A gradient decreasing from 51 to 38 pmol/min/mg protein (or 6 to 4 nmol/min/g wet weight) was shown in the distal direction. Specific metabolic activities in the small intestine and colon appeared to be equal to or even higher than those of liver slices [42 pmol/min/mg protein (De Kanter et al., 2002) or 4.5 nmol/min/g wet weight], being 50 to 140% and 90 to 120% of liver slice values. When converted to sulfation activity per amount of active cells, the cells in small intestine (14–38 nmol/min/g epithelial cell) and colon (25–34 nmol/min/g epithelial cell) have 1 to 3 times higher activity compared with hepatocytes (11 nmol/min/g hepatocyte).

Metabolizing activity of precision-cut slices prepared from small intestine (at 25–40 cm from the stomach) (♦) and colon (▪) during 3 h of incubation under standard conditions: A, 7EC into 7HC; B, testosterone into androstenedione; C, 7HC into 7HC-GLUC; and D, 7HC-SULF. Results are mean ± S.E.M. of four to six rats; in each experiment at least three slices were incubated per time point.
Inhibition Studies. Slices were preincubated with KT for 1 h, followed by 3 h of incubation with TT or 7EC. TT metabolism was only partially inhibited to 61 ± 4% (from 432.6 ± 194 to 255.3 ± 89 pmol/min/mg protein); in contrast, 7EC metabolism was totally blocked (from 22.7 ± 6.3 pmol/min/mg protein). Results are mean ± S.E.M. of three rats; in each experiment, three slices were incubated per treatment.
Induction Studies. Induction of 7EC metabolism in precision-cut slices of small intestine and colon was measured after 24 h of incubation with βNF (Fig. 4). A very strong induction of phase I metabolism of 7EC was found in both small intestine and colon. Directly after slicing, the metabolic rate of 7EC in slices prepared from small intestine was about 43 pmol/min/mg protein (“fresh slices”), but declined to nearly undetectable levels after 24 h of preincubation in control medium without βNF (p < 0.01). In contrast, when small intestinal slices were preincubated with βNF for 24 h, the metabolic rate increased strongly to 140 pmol/min/mg protein (p < 0.001 compared with fresh slices). The increase was independent of the βNF concentrations used; both concentrations (50 μM and 100 μM) showed a 3.5-fold induction of the metabolic rate compared with the rate of fresh slices.
In colon slices, phase I metabolism of 7EC could be induced by βNF as well. 7EC O-deethylation of colon slices was near the detection level in both fresh slices and slices after 24 h of preincubation. However, when incubated for 24 h with βNF, the metabolic rate increased tremendously to about 100 pmol/min/mg protein (p < 0.05).
Discussion
Recently, we presented rat small intestinal and colon precision-cut slices as a tool to study intestinal metabolism (de Kanter et al., 2005). In this study, we further characterized the viability and functionality of these slices. We assessed the gradient of several phase I and phase II enzyme activities along the intestine, as well as the applicability of slices to investigate drug interactions.
The metabolic rate of model compounds (7EC, 7HC, or TT) was constant, as was the AP retention in small intestinal slices during the first 3 h of incubation. In addition, hardly any signs of degradation of colon and intestinal slices were observed upon histomorphological examination. Moreover, the mucus layer, which presumably protects the epithelial lining in vivo (Allen et al., 1984), remained present. Altogether, these results imply the integrity of slices during 3 h of incubation.
The intestine is not homogeneous with respect to drug metabolism along its length (Takemoto et al., 2003) nor along the villi (Dubey and Singh, 1988). Generally, a decreasing gradient in the distal direction is reported (Kaminsky and Zhang, 2003). However, the preparation method of intestinal tissue has been found to be critical for the outcome of quantitative evaluation of intestinal metabolism (Plumb et al., 1987). Gradients of mRNA expression only allow speculation of protein expression gradients, whereas Western blotting and immunohistochemical staining of metabolizing proteins reveal only the enzyme distribution pattern along the tract. This might not necessarily correlate with the in vivo distribution of the metabolic activity, since other factors, such as cofactors and transporters, can be rate-limiting. Tissue slices may be preeminently suitable to study the distribution of metabolic activity along the intestine, since no isolation steps are required. In addition, cofactors and transporters are supposed to be present at physiological levels.

Metabolic activity along the intestinal tract. Precision-cut slices from small intestine were prepared at distinct distances from the stomach, incubated for 3 h with 7EC (A), testosterone (B), or 7HC (C and D). Phase II metabolism of 7HC is shown in two different graphs: the formation of 7HC-GLUC (C) and 7HC-SULF (D). Results are mean ± S.E.M. of seven rats. Significant differences toward small intestine (at 5 cm) are indicated by *, p < 0.05, **, p < 0.01; ***, p < 0.001. Significant differences between upper colon and ileum, middle colon, and lower colon were tested and indicated by #, p < 0.05; ##.

