


Abstract
Increasing reports of time-dependent inhibition of cytochrome P450 (P450) suggest further emphasis on interpreting the consequences, either from a pharmacokinetic or toxicological perspective. Two automated, time-dependent inhibition assays with a liquid chromatography-tandem mass spectrometric endpoint are presented. The initial assay utilizes human liver microsomes, a single concentration of inhibitor, and a single preincubation time of 30 min. Phenacetin, diclofenac, S-mephenytoin, bufuralol, and midazolam are used as substrates for CYP1A2, 2C9, 2C19, 2D6, and 3A4, and the assay differentiates between reversible and irreversible inhibition. The second assay uses individual recombinant human P450s, six inhibitor concentrations, and three time points to accurately define kinact and KI. A good correlation is demonstrated between kinact/KI and partition ratio, indicating that both terms are related in describing the efficiency of enzyme inactivation. Despite the single preincubation time point of 30 min used in the initial assay, a good relationship has been found to exist between the unbound IC50 estimated from this initial screen and the kinact/KI ratio derived from the more extensive subsequent single P450 assay. The higher throughput human liver microsomal assay can therefore generate IC50 values that can be used to predict the pharmacokinetic impact on cotherapies from the estimated kinact/KI ratio, predicted human dose, and pharmacokinetics.
Drug-drug interactions (DDIs) following drug therapy, which result in a high number of hospital admissions and even deaths (Lazarou et al., 1998; Kohler et al., 2000), are primarily caused by macromolecule binding of reactive species or drug cotherapy resulting in plasma concentrations of one of the coadministered drugs being elevated to toxic levels (Hollenberg, 2002). The mechanism is frequently competition at the active site of drug-metabolizing enzymes. Since multiple drug therapy is a very common practice, the possibility of DDI therefore exists in the majority of patients. This is a high profile issue for drug discovery and development programs, and there is even an on-line DDI database that shows the number of drugs currently implicated (Carlson et al., 2002). Great importance is now placed on in vitro studies as tools for predicting in vivo DDIs, particularly those resulting from cytochrome P450 (P450) inhibition (Lin and Lu, 1998), since the metabolic elimination of a large number of drugs is dependent on the P450 family of enzymes.
To date, 57 human cytochrome P450 genes have been identified (http://drnelson.utmem.edu/CytochromeP450.html), but remarkably, only three human P450s (3A4, 2C9, and 2D6) perform the majority of biotransformations involving pharmaceuticals (Smith et al., 1998). CYP1A2, 2B6, 2C8, 2C19, and 2E1 are also involved, but to a much lesser extent. Inhibition of P450-dependent metabolism can generally be classified into three categories: reversible, quasi-irreversible (when compounds complex the heme prosthetic group and leave the P450 functionally inactive), and irreversible or mechanism-based (compounds covalently bind to the heme or the surrounding protein) (Hollenberg, 2002). Reversible P450 inhibition screening has been commonplace within drug discovery functions for some time, but reports of time-dependent inhibition (TDI) are increasing in prevalence, indicating that more emphasis is now being placed on interpreting the effects, either from a pharmacokinetic or toxicological perspective. Moreover, irreversible and quasi-irreversible inhibition are often viewed as more serious than reversible inhibition, since the inhibitory effect remains after elimination of the parent drug from the body. TDI is an unusual occurrence with most enzymes, but it is observed at a higher frequency in P450-catalyzed reactions, perhaps due to the reactivity of the oxygenated species formed during the course of the oxygenation reactions (Hollenberg, 2002). There are examples of irreversible or quasi-irreversible P450 inhibition across many classes of therapeutic drugs, recreational drugs, and herbal medicines (Zhou et al., 2005), and all of the major drug-metabolizing P450s have been implicated (Kunze and Trager, 1993; Lopez-Garcia et al., 1994; Newton et al., 1995; Koenigs and Trager, 1998; Chun et al., 2001a,b; Ha-Duong et al., 2001; Palamanda et al., 2001; Bertelsen et al., 2003; Fan et al., 2003; Lu et al., 2003; Polasek et al., 2004; Zhou et al., 2004).
In a drug discovery setting, three different analytical endpoints are typically used for the study of P450 inhibition in vitro: liquid scintillation counting of radioactivity liberated during site-specific metabolism (Moody et al., 1999), selective analysis of fluorescent metabolites (Crespi and Stresser, 2000), and mass spectrometry (Yin et al., 2000; Weaver et al., 2003). Although all these formats are available in this laboratory, two automated time-dependent inhibition assays are presented that use an LC/MS-MS endpoint. The initial assay utilizes human liver microsomes, a single concentration of inhibitor, and a single incubation time. The assay uses industry-recommended P450 substrates (Bjornsson et al., 2003), and is shown to differentiate between reversible and irreversible inhibition and to give a good prediction of kinact/KI (kinetic parameters of time-dependent inhibition). The second assay uses single recombinant human P450s, six inhibitor concentrations, and three time points to define kinact and KI accurately.
Materials and Methods
Chemicals. Furafylline, ticlopidine, troleandomycin, mifepristone, 6β-hydroxy testosterone, testosterone, methylenedioxy-N-methylamphetamine (MDMA), β-nicotinamide adenine dinucleotide phosphate, reduced form (NADPH), phenacetin, and diclofenac (sodium salt), were purchased from Sigma-Aldridge (Poole, Dorset, UK). S-Mephenytoin, bufuralol (hydrochloride salt) and midazolam (hydrochloride salt) were purchased from Ultrafine Chemicals (Manchester, UK).
Dimethyl sulfoxide (DMSO), formic acid, acetonitrile, and methanol were purchased from Fisher Scientific (Loughborough, UK). Tienilic acid was synthesized at AstraZeneca Charnwood.
Source of Human Liver Microsomes and Cytochrome P450 Enzymes. The HLM pool (48 donors) was supplied by BD Gentest (Oxford, UK). Escherichia coli membranes coexpressing human P450 and human NADPH-P450 reductase (1A2LR, 2C9R, 2C19R, 2D6R, 3A4LR) were purchased from Cypex (Dundee, UK).
Substrate Selectivity. With the possible exception of phenacetin (O-deethylation), the P450 probe reactions used in this study are P450-selective when the substrate concentrations are equivalent to their Km values (Weaver et al., 2003). However, to minimize the impact of reversible inhibition [since IC50 = Ki × (1 + substrate concentration/Km) for competitive inhibition; Segel, 1975], substrate concentrations equivalent to 4 times the Km values were used for the time-dependent inhibition assays. Using these elevated concentrations (75 μM, 10 μM, 100 μM, 40 μM, and 10 μM for phenacetin, diclofenac, S-mephenytoin, bufuralol, and midazolam), the selectivity of each P450 probe reaction was investigated using individual P450s at enzyme concentrations intended to mimic those present in the human liver microsomal preparation (40, 60, 15, 5, and 100 pmol/ml for CYP1A2, 2C9, 2C19, 2D6 and 3A4, respectively; Soars et al., 2003). Incubations were performed in a 37°C water bath in glass vials containing NADPH (1 mM), recombinant human P450, substrate, and phosphate buffer (0.1 M, pH 7.4). After 10, 20, and 30 min, aliquots (100 μl) were removed and pipetted into 1.2-ml polypropylene tubes containing ice-cold methanol (100 μl). The samples were centrifuged and the supernatant was analyzed by LC/MS-MS, as described below.
TDI Assay Using HLMs. The fully automated TDI assay using human liver microsomes is performed using a Genesis robot (Tecan, Reading, UK) running Gemini software. The assay is designed to run 18 compounds (12 test compounds + 6 standard inhibitors). The Tecan bed layout contains a sample rack, two incubation plates, a reagent rack, and a quench rack. The sample rack consists of 1.2-ml polypropylene tubes in an 8 × 12 (96-well format) box (VWR International, Poole, UK). The tubes contain test and standard inhibitor stocks (100 times the initial incubation concentration in DMSO). The standard inhibitor stock solution concentrations are as follows: 1 mM furafylline, 0.05 mM tienilic acid, 0.5 mM ticlopidine, 0.5 mM MDMA, 0.15 mM troleandomycin, and 0.5 mM mifepristone. Initial investigations with test compounds use 1 mM stocks (10 μM preincubation concentration). The two incubation plates are 96-well polypropylene microplates (Corning Life Sciences, Schiphol-Rijk, The Netherlands). The reagent rack consists of two polypropylene inserts, each containing two reagent reservoirs (Biomek reservoirs, purchased from Beckman Coulter, Fullerton, CA) within an aluminum block heated to 37°C. The separate reservoirs contain HLMs, NADPH, and substrate solution. The quench rack consists of 1.2-ml polypropylene tubes in an 8 × 12 (96-well format) box (VWR International).
