


Abstract
An improved mass defect filter (MDF) method employing both drug and core structure filter templates was applied to the processing of high resolution liquid chromatography/mass spectrometry (LC/MS) data for the detection and structural characterization of oxidative metabolites with mass defects similar to or significantly different from those of the parent drugs. The effectiveness of this approach was investigated using nefazodone as a model compound, which is known to undergo multiple common and uncommon oxidative reactions. Through the selective removal of all ions that fall outside of the preset filter windows, the MDF process facilitated the detection of all 14 nefazodone metabolites presented in human liver microsomes in the MDF-filtered chromatograms. The capability of the MDF approach to remove endogenous interferences from more complex biological matrices was examined by analyzing omeprazole metabolites in human plasma. The unprocessed mass chromatogram showed no distinct indication of metabolite peaks; however, after MDF processing, the metabolite peaks were easily identified in the chromatogram. Compared with precursor ion scan and neutral loss scan techniques, the MDF approach was shown to be more effective for the detection of metabolites in a complex matrix. The comprehensive metabolite detection capability of the MDF approach, together with accurate mass determination, makes high resolution LC/MS a useful tool for the screening and identification of both common and uncommon drug metabolites.
The identification of drug metabolites, particularly metabolites formed through oxidation, reduction, or hydrolysis reactions, has become an integral part of the drug discovery and development process. These metabolites may have intrinsic pharmacological activity or display specific toxicity (Parkinson, 1996; Guengerich, 2000). In addition, most clinical drug-drug interactions are associated with oxidative biotransformation mediated by cytochrome P450 (Bjornsson et al., 2003). Although analytical sensitivity and the processing of data for liquid chromatography/mass spectrometry (LC/MS) have been tremendously improved in the last decade (Clarke et al., 2001; Kostiainen et al., 2003; Liu and Hop, 2005), the detection and identification of drug metabolites in complex biological matrices continue to be a challenge.
Traditionally, detection of common or expected metabolites has been conducted on LC/MS data by generating extracted or reconstructed ion chromatograms corresponding to the expected protonated molecules of drug metabolites (Plumb et al., 2003). Over the last decade, product ion scanning techniques that use rule-based algorithms to generate a list of potential metabolite masses have been developed and continuously improved for rapid screening for common metabolites (Yu et al., 1999; Gangl et al., 2002; Lafaye et al., 2003). The technique employs a survey mode to search for the metabolites that are listed in the acquisition method. Both the detection of expected metabolites and the acquisition of their product ion spectra can be accomplished in a single LC/MS analysis (Gangl et al., 2002; Xia et al., 2003; Mortishire-Smith et al., 2005). With the availability of comprehensive metabolite databases developed from a knowledge of biotransformations, the list-dependent product ion scan has been very successful in screening for predicted metabolites, especially in vitro metabolites (Anari et al., 2004). The disadvantage of these approaches is that only predictable metabolites can be detected, and therefore, the robustness of the metabolite prediction algorithms being used becomes a critical factor.
Detection of uncommon metabolites in complex biological matrices is more challenging, and is often carried out using precursor ion (PI) or neutral loss (NL) scanning techniques on a triple quadrupole mass spectrometer (Jackson et al., 1995; Clarke et al., 2001; Kostiainen et al., 2003). The detection of conjugates (e.g., glucuronide and sulfate) can usually be accomplished with a NL analysis because these conjugates often undergo common cleavages to generate specific neutral fragments under collision-induced dissociation conditions (Baillie and Davis, 1993; Lafaye et al., 2004). PI scanning can also be used to search for metabolites with common product ions that can be predicted from the patterns of the parent drug product ions (Xia et al., 2003; Liu and Hop, 2005). For a PI or NL analysis, however, one or a few expected neutral or charged fragments must be defined in a LC/MS/MS acquisition method. Metabolites that do not generate the expected fragments will not be detected (Xia et al., 2003).
The task of metabolite identification has been greatly facilitated by recent developments in high resolution LC/MS technology [e.g., time-of-flight (ToF) and Fourier transform (FT) mass spectrometers], which allow for the determination of molecular formulae (Corcoran et al., 2000; Zhang et al., 2000; Sundstrom et al., 2002) and product ion formulae (Sleno et al., 2005) with minimal uncertainty. In addition, the specificity of list-dependent acquisition of MS/MS data for expected metabolites is improved (Castro-Perez et al., 2002). Similarly, triple quadrupole mass spectrometry with improved mass resolution has provided improved selectivity in NL and PI analyses (Jemal et al., 2003). A combination of high resolution mass spectrometry and other types of LC/MS instruments has been recommended for metabolite identification, given the complementary capabilities of triple quadrupole, ion trap, and high resolution mass spectrometers (Clarke et al., 2001; Liu et al., 2002; Anari et al., 2004).
Taking advantage of the advances in modern high resolution LC/MS technology, we have developed a mass defect filter (MDF) technique for the selective detection of drug metabolites (Zhang et al., 2003). The MDF approach attempts to discriminate metabolite ions from matrix ions based on the similarity of the mass defect values of a drug and its metabolites. With a mass defect window set approximately ±50 mDa from that of the parent drug, the metabolite profile of a dog bile sample was obtained with the majority of interference ions removed. The filtered data facilitated the identification of both common and uncommon metabolites (Zhang et al., 2003). A similar approach was also included in a recent release of MetaboLynx software to reduce the number of false-positive entries in analyzing metabolites in liver microsomal incubations (Mortishire-Smith et al., 2005). However, those methods may not be able to selectively detect metabolites from dealkylation or hydrolysis since these metabolites can have mass defects significantly different from that of a parent drug. For example, the mass defect values of nefazodone (NEF; MH+ = 470.2323) and its major metabolite, 1-meta-chlorophenylpiperazine (m-CPP; MH+ = 197.0845), are 148 mDa apart.