Induction of 7EC metabolism in precision-cut slices prepared from small intestine (25–40 cm from stomach) (black columns) and colon (open columns). Slices were incubated for 24 h with 0, 50, or 100 μM βNF transferred to fresh medium and incubated with 500 μM 7EC for 3 h. Activity in fresh slices was determined directly after slicing without preincubation. (N.D., nondetectable.) Results are mean ± S.E.M. of three rats. In each experiment three slices were incubated per treatment. Significant differences toward the activities of fresh small intestinal slices are indicated by **, p < 0.01; ***, p < 0.001. Significant differences in colon metabolic activity toward fresh colon are indicated by #, p < 0.05.
Gradients of metabolic activity differed depending on the substrate used. Metabolic activity toward 7EC (CYP1A and 2B) was the highest in the proximal part of the small intestine, which is in accordance with the reported higher levels of CYP1A1 and 2B1 protein in duodenum compared with ileum (de Waziers et al., 1990). However, others described an initial increase followed by a decrease in metabolic activity toward 7EC in the distal direction along the intestinal tract (Lin et al., 1999). To our knowledge, data describing the 7EC metabolism gradient in colon have not been published before.
In rat, testosterone is metabolized by several P450 isozymes such as CYP1A1/2, 2C11, and 3A1/2 (Arlotto et al., 1991), and by 17β-hydroxysteroid dehydrogenase (17β-HSD) (Farthing et al., 1982). Androstenedione formation is mediated by 17β-HSD and several P450 isozymes (Sohlenius-Sternbeck and Orzechowski, 2004), as well. This was confirmed in the present study, where KT inhibited androstenedione formation by 40%. KT totally blocked 7EC metabolism as expected. In addition, the gradient of androstenedione formation found appeared to be similar to the 17β-HSD activity (Farthing et al., 1982) along the length of the intestine, indicating a large contribution of 17β-HSD to androstenedione formation. An increase of androstenedione formation in the distal direction, including colon, using S9 fractions was reported by others (Sohlenius-Sternbeck and Orzechowski, 2004). However, these activity rates were over 100-fold lower (0.35 nmol/min/g intestine) than ours, impeding good comparison. Besides androstenedione, no other metabolites of TT were found in the present study, contrary to the findings of Takemoto et al. (2003), who found considerable 6β-hydroxytestosterone formation (CYP3A). However, others reported nondetectable levels of CYP3A1 mRNA (Martignoni et al., 2004) or low levels of 6β-hydroxytestosterone formation (Sohlenius-Sternbeck and Orzechowski, 2004) in intestinal preparations. We found that when four slices per well were incubated, 6β-hydroxytestosterone formation was detected (unpublished observation). This indicates that CYP3A activity is present, but undetectable, due to the small amount of tissue used.
Phase II metabolism along the intestine was assessed using 7HC as a substrate, which is conjugated. For 7HC-GLUC formation, we observed a decreasing gradient in the distal direction. Sulfation, thought to be important in overall metabolism in small intestine, especially in comparison with total liver capacity (Lin et al., 1999), showed a small, but nonsignificant increase followed by a decrease.
The underlying cause of the gradients is unknown. A metabolic gradient could be explained by a gradient in the number of metabolizing cells and/or in enzyme activity per enterocyte along the intestine. The latter can be caused by enterocyte differences in enzyme expression, which may be induced by gradients of transcription factors or inducing components, or by differences in the presence of transporters. The number of enterocytes alone cannot explain the gradients, since activity patterns differed for the substrates used.
The rate of 7EC O-deethylation in proximal small intestine (4.4 nmol/min/g intestine) is over 100-fold higher than that reported by others (10.3 and 16.6 pmol/min/mg microsomal protein in mucosa, which corresponds to 25 and 40 pmol/min/g intestine) using, respectively, microsomes and everted sacs of proximal small intestine (Takemoto et al., 2003). Since microsomal activities are measured under optimal cofactor conditions, one would expect at least equal or higher activities in microsomes compared with intact cells in which cofactor concentrations may be suboptimal. Possibly, the lower activity found with microsomes is caused by enzyme inactivation by proteases or their low isolation yield, which does not play a role when slices are used.