The assay runs a preincubation (30 min, 1 mg/ml HLM incubation containing 1 mM NADPH and test inhibitor) and a secondary incubation (containing an aliquot from the preincubation, 1 mM NADPH, and substrates for CYP1A2, 2C9, 2C19, 2D6, and 3A4). The secondary incubation runs for 15 min. The substrates are prepared manually: aliquots of methanolic stocks of phenacetin (50 μl of a 8.96 mg/ml stock), diclofenac (50 μl of a 2.12 mg/ml stock), S-mephenytoin (50 μl of a 14.56 mg/ml stock), bufuralol (50 μl of a 7.94 mg/ml stock), and midazolam (50 μl of a 2.42 mg/ml stock) are pipetted into a 20-ml glass scintillation vial, evaporated to dryness under nitrogen gas, and stored at –20°C until required. On the day of the assay, the dried substrate cocktail is resuspended in phosphate buffer (20 ml, 0.1 M, pH 7.4) by vortexing and sonication (20 min in a sonic bath at 37°C).
The preincubation plate comprises HLMs (195 μl, 1.3 mg/ml microsomal protein) and aliquots (2.5 μl) of inhibitor stock solution or DMSO for the control incubations. The preincubation is initiated by the addition of NADPH (52.5 μl, 5 mM) to the left-hand half of the plate and phosphate buffer (52.5 μl, 0.1 M, pH 7.4) to the right-hand half of the plate. The preincubation plate is then moved into a Tecan MIO incubator set at 37°C. During this preincubation stage, the secondary incubation plate is prepared: the substrate cocktail (150 μl) is added to all wells and an additional aliquot of NADPH (50 μl, 5 mM) is added. At the end of the 30-min preincubation, an aliquot of this incubation (50 μl) is removed and added to the secondary incubation plate. The final secondary incubation concentration of the substrates is as follows: 75 μM phenacetin, 10 μM diclofenac, 100 μM S-mephenytoin, 40 μM bufuralol, 10 μM midazolam. The secondary incubation progresses for 15 min in a Tecan MIO incubator set at 37°C prior to quenching. An aliquot (50 μl) is removed from the incubation and pipetted into the tubes in the quench rack (containing 100 μl of methanol). Quenched samples are chilled at –20°C for 2 h, centrifuged at 2000g for 15 min, and the supernatants are transferred to microtiter plates for LC/MS-MS analysis.
Automated TDI Assay Using Individually Expressed Human Cytochrome P450 Enzymes to Determine KI and kinact. The fully automated KI and kinact TDI assay is designed to run three compounds (two test compounds plus one standard inhibitor). The incubation is performed in phosphate buffer (0.1 M, pH 7.4) and contains P450 (25 pmol/ml in the preincubation), NADPH (1 mM), substrate for the P450 being incubated, and test inhibitor in DMSO (1% v/v). The substrate is prepared as detailed before (“Automated TDI Assay using HLMs”), but only a single substrate is used in this assay. The first stage of the assay is a serial dilution of the test inhibitor stocks to give five secondary solutions for each compound over a 50-fold concentration range. Following the serial dilution, P450 membranes (195 μl, 32 pmol/ml) are added to the preincubation section of the incubation plate. Substrate (150 μl) and NADPH (50 μl, 5 mM) are added to the secondary incubation section. The inhibitor solutions are then spiked (2.5 μl) into the preincubations and incubation is initiated by the addition of NADPH (52.5 μl, 5 mM NADPH). The plate is incubated at 37°C for 10 min. At this time point, aliquots (50 μl) of the 10-min preincubates are transferred to secondary incubations containing substrate (150 μl) and NADPH (50 μl, 5 mM). After another 10 min (20-min preincubation), the procedure is repeated and, finally, the procedure is repeated for the 30-min preincubates. The secondary incubations are allowed to proceed for 15 min. At this time, aliquots (50 μl) are removed and added to the quench tubes containing methanol (100 μl). Quenched samples are chilled at –20°C for 2 h, centrifuged at 2000g for 15 min, and the supernatants are transferred to microtiter plates for LC/MS-MS analysis.
Manual KI and kinact Determinations for Standard Time-Dependent Inhibitors of CYP3A4. Preincubations and secondary incubations were performed in 96-well microtiter plates. Wells contained recombinant CYP3A4 (195 μl, 32 pmol) diluted from the stock (11 nmol/ml) in phosphate buffer (0.1 M, pH 7.4). The wells also contained test inhibitor (2.5 μl at 100 times the incubation concentration) in DMSO, or DMSO (2.5 μl) for solvent controls. The concentrations of the test inhibitors in the preincubation were 4, 10, 20, 40, 100, and 200 μM (erythromycin); 0.05, 0.125, 0.25, 0.5, 1.25, and 2.5 μM (troleandomycin); and 0.2, 0.5, 1, 2, 5, and 10 μM (mifepristone). A single plate was set up for each individual inhibitor investigated. The plates were prewarmed for 3 min in an incubator at 37°C. Prewarmed NADPH (52.5 μl, 5 mM), or phosphate buffer (52.5 μl) for zero time point samples, was added to wells A1 to H1 and A2 to H2, respectively, and the incubations continued. At the set time points of 0, 10, 30, 60, 150, and 300 s, aliquots (50 μl) were removed from the preincubation wells and added to wells containing prewarmed NADPH (50 μl, 5 mM) and midazolam dose solution (150 μl, 16.7 μM). After a 10-min incubation, aliquots from each well (50 μl) were removed and added to 1.2-ml polypropylene tubes containing ice-cold methanol (100 μl). The samples were centrifuged and analyzed by LC/MS-MS.
The Use of Testosterone as an Alternative CYP3A4 Substrate when Estimating KI and kinact. For studying reversible inhibition of CYP3A4, it is now commonplace to use several different substrates, since different binding modes or sites within the enzyme's active site can give rise to different inhibition profiles. To directly compare midazolam and testosterone as substrates and to avoid nonspecific binding to the enzyme preparation confounding the results, it was necessary to incubate identical P450 concentrations in the two assays. Since the rate of testosterone 6β-hydroxylation is much lower than that of midazolam 1-hydroxylation, each preincubation contained 1250 pmol of CYP3A4/ml. As a result of this high CYP3A4 concentration, the preincubation times were 2, 5, and 10 min. The secondary incubations in which midazolam was the substrate were performed for only 2 min (according to time linearity studies performed using the elevated CYP3A4 concentration; data not shown). The secondary incubations with testosterone as the substrate were performed for 15 min. With the exception of these details, the assay was performed as described above (“Manual KI and kinact Determinations for Standard Time-Dependent Inhibitors of CYP3A4”).
LC/MS-MS Analysis of Samples. LC/MS-MS analysis for the detection of paracetamol, 4-hydroxy diclofenac, 4-hydroxy S-mephenytoin, 1-hydroxy bufuralol, and 1-hydroxy midazolam was performed as previously described (Weaver et al., 2003).
HPLC analysis and UV detection of 6β-hydroxy testosterone was performed using a Waters 2795 separations module (Waters, UK, Elstree, Hertsfordshire, UK), coupled to a Waters 996 photodiode array detector unit (Waters, UK). Samples (10 μl) were injected from a 96-well plate onto a Gemini (5-μm) C18 column (50 × 2 mm; Phenomenex UK, Macclesfield, Cheshire, UK). Mobile phase consisted of solvent A (acetonitrile) and solvent B (0.1% formic acid in water), using a gradient of 5% to 95% (0–2.5 min), 100% A (2.51–3.0 min), 5% A (3.05 min), with a run time of 4 min and a flow rate 1 ml/min. 6β-Hydroxy testosterone was detected by measuring the absorbance at 254 nm (retention time of 2.9 min), using an authentic standard as a reference.
Analysis of the Data to Estimate KI and kinact. The natural log of the percentage control activity remaining after incubation with a single inhibitor concentration is plotted against the time of preincubation (for example, see Fig. 8). The slope, k, is the inactivation rate constant (describing the rate of inactivation at that inhibitor concentration). This is done for data from all inhibitor concentrations. Nonlinear regression analysis (WinNonlin; Pharsight, Mountain View, CA) is then used to determine KI and kinact from the function 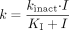 where I is the preincubation inhibitor concentration, k is the inactivation rate constant for a given I, kinact is the maximal inactivation rate constant, and KI is the inhibitor concentration, I, when the inactivation rate constant is half kinact.
where I is the preincubation inhibitor concentration, k is the inactivation rate constant for a given I, kinact is the maximal inactivation rate constant, and KI is the inhibitor concentration, I, when the inactivation rate constant is half kinact.