In this study, we explored an improved MDF approach for the identification of drug metabolites that have mass defects either similar to or significantly different from those of the parent drug. The effectiveness, selectivity, and sensitivity of such a MDF approach for the detection of common and uncommon metabolites were studied using NEF as a model compound. The results were compared with those from the PI and NL scan analyses performed on a triple quadrupole mass spectrometer. In addition, the capability of the MDF approach in removing endogenous interferences from more complex biological matrices was investigated by analyzing omeprazole metabolites in human plasma.
Materials and Methods
Chemicals. Pooled human liver microsome (HLM) preparations were purchased from BD Biosciences (Woburn, MA). Nefazodone and its metabolite standards, para-hydroxy-nefazodone (M2), triazoledione (M4), and hydroxynefazodone (M5), were synthesized at Bristol-Myers Squibb (Princeton, NJ). Omeprazole and m-CPP (M14) were purchased from Aldrich Chemical Company (Milwaukee, WI). Control human plasma was purchased from Lampire Labs (Pipersville, PA).
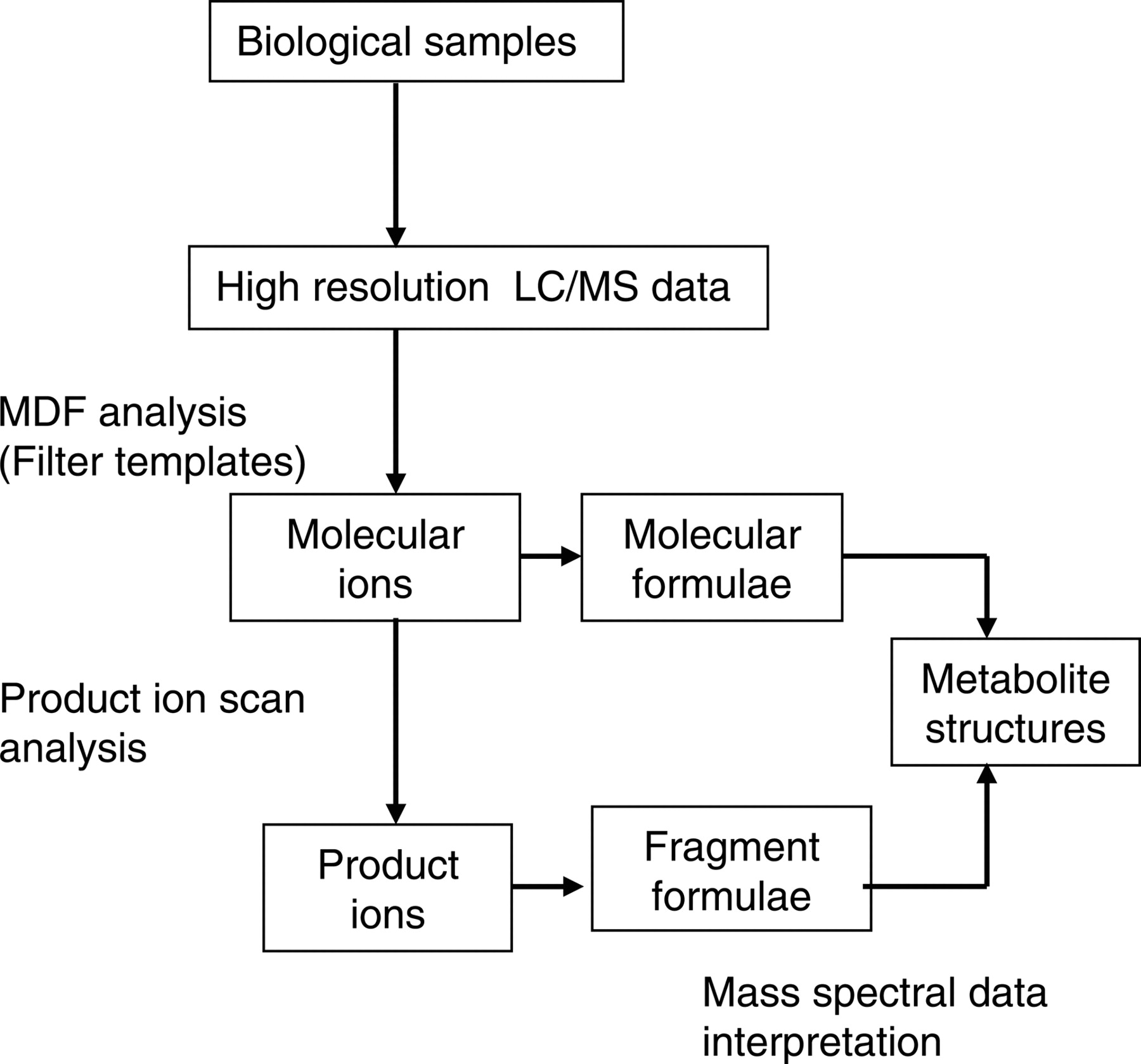
Strategy for detection and characterization of metabolites that formed through oxidation, reduction, or hydrolysis using high resolution LC/MS and MDF.
In Vitro Metabolism in HLMs. Omeprazole (50 μM) and nefazodone (20 μM) were incubated separately with pooled HLMs (1.0 mg/ml) and NADPH (1 mM) in 100 mM sodium phosphate buffer (pH 7.4) for 10 and 60 min, respectively. The biotransformation reactions were initiated by the addition of an NADPH solution after a 3-min preincubation, and were stopped by the addition of either an equal volume of ice-cold acenonitrile for the omeprazole incubation or 2 volumes of methanol for the nefazodone incubation. Control HLM incubations were carried out in the same fashion without the addition of NADPH. The suspensions were centrifuged at 13,000 rpm for 10 min. Aliquots of the supernatants were either analyzed directly by LC/MS or spiked into human plasma extracts followed by LC/MS analysis.