Small intestinal specific activity per total protein was about 4 to 23% of the hepatic 7EC O-deethylation, confirming the results of Shirkey et al. (1979) with microsomes. However, when expressed per metabolically active epithelial cell, the activity is estimated to amount to 8 to 47% of hepatocytes, indicating the high metabolic competence of epithelial cells.
The metabolic activities toward TT found in our system (proximal, 360; and distal, 42 nmol/min/g epithelial cell) are comparable with the androstenedione formation rates in scraped mucosa homogenates: 595–29-157 nmol/min/g intestinal mucosa (duodenum-ileum-colon) reported by others (Farthing et al., 1982). In slices, epithelial cells of small intestine and colon are estimated to process androstenedione formation about 0.2 to 2 times faster than hepatocytes. However, the competence of colon epithelial cells is probably underestimated, since the scaling factor for small intestinal epithelial protein is used due to lack of colon data. The colon epithelial surface area is smaller than that of small intestine, whereas the muscle layer in colon is thicker. In contrast, Sohlenius-Sternbeck and coworkers found in small intestinal S9 fractions only 0.9 to 5.2% of the androstenedione formation measured in liver (Sohlenius-Sternbeck and Orzechowski, 2004).
7HC glucuronidation (36–72 nmol/min/g intestine) and sulfation in slices (2–6 nmol/min/g intestine) is about 1 to 5 times higher than in isolated epithelial cells [13.1 and 2.3 nmol/min/g intestine (Koster and Noordhoek, 1983)]. Interestingly, the rates of glucuronidation and sulfation in small intestinal and colon epithelial cells were estimated to be 1 to 3 times higher than the estimated rates of hepatocytes measured in liver slices.
The high metabolic rate in intestinal epithelial cells in comparison with the hepatocytes indicates the important role of the intestine in drug metabolism, its ability to contribute significantly to the in vivo first-pass metabolism, and the risk of site-specific toxicity caused by toxic metabolites formed in the gut.
Up to now, an adequate in vitro model to study enzyme induction in the intestine was lacking. To mimic enzyme induction in vitro, an intact cellular machinery is required. Existing in vitro systems such as everted sacs and Ussing chambers have never been proven to survive long enough to allow incubation with an inducer for 24 h.
In the present study, we found a clear induction of the metabolic activity of slices toward 7EC after incubation for 24 h with βNF [induction via aryl hydrocarbon receptor (Lin and Lu, 1998)]. Administered in vivo, βNF was reported to significantly induce CYP1A1 mRNA, protein, and activity (Zhang et al., 1997) and 7EC O-deethylation in small intestine (Borm et al., 1983) and colon (McDanell and McLean, 1984). Our finding is also consistent with the induction of CYP1A1/1A2 and 2B1 mRNA by βNF in intestinal slices reported by Martignoni et al. (2004).
These data, showing a prominent induction of metabolic activity in slices after 24 h of incubation, are a strong indication that the low basal metabolism of 7-EC after 24 h of preincubation cannot be due to loss of viability only. After all, viability is indirectly indicated by inducibility and by a metabolic activity after induction being much higher than in the control slices at the first 3 h of incubation. This decrease may, rather, indicate some kind of down-regulation, due to a lack of physiological stimuli, or to the loss of epithelial cells during incubation. Further study is required to confirm this hypothesis.
In conclusion, the present study has shown that the intestinal slice model can serve as a tool to study drug metabolism in the intestine in vitro, since it was stable during at least 3 h, and comparable or even higher metabolic activities than described for other methods were observed. In addition, the model is able to detect gradients of metabolic activity along the intestine, inhibitory drug-drug interactions, and induction responses. We conclude that the slice model is a promising tool to study these phenomena in small intestine and colon in vitro, as well as in human tissue in future.
Acknowledgments
Dr. J. Nieken is acknowledged for help with histomorphological assessment, J. Visser for help with HPLC analysis, and A. van Loenen-Weemaes for help with animal handling.
Footnotes
-
This study is supported by the Technology Foundation STW, Applied Science Division of NWO and the Technology Programme of the Ministry of Economic Affairs, and Yamanouchi Europe.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005686.
-
ABBREVIATIONS: AP, alkaline phosphatase; TT, testosterone; TOH, hydroxytestosterone; 7HC, 7-hydroxycoumarin; 7HC-GLUC, 7-hydroxycoumarin glucuronide; 7HC-SULF, 7-hydroxycoumarin sulfate; 7EC, 7-ethoxycoumarin; 17β-HSD, 17β-hydroxysteroid dehydrogenase; βNF, β-naphthoflavone; DMSO, dimethyl sulfoxide; KT, ketoconazole; HPLC, high-performance liquid chromatography.
- Received May 24, 2005.
- Accepted July 27, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}