Analysis of the Data to Estimate Partition Ratio. The partition ratio was determined according to the method described by Knight and Waley (1985) and Waley (1985) as follows:  where E is the active enzyme, I is the test inhibitor, P represents the products of the reaction that escape reactive binding in the active site, and EI is the initial complex formed between the enzyme and inhibitor. Einactive-MI is the irreversible complex formed between the enzyme and the (irreversibly) bound reactive metabolite of the inhibitor.
where E is the active enzyme, I is the test inhibitor, P represents the products of the reaction that escape reactive binding in the active site, and EI is the initial complex formed between the enzyme and inhibitor. Einactive-MI is the irreversible complex formed between the enzyme and the (irreversibly) bound reactive metabolite of the inhibitor.
The fate of the intermediate, EI, is governed by the partition ratio: the subsequent reaction can yield a metabolite that irreversibly binds to the enzyme or a metabolite that does not bind to the enzyme and escapes the active site. Accordingly, the partition ratio is the number of molecules of inhibitor (substrate) metabolized to product for each molecule that inactivates the enzyme (= k+3/k+4). (Partition ratio + 1) can be determined from the x-axis intercept of a plot of enzyme activity remaining (y-axis) against inhibitor concentration/enzyme concentration (x-axis). This analysis can only provide a true partition ratio if all the inhibitor is metabolized. Often, this plot is nonlinear (Fig. 1), and in these cases, an estimation of the partition ratio can be best obtained by extrapolating to the x-axis intercept from the linear (low I/E ratio) portion of the plot. It must be remembered that under these circumstances, the data provide only an estimate of partition ratio since inhibitor consumption may not be complete and the partition ratio may be overestimated. The following equations demonstrate these points:  therefore,
therefore, 
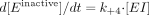 therefore,
therefore, 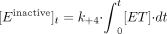 [I] = [Einactive]t + [P]t if we assume [I]t = 0 (i.e., all the inhibitor has been metabolized), where [I]0 is the inhibitor concentration at time 0 and [I]t is the inhibitor concentration at time, t.
[I] = [Einactive]t + [P]t if we assume [I]t = 0 (i.e., all the inhibitor has been metabolized), where [I]0 is the inhibitor concentration at time 0 and [I]t is the inhibitor concentration at time, t.
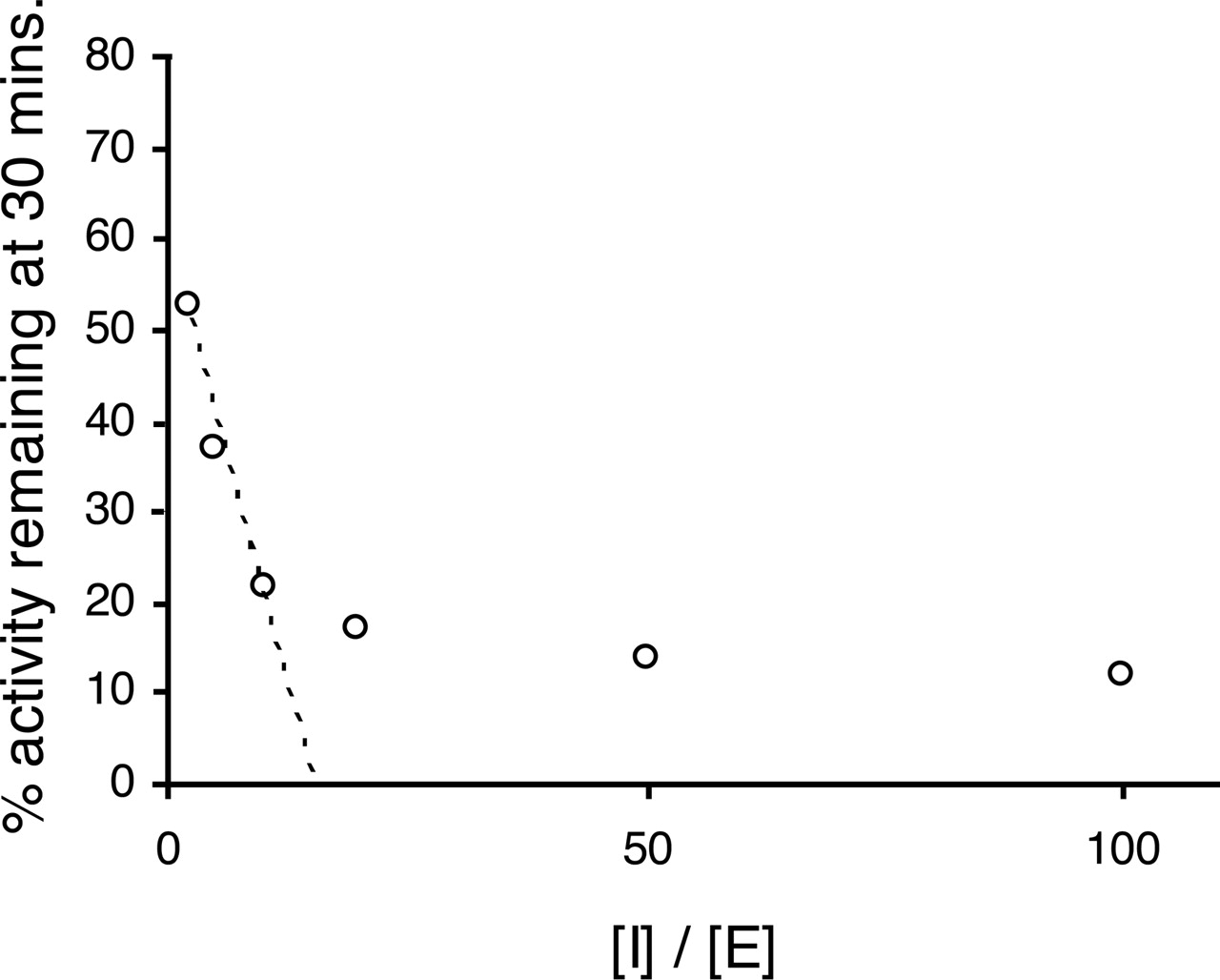
Plot of remaining enzyme activity against inhibitor concentration/enzyme concentration for troleandomycin and recombinant CYP3A4. The data were generated using the automated three-time point, six-inhibitor concentration, time-dependent inhibition assay, with midazolam 1-hydroxylation as a measure of CYP3A4 activity. The dashed line is the regression line used to estimate the partition ratio from the x-axis intercept (see Materials and Methods).
It follows that: 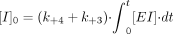 At the x-axis intercept, the enzyme activity remaining = 0; therefore, [E]0 = [Einactive]t, and it follows that
At the x-axis intercept, the enzyme activity remaining = 0; therefore, [E]0 = [Einactive]t, and it follows that 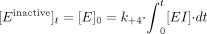 Therefore,
Therefore, 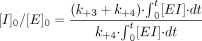
where the partition ratio = k+3/k+4.
So, (partition ratio + 1) = [I]0/[E]0.
Results
Substrate Selectivity. Recombinant human P450s were individually incubated with the five P450 substrate cocktails (concentrations approximately equivalent to 4 times Km). The P450 concentrations used were chosen to mimic those present in the human liver microsomal preparation used in the time-dependent inhibition assay (Soars et al., 2003). Under these conditions, only CYP1A2 catalyzed the phenacetin O-deethylation reaction, only CYP2C9 catalyzed the diclofenac 4-hydroxylation reaction, only CYP2C19 catalyzed the S-mephenytoin 4-hydroxylation reaction, and only CYP3A4 metabolized midazolam to the 1-hydroxy metabolite. Additionally, the amount of metabolite formed was linear with respect to time over the 30-min incubation period. CYP2D6-dependent bufuralol 1-hydroxylation was also linear with respect to time over the 30-min incubation period. Bufuralol 1-hydroxylation was, however, catalyzed by CYP2C19 as well as CYP2D6. The HLM pool was assumed to contain 15 and 5 pmols of CYP2C19 and CYP2D6 per mg microsomal protein, respectively. Using these amounts of P450 in 1-ml incubations gave a CYP2C19-dependent rate of bufuralol 1-hydroxylation approximately equal to 40% of the CYP2D6-dependent bufuralol 1-hydroxylation.