Preparation of Model Human Plasma Sample. An aliquot of the supernatant from the omeprazole incubation was spiked into pooled human plasma at a ratio of 1:50 (v/v). The human plasma sample was loaded onto a solid phase extraction cartridge (Oasis Extraction Cartridge; Waters, Milford, MA), followed by elution with water and methanol. The methanol fraction was collected and was dried under a stream of nitrogen. The dried sample was reconstituted in HPLC solvents before LC/MS analyses.
HPLC. For the analysis of nefazodone metabolites, the HPLC analysis was performed using a Luna Phenyl-hexyl column (4.6 × 100 mm; Phenomenex, Torrance, CA). A gradient with buffer A (5 mM ammonium formate, pH 3.0) and buffer B (acetonitrile) was used at a flow rate of 1.0 ml/min. The gradient was linearly increased from 0 to 5% B over 5 min, then from 5 to 60% B over 25 min, from 60 to 90% B over 5 min, and then from 90 to 0% B over 2 min, followed by reequilibration at 0% B for 3 min.
For analyzing omeprazole metabolites, separation was performed using a YMC ODS AQ column (2.0 × 150 mm; Waters). A gradient with buffer A (water with 0.1% trifluoroacetic acid) and buffer B (acetonitrile with 0.1% trifluoroacetic acid) was used at a flow rate of 0.25 ml/min. Buffer B was maintained at 5% for the first 2 min and then linearly increased to 50% in 40 min. After holding at 50% for 1 min, the gradient returned to the initial conditions in 1 min and was held for 8 min before the next injection.
Triple Quadrupole Mass Spectrometry. Precursor ion, neutral loss, and product ion scan analyses of nefazodone metabolites were carried out using a Finnigan TSQ Quantum triple quadrupole mass spectrometer (Thermo Electron, Waltham, MA). The mass spectrometer was operated in the positive ion electrospray mode with a capillary temperature of 350°C. The MS/MS analysis was performed using argon as the collision gas (1.5 mTorr), with the collision energy being set at 40 eV and the scan range set for m/z 50 to 600.
Ion Trap Mass Spectrometry. The MSn spectra of nefazodone metabolites were obtained using a Finnigan LCQ-Deca XP ion trap mass spectrometer (Thermo Electron). The mass spectrometer was operated in the positive ion electrospray mode with a capillary temperature of 300°C. The MSn analyses were performed with a relative collision energy of 35%.
Q-ToF Mass Spectrometry. For analyzing omeprazole metabolites, a Micromass (Manchester, UK) Q-ToF Ultima API mass spectrometer was used. The mass spectrometer was operated in the positive ion electrospray mode at a desolvation temperature of 350°C. A 2.0 ng/ml leucine enkephalin solution in 50% acetonitrile with 0.1% trifluoroacetic acid was delivered to the lockspray channel of the electrospray source. The Q-ToF instrument was tuned to a resolving power of 18,000 at half peak height. The LC/MS data were converted to centroid data using the “all file accurate mass measurement” feature of the MassLynx software without applying any lock mass correction before processing with the mass defect filter.
FT Mass Spectrometry. The high resolution LC/MS data for the MDF analyses of nefazodone metabolites were obtained using a Finnigan LTQ FT mass spectrometer (Thermo Electron). The mass spectrometer was operated in the positive ion electrospray mode with a capillary temperature of 320°C. The FT-MS data were acquired at a resolving power of 50,000 in a range of m/z 85 to 850. The ion trap MS/MS analysis was performed on the same instrument with a relative collision energy of 25%.
Mass Defect Filter Methods. The mass defect filtering utility was implemented using the Python programming language (available at http://www.python.org) with the Scientific Python Module (available at http://starship.python.net/~hinsen/ScientificPython). Data files were input in the universal NetCDF format (information available at http://www.unidata.ucar.edu/packages/netcdf). A centroid LC/MS data file was converted to NetCDF format using either the DataBridge utility in MassLynx for the Q-ToF data or the File Converter tool in Xcalibur (Thermo Electron) for the LTQ FT data. One can define the mass defect window for the filter, determine the mass range over which a mass defect filter applies, or implement multiple mass defect filters over different mass ranges. For the NEF metabolite analysis, the mass defect filter windows were set to ±0.040 Da around the mass defect of NEF or its core substructures over a mass range of ±50 Da around the masses of filter templates applied. For the analysis of omeprazole metabolites in human plasma, the mass defect filter window was set from –0.040 to +0.020 Da around the mass defect of omeprazole over a mass range of ±50 Da around the mass of omeprazole. When applied to an LC/MS data file, the filter retains ions whose mass defect falls within the specified boundaries while rejecting ions whose mass defect does not. After processing, the output file can be converted from the NetCDF format back to its native data file format using the original file conversion utility to facilitate comparison to the original data.

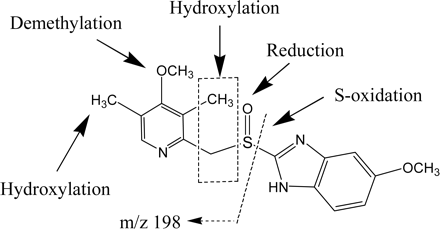
Proposed structures and biotransformation pathways of NEF metabolites in HLM.
Results
Metabolite Detection and Identification Strategy. A strategy for the screening and structural characterization of metabolites using MDF is illustrated in Fig. 1. High resolution LC/MS data are used for the detection of metabolite ions. The identities of the detected metabolites are determined based on the empirical formulae of their protonated molecules and the interpretation of their product ion spectra. MDF processing using both drug and core structure filter templates is the critical step in this metabolite identification strategy. The drug filter template is designed to retain metabolites that have undergone relatively minor changes in their molecular formulae (Table 1). Most of these metabolites are generated via oxidation, reduction, and dealkylation of small aliphatic groups. The core structure filter template, which follows one or a few core substructures of a drug, instead of the drug itself, is designed to select metabolites that are formed via metabolic reactions that lead to either a loss of a halogen (Table 2) or a cleavage of a parent drug into two significantly smaller molecules (Table 3). These metabolites (Tables 2 and 3) have nominal masses significantly lower than that of a parent drug, and consequently, their mass defects can be substantially different. The filter templates selected for the model compound nefazodone are listed in Table 4.
Mass and mass defect shifts for metabolites that have structures similar to those of the parent drugs
Mass and mass defect shifts for metabolites formed via typical dehalogenation reactions
Metabolites formed via dealkylation, hydrolysis or other metabolic reactions that metabolize parent molecules into significantly lower molecular masses
Summary of NEF metabolites in HLM detected by the MDF method with different templates
Screening for Nefazodone Metabolites Using MDF. An HLM incubation of NEF was analyzed by LC/FT-MS. The resultant high resolution/accurate mass LC/MS data file was then processed by the MDF method using drug and core structure filter templates. The base peak chromatogram (BPC) profile of the LC/MS data processed with the drug filter template revealed seven NEF metabolites (M1–M7) (Fig. 2) that have minor changes to their molecular formulae (Fig. 3A; Table 4). Most of these metabolites were mono- or dihydroxylated NEF except for an uncommon metabolite, M4, which was 12 Da less than that of NEF (Fig. 2). M7, a minor monohydroxylated metabolite, was displayed in the MDF-filtered BPC (Fig. 3A; Table 4), but not in the unprocessed BPC (data not shown).
The chromatographic profiles of the processed data based on the core structure from a loss of the chlorophenyl ring via N-dealkylation displayed three metabolites (Fig. 3B; Table 4). These metabolites were dechlorophenyl NEF (M9) and its derivatives (M8 and M10) (Fig. 2). The base peak chromatogram obtained from MDF processing using the core structure resulting from a loss of the m-CPP moiety showed metabolites M11 and M12 (Fig. 3C; Table 4). M11 and M12 were formed from the reduction and oxidation, respectively, of the aldehyde product of the NEF N-dealkylation reaction (Fig. 2). The MDF analysis using m-CPP as a filter template detected two metabolites, M14 (m-CPP) and M13 (a monohydroxy m-CPP) (Figs. 2 and 3D; Table 4). MDF processing following the core structures derived from potential O-dealkylation, and dechlorination reactions did not reveal any metabolites (data not shown); this is consistent with literature findings (Mayol et al., 1994; von Moltke et al., 1999; Kalgutkar et al., 2005; Peterman et al., 2006). Figure 3E is a chromatographic profile of NEF metabolites from data processed with a combination of the four filters as described above (Table 4). It showed, with few minor interference peaks, all the NEF metabolites present in the sample except for the minor monohydroxylated metabolite M7 (Fig. 4). Once the NEF metabolite ions were determined, their structures were further characterized by MS/MS or MSn analysis. The structures of M2, M4, M5, and M14 were confirmed based on comparisons of their LC retention times and product ion spectra with those of metabolite standards. The tentative structures of M8, M9, and M10 were postulated based on the comparison of the MSn spectra (data not shown) with those of the same metabolites reported in the literature (Kalgutkar et al., 2005). The proposed structures and formation pathways of NEF metabolites in HLMs are displayed in Fig. 2.

Metabolite profiles of NEF in HLMs determined by LC/FT-MS and MDF method. Base peak chromatograms of NEF metabolites in HLMs after MDF process using various filter templates (±0.040 Da), including: A, NEF as a filter template (MDF 0.1923–0.2723 Da; nominal mass 420–520 Da); B, the core structure from a loss of the chlorophenyl group as a filter template (MDF 0.1999–0.2799 Da; nominal mass 310–410 Da); C, the core structure from a loss of m-CPP as a filter template (MDF 0.1105–0.1905 Da; nominal mass 240–340 Da); D, the m-CPP as a filter template (MDF 0.0445–0.1245 Da; nominal mass 147–247 Da); E, a combination of all the above filter templates. Structures of these filter templates are presented in Table 4. Structures of NEF metabolites are presented in Fig. 2.
Screening for Nefazodone Metabolites by PI and NL Scan Analyses. For comparison, the NEF metabolites in HLMs were also analyzed using precursor ion and neutral loss scanning on a triple quadrupole mass spectrometer. The results are summarized in Table 5. NEF generated a major product ion at m/z 274 due to a loss of 3-chlorophenylpiperazine (a neutral loss of 196 Da; Fig. 5A). It also produced several minor product ions, including an ion at m/z 246 from a neutral loss of 224 Da. The ion current chromatogram (Fig. 6A) obtained from the PI scan of m/z 274 revealed three major drug-related components (M9, M2, and NEF) and two minor metabolite peaks (M3 and M7). All of the four metabolites detected were derived from the metabolic modification on the 3-chlorophenylpiperazine moiety (Fig. 2). A second PI scan that followed the product ion at m/z 246 (Fig. 6B) was able to detect two additional metabolites, M11 and M12, that were not found in the PI scan of m/z 274 (Fig. 6A). These two metabolites were derived from a loss of the 3-chlorophenylpiperazine moiety via N-dealkylation (Fig. 2). A third PI scan of m/z 290 (274 + 16) was also carried out to search for possible mono-oxidation metabolites. As a result, metabolites M1, M5, M6, and M8 (Fig. 6C) were detected, none of which were shown in the PI scan analyses of the m/z 274 or m/z 246 ions (Table 5). NL scan analyses were conducted by following the neutral losses of 196, 212 (i.e., 196 + 16), and 224 Da, respectively. These analyses detected five NEF metabolites in total: M4, M5, M1, M2, and M7 (Figs. 6, D–F).