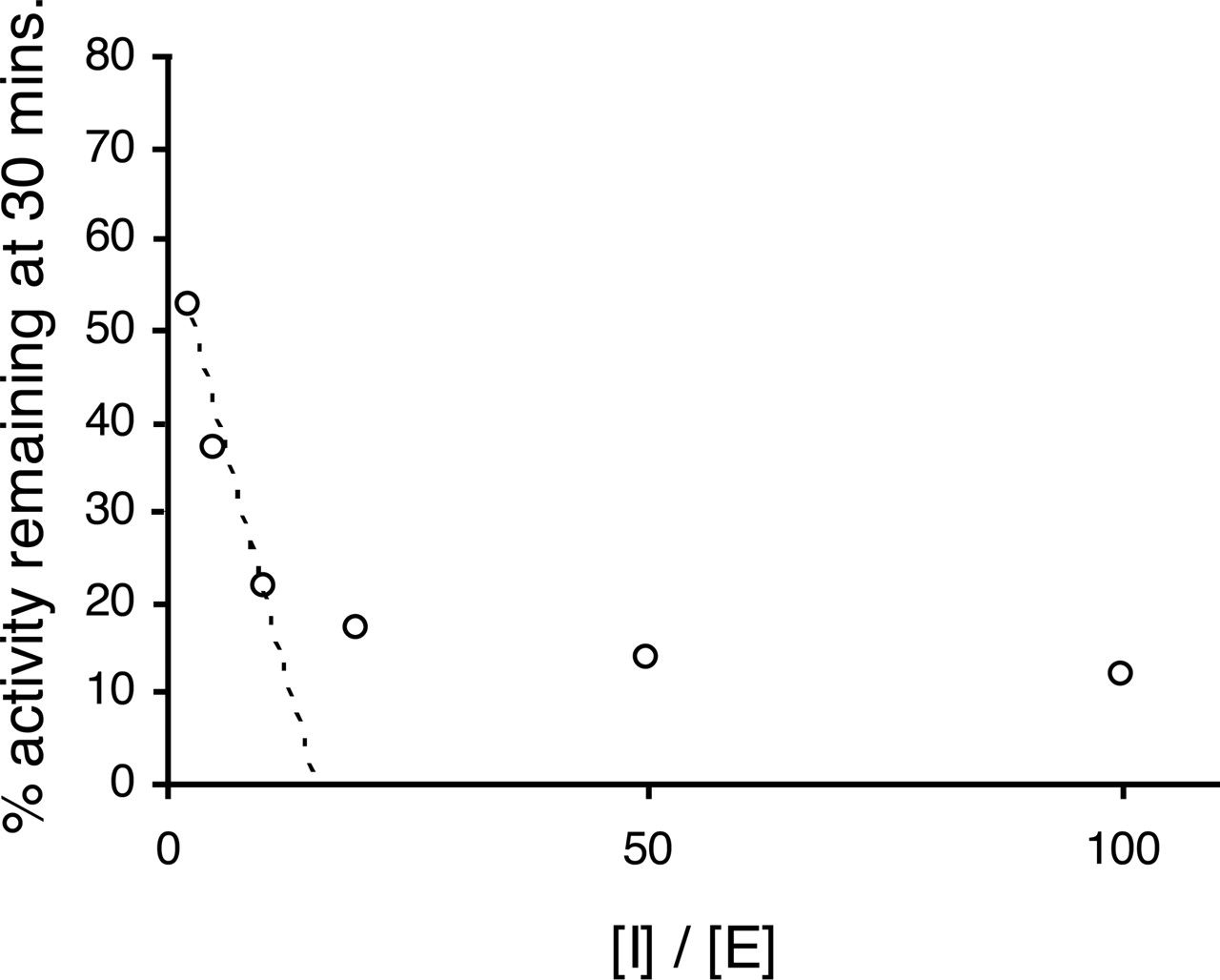
Selectivity of Time-Dependent Inhibitors. Time-dependent inhibitors of CYP1A2 (furafylline), CYP2C9 (tienilic acid), CYP2C19 (ticlopidine), CYP2D6 (MDMA), and CYP3A4 (troleandomycin and mifepristone) were chosen as standard inhibitors to validate each run of the automated single-time point, human liver microsomal time-dependent inhibition assay. The inhibitors were apparently P450-selective when incubated at 10 μM, 0.5 μM, 5 μM, 5 μM, 1.5 μM, and 5 μM, respectively (Table 1). The fact that the inhibitors were selective in the human liver microsomal assay confirms that the P450 probe reactions themselves must be specific for the individual P450s. For example, if the phenacetin O-deethylation reaction had been performed by CYP2C19 as well as CYP1A2, a time-dependent inhibition of the reaction would have been observed when ticlopidine was used as an inhibitor. It would therefore have appeared as if ticlopidine was an inhibitor of CYP1A2. The CYP2C19 time-dependent inhibitor ticlopidine did not show a strong inhibition of bufuralol 1-hydroxylase activity, indicating that the lack of selectivity of this probe reaction does not impact significantly on the interpretation of the results using this HLM pool in the single-time point HLM TDI assay (Table 1).
Inhibition of the P450 probe reactions, phenacetin O-deethylation (POD), diclofenac 4-hydroxylation (D4OH), S-mephenytoin 4-hydroxylation (SM4OH), bufuralol 1-hydroxylation (B1OH), and midazolam 1-hydroxylation (M1OH) by standard time-dependent inhibitors
Percentage TDI was calculated from the ratio of R + INADPH/R – INADPH and R + INO NADPH/R – INO NADPH as detailed under Results (eq. 2). Projected IC50 was calculated according to eq. 1 (Results). Inhibitor concentrations were as follows: Furafylline (FUR),10 μM; tienilic acid (TA), 0.5 μM; ticlopidine (TIC), 5 μM; MDMA, 5 μM; troleandomycin (TAO), 1.5 μM; mifepristone (MIF), 5 μM. Data are the mean of 30 experiments. For each inhibitor, the coefficient of variance (CV) was less than 5% for percentage TDI values and less than 20% for projected IC50 values.
Time-Dependent (Irreversible) IC50 Determination Using the Automated Single-Time Point, Human Liver Microsomal Time-Dependent Inhibition Assay. Furafylline, tienilic acid, ticlopidine, MDMA, troleandomycin, and mifepristone were separately incubated at five concentrations, using the automated single-time point human liver microsomal assay. The results (Fig. 2) demonstrate that each inhibitor decreased the control activity approximately 80% (from 90% to 10%) over an inhibitor concentration range of 2 log units (100-fold). Therefore a 1% change in control activity is brought about by a 0.025-log unit change in inhibitor concentration. Assuming this to be true for all time-dependent inhibitors, eq. 1 was written to project an IC50 value from a single inhibitor concentration and percentage inhibition relative to control. 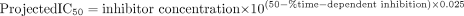 where the percentage time-dependent inhibition is calculated as follows:
where the percentage time-dependent inhibition is calculated as follows: 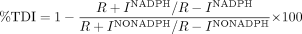 R +INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor and NADPH. R – INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) but in the presence of NADPH. R + INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor but the absence of NADPH. R – INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) and NADPH. IC50 values for all the standard inhibitors are shown in Table 1.
R +INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor and NADPH. R – INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) but in the presence of NADPH. R + INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor but the absence of NADPH. R – INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) and NADPH. IC50 values for all the standard inhibitors are shown in Table 1.