Detection of NEF metabolites in HLM by neutral-loss and precursor ion scans using a triple quadrupole mass spectrometer

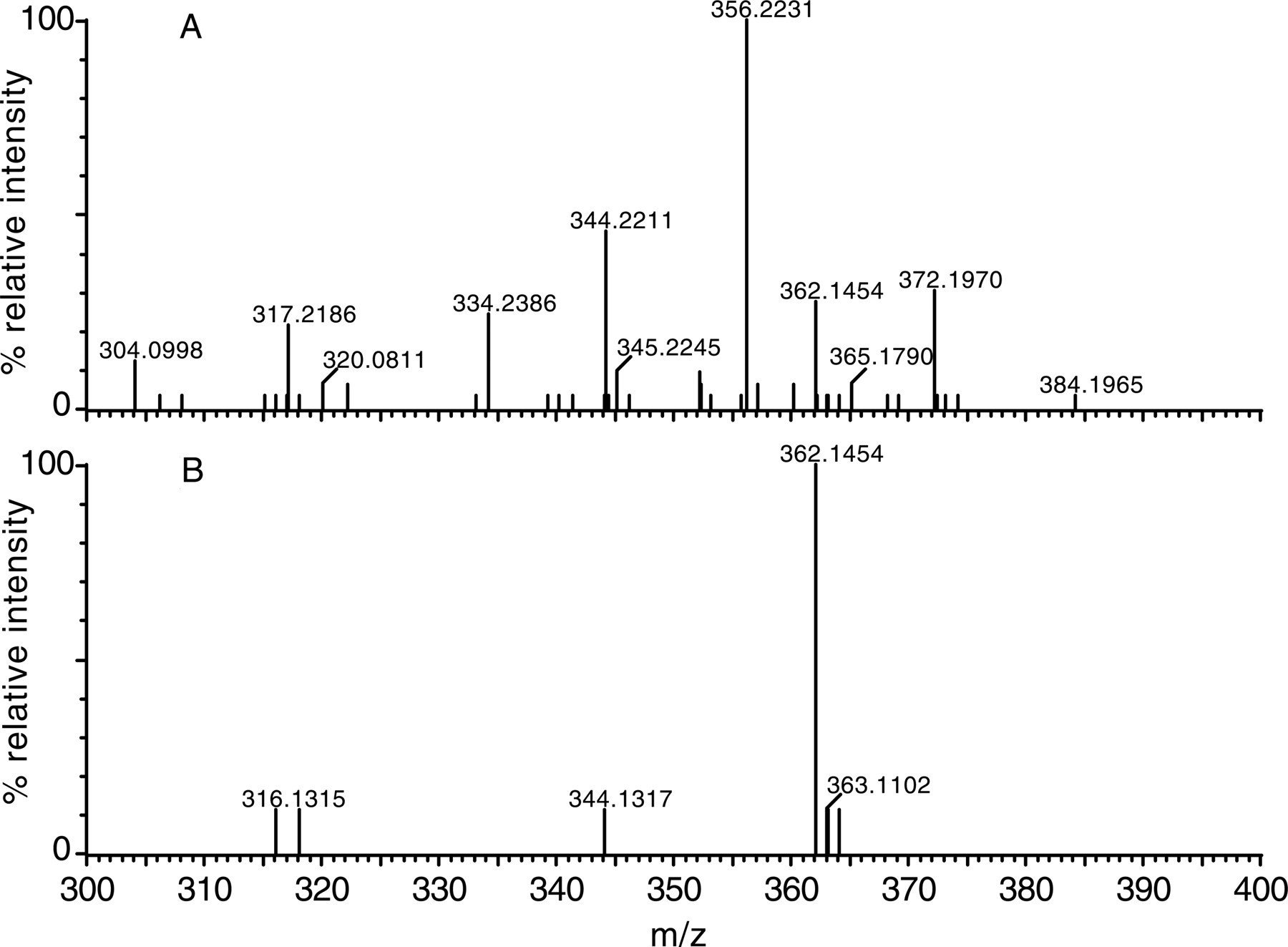
Mass spectra of monohydroxylated NEF (M7) from an HLM incubation, determined by LC/FT-MS with and without MDF processing. The spectrum of M7 was acquired by analyzing NEF metabolites in HLM using high resolution LC/FT-MS. A, spectrum of M7 obtained from the unfiltered data; B, spectrum of M7 obtained from MDF-processed data using NEF as a filter template (mass defect 0.1923–0.2723 Da, and nominal mass 320–620 Da).
Detection of Omeprazole Metabolites in Plasma by MDF. Q-ToF LC/MS analysis of human liver microsomal incubations of omeprazole showed several metabolites, including three mono-oxygenated metabolites, a reduction product, and a metabolite from a loss of 30 Da (Table 6). The possible modification sites of omeprazole are illustrated in Table 6, based on the major diagnostic product ion of each metabolite. Many of these metabolites were previously observed from in vitro incubations and/or in rats (Hoffmann, 1986; Weidolf and Covey 1992; Andersson et al., 1993). The TIC profile of omeprazole metabolites in plasma without MDF filtering displayed significant endogenous interferences with no apparent metabolite peaks (Fig. 7A). However, after MDF filtering, the TIC exhibited all metabolite peaks (M1–M5, Table 6) and minimal endogenous peaks (Fig. 7B). In addition, mass spectra of the metabolite peaks were greatly simplified after MDF processing. As a result, the mass spectra of metabolite peaks show predominantly the protonated molecules of the metabolites (e.g., Fig. 8). Furthermore, the accurate mass data also allow for the elimination of false-positive peaks. For example, peak E (Fig. 7B) shows predominantly an m/z 305 ion which does not generate any meaningful omeprazole-related empirical formulae (Table 6). A demethylated metabolite (MH+ = 332 Da), originally present in HLMs (data not shown), was not found in the plasma data (Fig. 7B), possibly due to ion suppression of coeluted plasma components.