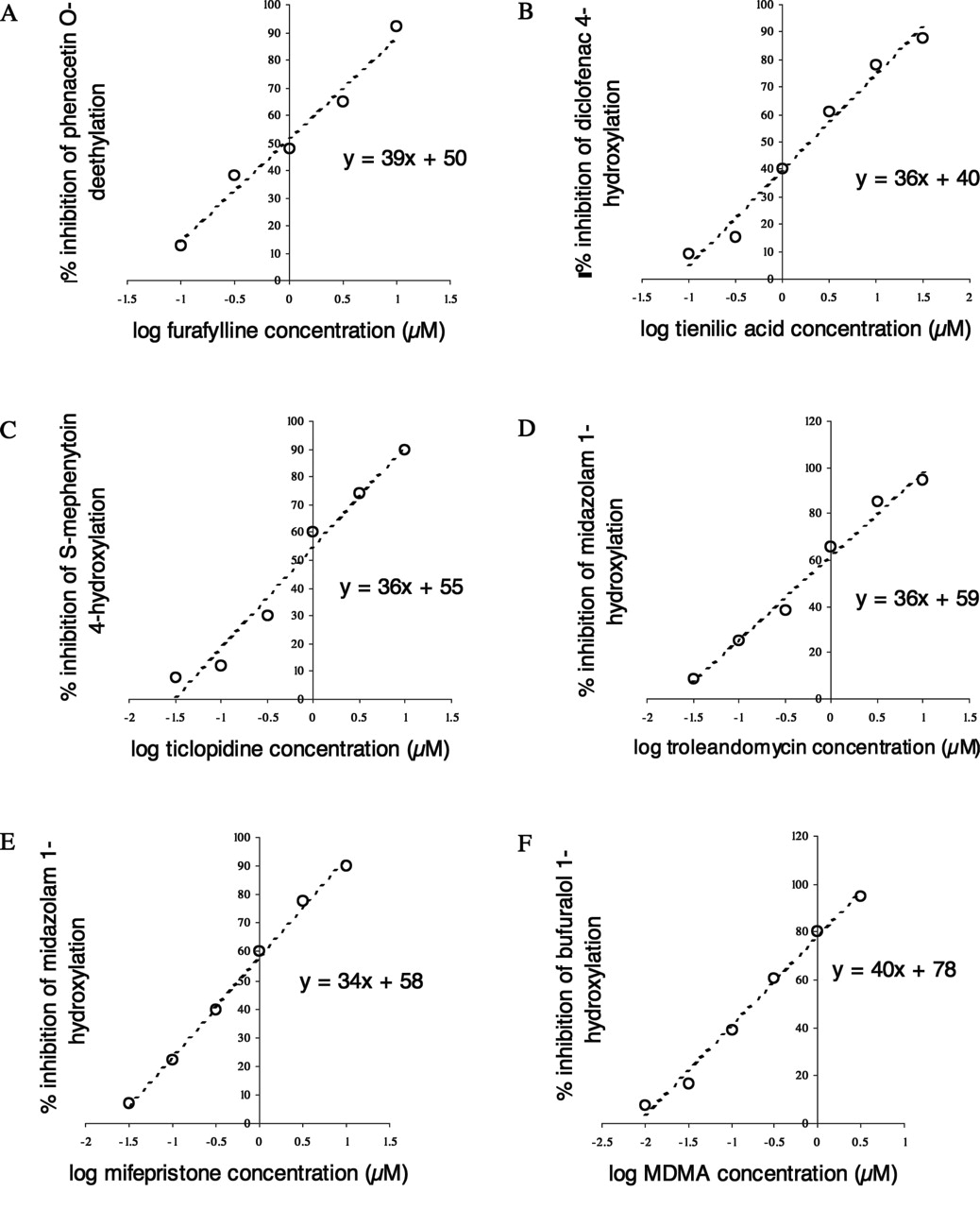
Inhibition of the P450 probe reactions, phenacetin O-deethylation (CYP1A2) (A), diclofenac 4-hydroxylation (CYP2C9) (B), S-mephenytoin 4-hydroxylation (CYP2C19) (C), midazolam 1-hydroxylation (CYP3A4) (D and E), and bufuralol 1-hydroxylation (CYP2D6) (F) by standard time-dependent inhibitors in the single-preincubation time point (30 min), human liver microsomal time-dependent inhibition assay.
KI and kinact Determinations for Known Time-Dependent Inhibitors Using the Automated Three-Time Point, Six-Concentration, Recombinant P450 TDI Assay. The automated TDI assay was used to generate kinetic parameters for time-dependent P450 inhibitors selected from the literature. An example of the data generated is shown in Fig. 3.

Plot of inactivation rate constant (k) and inhibitor concentration (I) for troleandomycin. The data were generated using recombinant human CYP3A4 in the automated time-dependent inhibition assay. The natural log of the percentage control activity remaining following incubation with each inhibitor concentration was plotted against the time of preincubation. The slopes, k (the inactivation rate constants), were used with inhibitor concentration to determine KI and kinact by nonlinear regression analysis, applying the equation k = (kinact · I)/(I + KI).
The comparison with kinact and KI values taken from the literature is presented in Table 2. Generally there is good agreement between our values and those quoted in the literature. The only literature kinact and KI values available for tienilic acid and ticlopidine were generated in the same laboratory using microsomes prepared from yeast-expressing human P450. The expression levels of CYP2C9 and CYP2C19 per milligram of microsomal protein were very low, and the experiments consequently used high microsomal protein concentrations. This may be the reason why the kinact values from our assay are similar to the literature values, but the KI values we report are lower.
Comparison of literature values for KI and kinact with values estimated from data generated using the automated time-dependent inhibition assay described in this article
Manual KI and kinact Determinations for Standard Time-Dependent Inhibitors. Three of the time-dependent CYP3A4 inhibitors profiled in the automated assay were chosen for manual kinact and KI determination. The manual assay was set up to replicate the automated assay in all features except that the preincubation times were 0, 10, 30, 60, 150, and 300 s. The purpose of this was to investigate the appropriateness of the 10-, 20-, 30-min preincubation times used in the automated assay. The results (Table 3) show that the automated assay conditions are effective for robust and accurate KI and kinact determinations.
Comparison of the kinetic constants of reversible and time-dependent inhibition (TDI) determined from manual and automated assays for troleandomycin (TAO), mifepristone (MIF), and erythromycin (EMC)
The reversible inhibition IC50 values derived from the TDI data were calculated as described in Fig. 8. The reversible inhibition screen was performed as described by Weaver et al. (2003). Manual and automated experiments were n = 3 with CV values less than 10% for KI and kinact.
The Use of Testosterone as an Alternative CYP3A4 Substrate when Estimating KI and kinact. The high CYP3A4 concentrations used for this comparison meant that the preincubation times had to be short (2, 5, 10 min) and the secondary incubation with midazolam was only 2 min (to maintain time-linearity of the 1-hydroxy metabolite formation). Data presented here suggest that there is little difference in the KI and kinact values determined using testosterone and midazolam as CYP3A4 substrates for those compounds tested (Table 3).
Relationship Between Time-Dependent IC50 Determined Using the Automated Single-Time Point Human Liver Microsomal Assay and kinact/KI Determined Using the Automated Three-Time Point, Six-Concentration Recombinant P450 Assay. To put results from the initial HLM assay into context, a relationship between the irreversible IC50 and the kinetic constants of time-dependent inhibition (kinact, KI) needs to exist. Since increasing IC50 values are associated with decreasing potency of inhibition, the reciprocal of the unbound IC50 values were plotted against kinact/KI (a measure of the efficiency of P450 inactivation) for 28 compounds shown to be time-dependent inhibitors of CYP3A4. The unbound IC50 value was calculated by multiplying the IC50 value by the fraction of inhibitor unbound in the 1 mg/ml hepatic microsomal preincubation [fuinc, calculated from the equation described by Austin et al. (2002)]. fuinc for the inhibitor in the automated three-time point, six-concentration recombinant P450 assay was always assumed to be 1. Figure 4 demonstrates that the initial HLM screen is effective in providing an estimate of kinact/KI.