Summary of omeprazole metabolites in human plasma detected by the MDF method
Discussion
In any metabolite identification study using LC/MS, the detection of drug metabolite ions is a critical step. The MDF approach, using both drug and core structure filter templates, greatly facilitates this process. MDF is a way of differentiating ions of interest from interference ions in an entire LC/MS data set by selecting ions at each nominal mass that have the predefined mass defect values. The mass defect of a compound is a characteristic associated with its empirical formula. By carefully evaluating the possible scenarios of mass defect and nominal mass changes in common oxidative metabolites, we concluded that these metabolites can be categorized into three groups: A, metabolites that are similar to the parent drug with respect to structure and exact mass (Table 1); B, metabolites derived from dehalogenation reactions (Table 2); and C, metabolites that are formed via the cleavage of the parent drugs and have significantly lower molecular masses (Table 3). For the majority of common biotransformation reactions, as listed in Tables 1, 2, and 3, the products have mass defects that are similar to, and are within a limited mass range of, those of either the parent drug or a core substructure of the parent drug. These predictable, narrow ranges for changes in mass defects and nominal masses are the basis for designing a mass defect filter strategy for drug metabolite detection in complex biological matrices (Fig. 1).

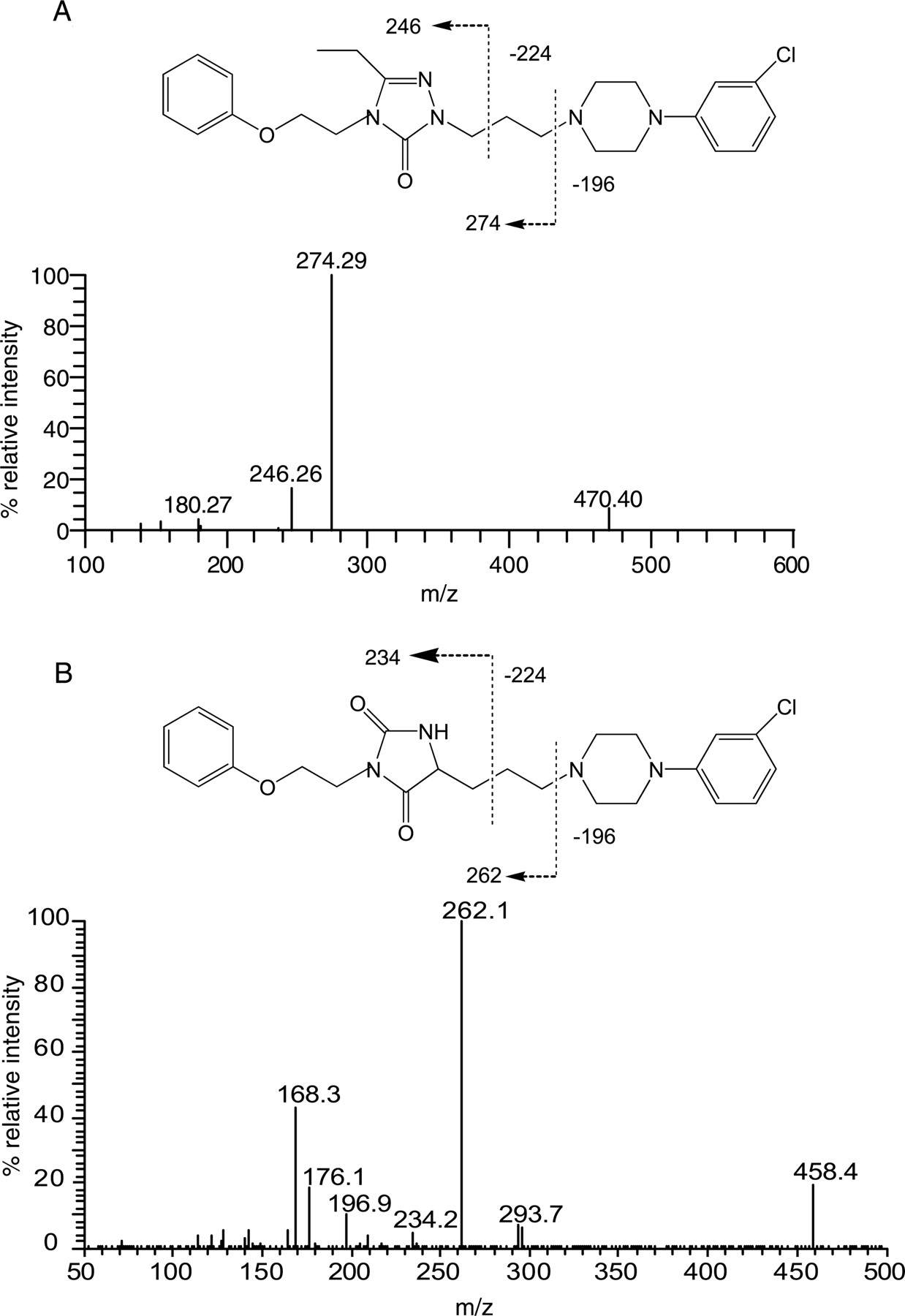
Triple quadrupole product ion spectra of A, NEF (m/z 470), and B, its triazoledione metabolite, M4 (m/z 458).
The drug filter template is designed to detect metabolites listed in group A (Table 1). Although these metabolites represent a variety of changes in the structures of the parent drugs, the changes in the mass defects and nominal masses of the metabolites are no more than 0.040 Da and 50 Da, respectively. Therefore, a drug filter template from –0.042 to +0.035 Da around the mass defect of a drug should be able to select for all the metabolites listed in Table 1. The utility of the drug filter template is demonstrated in the search of NEF (Table 4) and omeprazole (Table 6) metabolites that have mass defects similar to those of the parent drugs. For selective detection of a certain group of metabolites of interest, a specific mass defect range around that of the parent drug can be used. For example, a narrow mass defect filter from –0.0160 to –0.0050 Da around the mass defect of a drug may detect mono-, di-, and trihydroxylated metabolites. In the same way, for the screening of sequential oxidation products of a metabolite of interest, that metabolite may be used as a filter template.
The core structure template is designed to detect metabolites formed from biotransformation reactions that cleave a parent drug into two significantly smaller molecules that may have nominal masses and mass defects significantly different from those of the parent drug. Table 3 summarizes the metabolites from such metabolic cleavage reactions, including reduction of a disulfide, hydrolysis of amide or ester bonds, and N-, O-, and S-dealkylation reactions. Each of these metabolites can be used as a core structure template in search of the corresponding metabolites and their derivatives. In the NEF example, the use of core structure templates following potential N- and O-dealkylation products allowed for the detection of several N-dealkylated metabolites (M11–M14 in Fig. 3, C and D). These metabolites had significantly smaller mass defect values than NEF (Table 4) and were not detectable with the drug filter template (Fig. 3A). Another potential application of using core structures as filter templates is for the detection of dehalogenated metabolites (Table 2). The nominal mass changes of common dehalogenated metabolites vary significantly, from –160 Da to –2 Da, due to the loss of halogen-containing functional group(s) from a parent drug (RCHnXm, where X = F, Cl, or Br, n = 0, 1, or 2, and m = 1, 2, or 3). However, all the changes lead to metabolites that are similar to a core structure of RCH3, which is derived from the simple substitution of any halogen present in the structure with a hydrogen. Correspondingly, all these dehalogenated metabolites share mass defects and nominal masses similar to that of RCH3 (Table 2). Therefore, the use of RCH3 as a core structure template would select dehalogenated metabolites and their derivatives. In addition, a core structure template search would be useful for detecting metabolites of a prodrug by using the active metabolite as a filter template since most metabolites of a prodrug are further biotransformation products of the active metabolite.
The sensitivity and selectivity of the MDF approach for metabolite ion detection were demonstrated in the NEF example. NEF is known to undergo extensive cytochrome P450-mediated hydroxylation, N-dealkylation, and oxidation to multiple metabolites (Fig. 2), including those formed from uncommon reactions (the formation of M4) or via sequential metabolic pathways (the formation of M8, M10, and M13). After MDF processing with parent drug and core structure filter templates, a total of 14 metabolites in HLMs were revealed at the BPC levels (Fig. 3; Table 4). Most of the 14 metabolites, including M2, M4, M5, and M7 to M14, were previously observed in metabolism studies in vitro and/or in vivo (Mayol et al., 1994; von Moltke et al., 1999; Kalgutkar et al., 2005; Peterman et al., 2006). Two of the three dihydroxylation products, M1, M3, and M6, were not previously reported. Using extracted ion chromatograms from the raw data of NEF metabolites reported in the literature, it was verified that no known NEF metabolite peak was overlooked because of MDF processing.
Compared with the MDF approach, the PI and/or NL scanning methods detected fewer NEF metabolites. Monitoring three different precursor ions (i.e., m/z 274, 246, and 290), four NEF metabolites (M4, M10, M13, and M14; Table 5) were not detected because they did not generate the prescribed product ions under the collision-induced dissociation conditions chosen. Of course, M13 and M14 are common metabolites derived from the N-dealkylation reaction, and can be predicted and easily detected using a list-dependent analysis approach (Fig. 2). NL scan LC/MS analysis provides a complementary data set to the PI scan method. For example, M4, an uncommon metabolite with a protonated molecule 12 Da less than NEF (Fig. 5), was detected in a NL scan of 196 Da (Fig. 6D). However, the NL scan analyses alone missed the other nine NEF metabolites in the same sample (Fig. 6), possibly because of either the poor sensitivity or the lack of neutral loss fragment targets (Table 5). The results of NEF metabolite screening by the three techniques indicate that the MDF method allowed for the straightforward identification of a broader range of metabolites than PI and NL scan analyses. The sensitivity of the MDF method was comparable to that of the PI scan and was better than that of the NL scan.