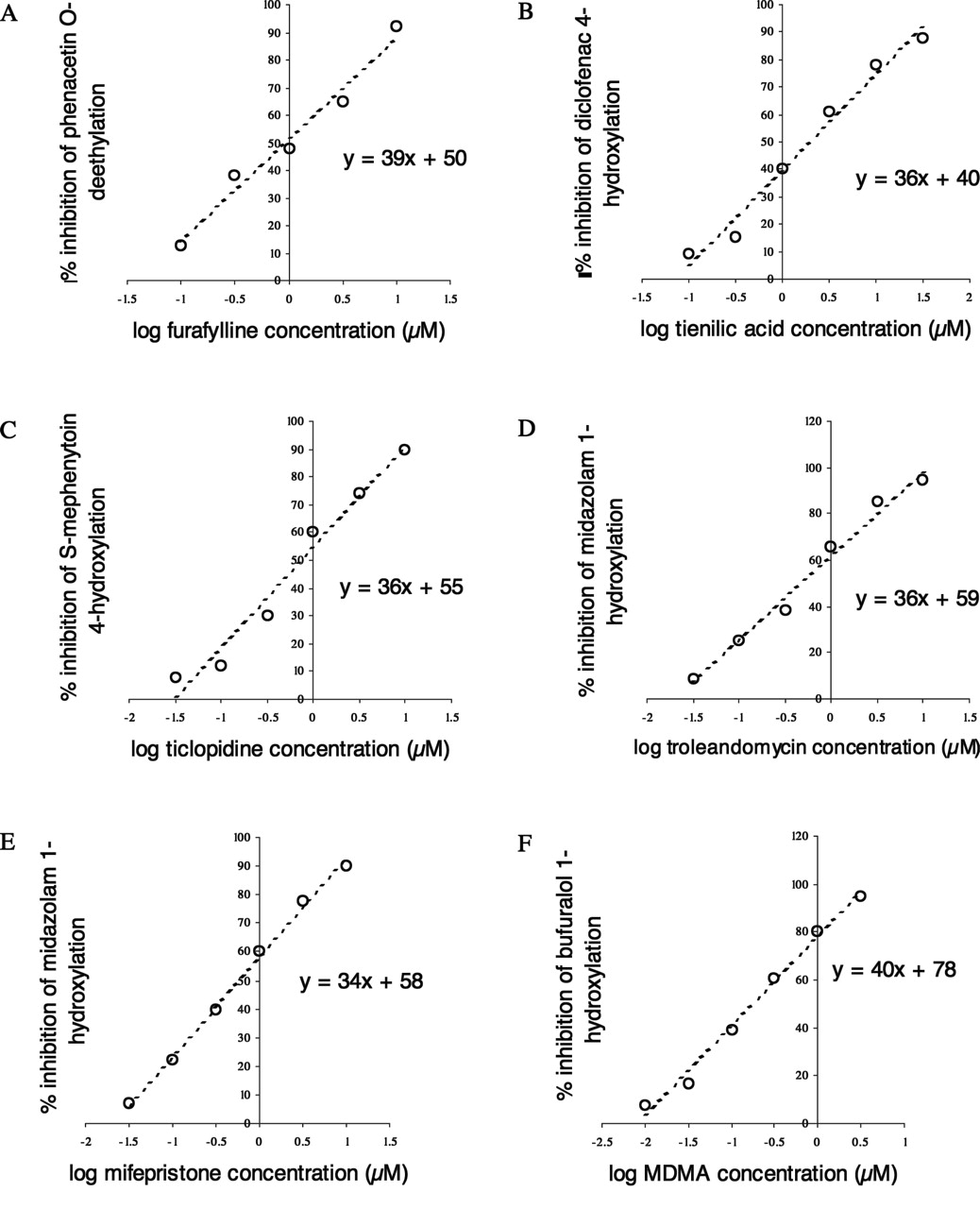
Relationship Between kinact/KI and Partition Ratio. The partition ratio is sometimes used to express the efficiency of enzyme inactivation, since it describes the number of molecules of inhibitor (substrate) metabolized to product for each molecule that inactivates the enzyme. The data generated in the automated time-dependent inhibition assay from 28 compounds shown to be time-dependent inhibitors of CYP3A4 were used to determine partition ratio, KI, and kinact. Figure 5 shows that a good relationship exists between kinact/KI and partition ratio. The equation of the line of best fit shows that for this data set, a partition ratio of 10 is equivalent to a kinact/KI ratio of 0.79 ml/min · nmol.
TDI Screening Strategy. Since the initial HLM screen is effective in providing an estimate of kinact/KI, which is itself closely related to the partition ratio as a measure of TDI potency, a proposed screening strategy is to use the higher throughput HLM assay to generate an IC50 for assessment of the pharmacokinetic impact on cotherapies using the estimated kinact/KI ratio. When necessary, for compounds in which a DDI is deemed likely, kinact and KI values can be determined accurately using the single P450 automated assay. This paradigm is depicted in Fig. 6.
Use of the Reversible Inhibition Data Obtained from the TDI Assays as “Quality Control Measures”. Both the TDI assays are designed to discriminate between reversible and irreversible P450 inhibition. Compounds are screened in the reversible P450 inhibition assay (described by Weaver et al., 2003) prior to incubation in the TDI assays, and reversible inhibition IC50 values can therefore be used to “quality control” and cross-check the data from the TDI assays. The following sections describe how these checks are made.
Reversible IC50 Determination Using the Automated Single-Time Point, Human Liver Microsomal Time-Dependent Inhibition Assay. The assay was designed to discriminate between reversible and irreversible P450 inhibition since the preincubation step is performed in the presence and absence of NADPH. IC50 values for any reversible inhibition detected can be calculated in a manner similar to that described for irreversible time-dependent inhibitors (eq. 1, since reversible inhibitors decrease control activity from 90% to 10% over an inhibitor concentration range of 2 log units; Segel, 1975). 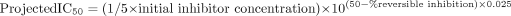 where the percentage reversible inhibition is calculated as follows:
where the percentage reversible inhibition is calculated as follows: 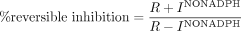 R + INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor but the absence of NADPH. R – INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) and NADPH.
R + INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor but the absence of NADPH. R – INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) and NADPH.

Plot of log 1/unbound IC50 against log kinact/KI for 28 time-dependent inhibitors of CYP3A4. IC50 values were generated using the automated single-time point, single-inhibitor concentration human liver microsomal time-dependent inhibition screen. These values were adjusted for incubational binding to give unbound IC50 values (according to the mathematical model of Austin et al., 2002). KI and kinact were estimated using the three-time point, six-inhibitor concentration, recombinant P450 automated time-dependent inhibition assay. The line of best fit is shown with dashed lines representing 3-fold deviation from the line.
The 5-fold dilution factor is included in eq. 3 because an aliquot of the initial preincubation (50 μl) is diluted into the final incubation with the P450 probe substrates (250 μl). This is a key difference compared to determining IC50 values for the irreversible, time-dependent P450 inhibition (eq. 1). It is necessary because the reversible P450 inhibition occurs in the second incubation, whereas the time-dependent inhibition occurs in the preincubation, when inhibitor concentrations are 5-fold higher.

Plot of log partition ratio against log kinact/KI for 28 time-dependent inhibitors of CYP3A4. Data were generated using the three-time point, six-inhibitor concentration, recombinant P450 automated time-dependent inhibition assay.
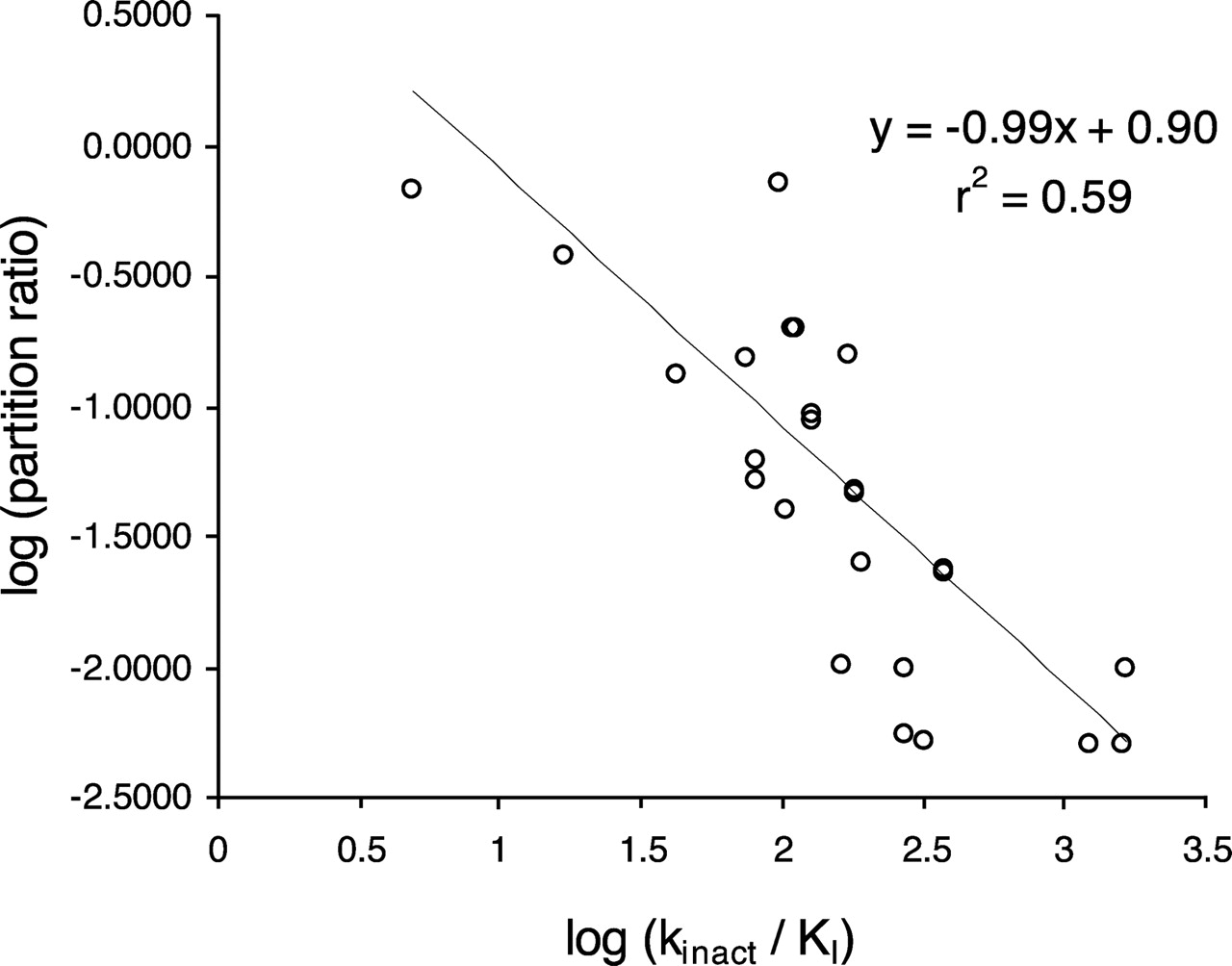
To compare the reversible P450 inhibition IC50 values calculated in this way with IC50 values generated from the reversible P450 inhibition screen (Weaver et al., 2003), the discrepancy in substrate concentration between the two assays has to be taken into account (P450 probe substrate is equal to Km in the reversible P450 inhibition screen but approximately equal to 4 times Km in the time-dependent inhibition assay). Therefore, since a competitive IC50 is equal to Ki (1 + [S]/Km), the following relationship should exist: 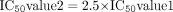 where IC50 value 1 is estimated from the normal reversible P450 inhibition screen and IC50 value 2 is the reversible inhibition IC50 estimated from the time-dependent inhibition assay. Accounting for nonspecific binding differences (due to the fact that the reversible P450 inhibition screen uses E. coli membrane preparations containing recombinant human P450, and the assay described here uses HLMs) was deemed to be largely unimportant, since the secondary incubation of the TDI assay contains only 0.2 mg/ml microsomal protein. Obviously the relationship described in eq. 5 does not hold for noncompetitive inhibitors, where IC50 is independent of substrate concentration. Figure 7 shows that there is a good correlation between the IC50 values determined in both assays. This type of interrogation of the data generated in the TDI screen acted as a quality control of the assay.
where IC50 value 1 is estimated from the normal reversible P450 inhibition screen and IC50 value 2 is the reversible inhibition IC50 estimated from the time-dependent inhibition assay. Accounting for nonspecific binding differences (due to the fact that the reversible P450 inhibition screen uses E. coli membrane preparations containing recombinant human P450, and the assay described here uses HLMs) was deemed to be largely unimportant, since the secondary incubation of the TDI assay contains only 0.2 mg/ml microsomal protein. Obviously the relationship described in eq. 5 does not hold for noncompetitive inhibitors, where IC50 is independent of substrate concentration. Figure 7 shows that there is a good correlation between the IC50 values determined in both assays. This type of interrogation of the data generated in the TDI screen acted as a quality control of the assay.

A general paradigm for TDI screening using the two automated assays described in the text. Since both assays differentiate between reversible and irreversible P450 inhibition, the reversible inhibition data from the TDI assays can be cross-checked with data from the standard reversible P450 inhibition screen (described by Weaver et al., 2003) as a quality control measure for the TDI assay data.
The Effect of Reversible P450 Inhibitors in the Automated Single-Time Point, Human Liver Microsomal Time-Dependent Inhibition Assay. To demonstrate that the automated human liver microsomal time-dependent inhibition screen could discriminate between reversible and irreversible P450 inhibition, potent reversible inhibitors of CYP2C9 (sulfaphenazole, 1 and 10 μM), CYP2D6 (quinidine, 0.025 and 0.25 μM), and CYP3A4 (ketoconazole, 0.005 and 0.05 μM) were incubated. Sulfaphenazole reduced the diclofenac 4-hydroxylation activity by the same amount when preincubations were performed in the presence and absence of NADPH, indicating reversible but not time-dependent inhibition. The same was true for quinidine (inhibition of bufuralol 1-hydroxylation) and ketoconazole (inhibition of midazolam 1-hydroxylation). Equation 3 was used to calculate reversible IC50 values from the single inhibitor concentrations used. These values compare well to IC50 values generated in the reversible P450 inhibition screen (Table 4).
Comparison of reversible IC50 values generated from the reversible P450 inhibition screen ( Weaver et al., 2003 ) for sulfaphenazole (SPZ), quinidine (QUI), and ketoconazole (KCZ) and those projected from a single inhibitor concentration in the human liver microsomal, single-time point time-dependent inhibition assay
R + INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor and NADPH. R – INADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the absence of inhibitor (i.e., solvent control) but the presence of NADPH. R + INO NADPH is the rate of P450 probe reaction when the initial (pre-) incubation is performed in the presence of inhibitor but the absence of NADPH.

Comparison of reversible inhibition IC50 values (micromolar) generated in the reversible P450 inhibition screen (set up as described in Weaver et al., 2003) and reversible inhibition IC50 values projected from the single-concentration human liver microsomal time-dependent inhibition screen. These IC50 values are calculated according to eqs. 3 to 5 (Results).
Reversible IC50 Determination Using Data from the Automated Three-Time Point, Six-Concentration, Recombinant P450 TDI Assay. The data from this assay are used to generate KI and kinact values (described under Materials and Methods). The first step is to plot the natural log of the percentage control activity remaining, following incubations with a range of inhibitor concentrations, against the time of preincubation (Fig. 8). It is obvious from the data that the straight lines of best fit do not always back-extrapolate to 4.6 (i.e., 100% control activity), even when very short incubation times are used. Clearly, the back-extrapolated percentage control activity represents the inhibition that occurs when no preincubation has taken place. This inhibition may be a mixture of reversible and irreversible inhibition occurring during the final 15 min of coincubation with P450 substrate. Thus, the time 0 percentage control activity for a given inhibitor concentration should be equivalent to the percentage control activity determined in the reversible P450 inhibition screen described previously (Weaver et al., 2003). As detailed in Fig. 8, these time 0 percentage control activities can be used with the respective inhibitor concentrations (once the 5-fold dilution from the preincubation had been accounted for) to determine an IC50 value for the reversible inhibition component. Table 3 shows that the data from the time-dependent inhibition assay match well with the data generated in the reversible P450 inhibition screen. A good fit of the ln percentage control activity/time plot coupled with this check on the y-intercept values serve as valuable quality control assessments for the assay. Although this exercise offers a way to compare the P450 inhibition data from two different assays, it is not ideal to have extensive reversible inhibition in the TDI assay. Clearly, 10-fold or greater dilution factors from the first to the second incubation in the TDI assay can reduce the impact of this complication.
Discussion
Many pharmaceutical companies utilize microtiter plate methods with real-time detection of fluorescent metabolites to facilitate high throughput screening for reversible P450 inhibition. A simple modification to this method facilitates an early time-dependent inhibition screen (detection of a time-dependent shift in IC50) (Yamamoto et al., 2002; Naritomi et al., 2004). Although large numbers of compounds can be studied with such methodology, interpretation of the IC50 data may be confounded if the opposing effects of TDI (decreasing IC50) and significant depletion of inhibitor concentration (increasing the reversible inhibition IC50) are manifested. Furthermore, since several reactions may contribute to many TDI episodes (Bensoussan et al., 1995), it is conceivable that one particular P450 may be involved in generating metabolites that are then further metabolized to reactive species in the active site of a second P450. Since the fluorescent probes used in these assays are generally not P450-specific, the experiments necessitate the use of single P450s, rather than HLM or P450 cocktails. It is therefore not inconceivable that false negative results could occur. Moreover, the suitability of fluorescent probes for making in vivo projections from in vitro reversible P450 inhibition experiments has recently been challenged (Cohen et al., 2003). Industry guidelines for in vitro drug-drug interaction studies recommend the use of P450 probe substrates that may be said to be more “drug-like,” although the list of substrates was not chosen with this specific intention (Tucker et al., 2001).
Recently, this laboratory implemented a reversible P450 inhibition assay using a cocktail of the recommended CYP1A2, 2C9, 2C19, 2D6, and 3A4 substrates at concentrations equivalent to their respective Km values. LC/MS-MS was used to follow the formation of specific metabolites (Weaver et al., 2003). The same endpoint was used in the assays described here, although the substrate concentrations were approximately 4 times their respective Km values. Analysis may be less rapid than with fluorescence-based assays, and the initial time-dependent inhibition assay described here does not easily afford screening on a large scale. However, given the fact that the functional groups and mechanisms involved in the majority of irreversible P450 inhibition cases are well known (Franklin, 1977; Ortiz de Montellano, 1988; Bensoussan et al., 1995), compounds can be rationally chosen based on structural features and, to avoid overlooking potentially inhibitory compounds, how well they represent chemistry that is current within a project.
To determine the P450 selectivity of the reactions in the HLM incubations using the elevated substrate concentrations, individual P450s were incubated with each substrate. P450 concentrations were chosen based on a calculation of the likely concentration in the HLM pool. Under these conditions, the five reactions were deemed to be appropriate P450-selective reactions for the single-time point HLM assay. Bufuralol 1-hydroxylation was catalyzed by CYP2C19 at a rate approximately equal to 40% of the CYP2D6-catalyzed reaction. In this case, CYP2D6 would only account for 70% of the total bufuralol 1-hydroxylase activity and, in theory, the potency of a CYP2D6 time-dependent inhibitor could be underestimated by a factor of 3. However, studies with the CYP2C19 time-dependent inhibitor ticlopidine indicated that the lack of P450 selectivity of the bufuralol 1-hydroxylation reaction may not have a significant impact on the interpretation of the TDI results.
The assay for determining KI and kinact for a single recombinant P450 uses three time points. Ideally, the assay would use more time points and ensure that these adequately span the inactivation half-life. However, a pragmatic approach of six inhibitor concentrations preincubated for 10, 20, and 30 min was adopted to reduce sample numbers. To validate this automated approach, more comprehensive manual assays were performed and the TDI kinetic parameters generated were compared with those generated from the three-time point automated assay. This comparison demonstrated the effectiveness of the automated assay for describing the enzyme inactivation by inhibitors with a range of kinact/KI values. The kinact and KI values determined in the automated TDI assay described here compare well with previously published results (Table 2). An additional concern was the capability of the assay to generate data uncontaminated by reversible P450 inhibition. Both the automated assays described here use a 5-fold dilution of the preincubation into the final incubation with the P450 probe substrates, whereas 10-fold or greater dilutions are commonly used. Figure 8 clearly demonstrates that the 5-fold dilution can give rise to concentrations of inhibitor that give considerable reversible inhibition in the second incubation. This should not greatly alter the slopes of natural log percentage control activity-time plots and, therefore, should not alter the kinetic parameters of inhibition estimated. However, the slopes may be easier to determine if the percentage inhibition is not swamped by the reversible P450 inhibition and, therefore, it is advisable to use a 10-fold or greater dilution factor if sufficient analytical sensitivity (detecting 1-hydroxy midazolam formation) can be achieved. This is of particular importance when test inhibitors are reasonably potent reversible P450 inhibitors but moderate to weak time-dependent inhibitors.

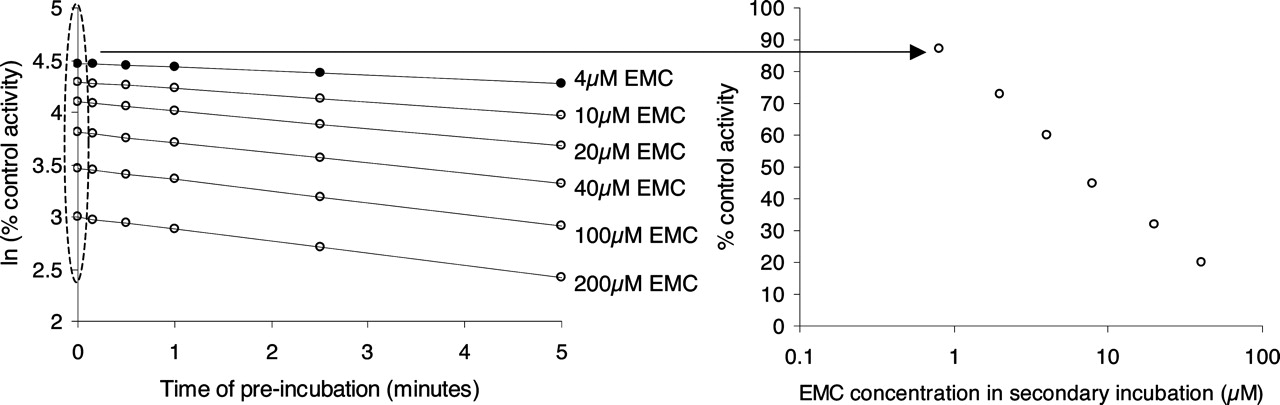
Inhibition of recombinant CYP3A4-dependent midazolam 1-hydroxylation by the time-dependent inhibitor erythromycin (EMC). The assay was performed manually using EMC preincubation concentrations of 4 to 200 μM, as described under Materials and Methods. Zero-minute preincubation time point samples were removed and added to the secondary incubation with NADPH and midazolam (10 μM). The formation of 1-hydroxy midazolam was monitored. The percentage of midazolam 1-hydroxylation activity relative to control (no EMC present in the preincubation) was plotted against EMC concentration in the secondary incubation (1/5 × the preincubation concentrations) and an IC50 value was estimated.
It is now commonplace to use several different substrates for reversible CYP3A4 inhibition assays, since different substrate binding sites within the active site of the enzyme can confound in vivo projections (Kenworthy et al., 1999). The majority of TDI studies use a preincubation step involving inhibitor and enzyme followed by the addition of substrate once the enzyme has been inactivated. This experimental design suggests that data for different CYP3A4 substrates would be comparable if inactivation occured as a result of heme binding. Substrate differences have been demonstrated for time-dependent inhibition studies, using midazolam as a time-dependent inhibitor (Schrag and Wienkers, 2001). However, midazolam oxidation results in protein adduct formation in the CYP3A4 active site, which could disparately influence the binding of diverse substrates. The substrate comparison described here was not straightforward, due to the very different rates of metabolism of the two substrates: a high concentration of enzyme was required (1250 pmol/ml) for the testosterone incubations. To facilitate the comparison by avoiding differences in nonspecific binding, the same enzyme concentration was used for the assay when midazolam was used as a substrate (a 40-fold increase in CYP3A4 concentration over the standard time-dependent inhibition assay). Short preincubation times and a short incubation with midazolam were therefore required. There was little difference in the KI and kinact values determined using testosterone and midazolam as CYP3A4 substrates for the compounds studied. It is interesting to compare the results from this experiment, when midazolam was used as a CYP3A4 substrate, with the results from the standard automated assay for kinact and KI determination using midazolam as a substrate. The kinact values determined for mifepristone in the two assays were very similar (0.08 min–1 and 0.07 min–1). The same was found to be true for erythromycin (0.12 min–1 and 0.09 min–1) and troleandomycin (0.12 min–1 and 0.16 min–1). The KI values for mifepristone were 0.6 μM and 2.8 μM, for erythromycin 8.8 μM and 8.3 μM, and for troleandomycin 0.3 μM and 0.8 μM. The differences in log P and therefore nonspecific binding most likely account for differences in observed (apparent) KI (Austin et al., 2002).
The results of the studies detailed above demonstrate that both automated assays are appropriate for the generation of creditable and robust data on the time-dependent inhibitory potency of test compounds. The potent reversible inhibitors sulfaphenazole, ketoconazole, and quinidine did not cause TDI, but significant reversible inhibition was detected. The IC50 values estimated for this reversible inhibition component (albeit projected off single inhibitor concentrations; eq. 3) matched well with those determined in the separate reversible P450 inhibition screen (Weaver et al., 2003). This has also consistently been shown to be true for a range of compounds that have been assayed in both screens (Fig. 7) and serves as a good quality control check for each experiment.
IC50 values are generated for the TDI component (eq. 1); but, because HLMs are used in this initial assay, the IC50 values can only be treated as apparent values. Unbound IC50 values are calculated using a lipophilicity term (log P for bases, log D for acid and neutral compounds) to estimate the extent of nonspecific binding in vitro (Austin et al., 2002). Despite the single time point of 30 min used in the initial screen, a good relationship exists between the unbound IC50 estimated in this format and the kinact/KI derived from the more extensive subsequent single P450 assay. This means that the initial screening assay generates an inhibitory potency term (IC50) that has a meaningful value. The kinact/KI itself was shown to relate well to the partition ratio as a measure of inhibitory potency or efficiency of enzyme inactivation (Fig. 5). The screening strategy proposed would therefore use the higher throughput HLM assay to generate an IC50 for assessment of the pharmacokinetic impact on cotherapies using the estimated kinact/KI ratio, predicted human dose, and pharmacokinetic parameters (clearance, volume of distribution, and plasma protein binding data). This scheme is depicted in Fig. 6. When necessary, for compounds in which a DDI is deemed likely, kinact and KI values can be determined accurately using the single P450 automated assay.
Acknowledgments
We thank Dr. Rob Riley and Dr. Douglas Ferguson for scientific discussion and advice.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005579.
-
ABBREVIATIONS: DDI, drug-drug interaction; P450, cytochrome P450; TDI, time-dependent inhibition; LC/MS-MS, liquid chromatography-tandem mass spectrometry; MDMA, methylenedioxy-N-methylamphetamine; DMSO, dimethyl sulfoxide; HLM, human liver microsome.
- Received May 23, 2005.
- Accepted July 26, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}