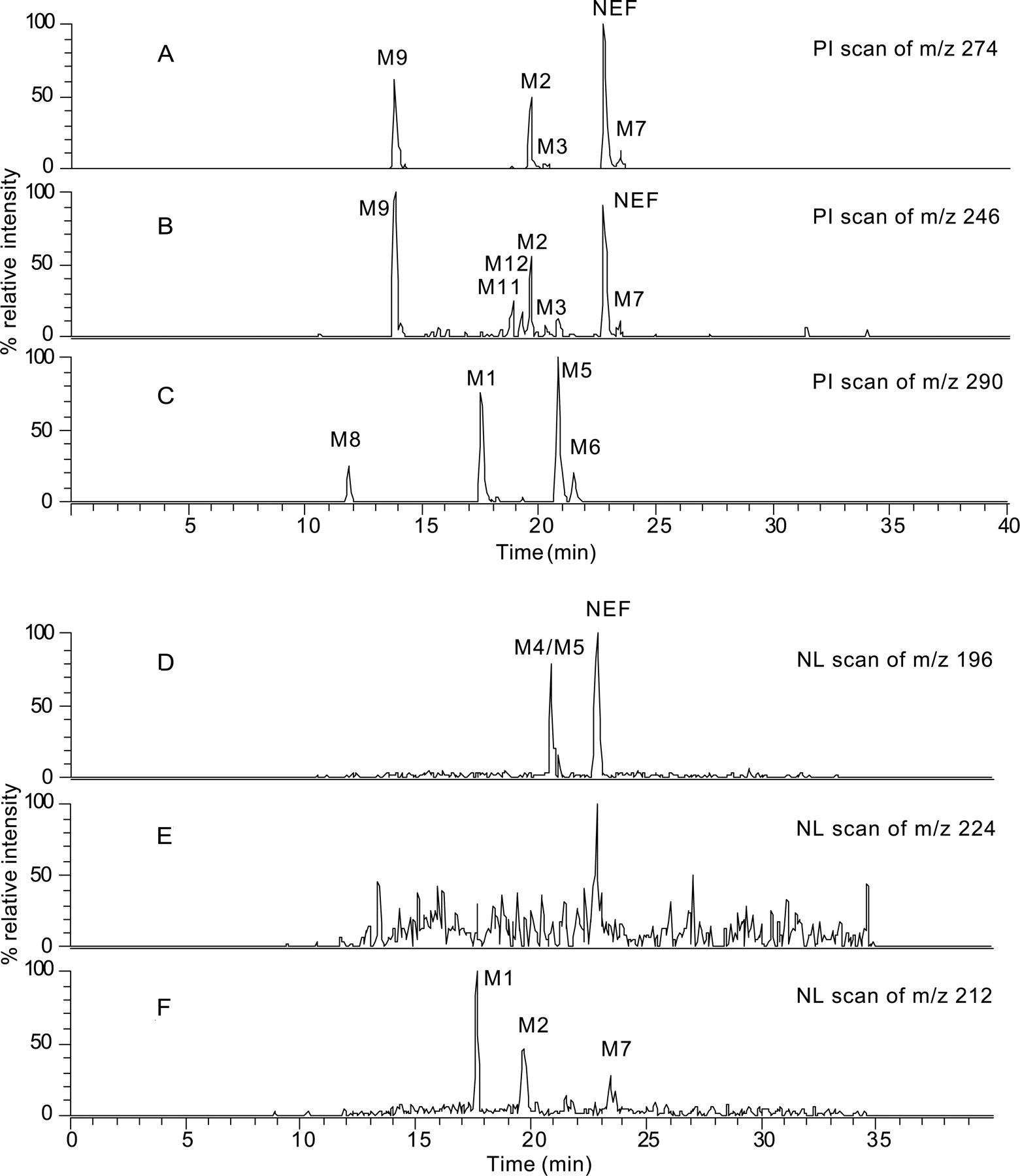
Analysis of NEF metabolites in HLMs by triple quadrupole LC/MS. A, B, and C are base peak chromatograms of precursor ion scans of m/z 274, 246, and 290, respectively; D, E, and F are base peak chromatograms of neutral loss scans of 196, 224, and 212 Da, respectively.
It is important to note that MDF is a post-acquisition approach. The MDF templates described here are flexible and can be refined as more information becomes available regarding a compound's metabolite profile, and the data can be reinterrogated without the need for further LC/MS analyses. Moreover, the data set required is straightforward high resolution, accurate mass LC/MS data, which has become routine on modern ToF and FT MS instruments. The experimental setup is simple, it does not change significantly from compound to compound, and minimal information about the compound under study is needed. In contrast, the PI and NL approaches require a detailed interpretation of the parent compounds MS/MS spectra, and the appropriate precursor ions and neutral losses need to be determined. The data set acquired is highly specific: if the metabolic modifications to the parent compound alter the fragmentation pathways, then the metabolites could be missed, and any adjustments an analyst might make to accommodate for this would require additional LC/MS acquisitions.
The effectiveness of the MDF approach for detecting metabolite ions in complex biological samples is illustrated in the analysis of omeprazole metabolites in human plasma. With MDF filtering, a significant number of endogenous interferences from a 1-ml human plasma sample were removed, rendering a drug metabolite profile essentially equivalent to that of a matrix-free sample (Fig. 7A versus Fig. 7B). This is the first attempt at the application of the MDF technique to the detection of metabolites in plasma samples provided that the metabolite ions present in the data. The capability of detecting drug metabolites in human plasma is especially valuable for first-inman studies in drug development, where nonradiolabeled drug candidates are typically administered. The metabolism and disposition studies of radiolabeled compounds in humans are often not conducted until late phase I to phase III clinical trials. An earlier assessment of the exposure of metabolites in humans would provide additional time for addressing potential issues associated with unique human metabolites that may not have been observed in the preclinical species, or metabolites with suspect toxic structures such as acyl glucuronides (Baillie et al., 2002). Some of these metabolites are often required to be quantitatively analyzed in toxicokinetic studies in animals and clinical pharmacokinetic studies.
The usefulness of the MDF process in simplifying the spectra of metabolites is also demonstrated in the presented examples. As shown in the unprocessed spectra, the protonated molecules of NEF metabolite M7 in HLMs (Fig. 4A) and omeprazole metabolite M3 in plasma (Fig. 8A) were obscured by many interference ions. Determining which of the many ions in these spectra are related to drug metabolites often takes significant effort. However, after MDF processing, all interference ions that have mass defects outside the filter windows were removed. Consequently, the protonated molecules of the metabolites of interest became predominant in the spectra (Figs. 4B and 8B). Not surprisingly, a few interference ions that have mass defects that fall within the filter windows still remain in the MDF-processed spectra, but they can often be excluded based on their molecular formulae. For example, there was an interference ion at m/z 516.2035 displayed in the MDF-processed spectrum of NEF metabolite M7 (Fig. 4B). The five most probable empirical molecular formulae of the molecule, including C31H26O3N5, are significantly different from the NEF molecular formula (C25H33O2N5Cl), indicating that it is not NEF-related.

Chromatographic profiles of omeprazole samples analyzed by Q-ToF LC/MS and MDF approach. A and B are TIC profiles of plasma spiked with omeprazole metabolites obtained without and with MDF processing, respectively.
As demonstrated in the NEF and omeprazole examples, the MDF processing technique uses high resolution LC/MS for the detection of both common and uncommon metabolites. However, one limitation of the current MDF method is that to effectively use core substructures as filter templates, an understanding of the basic biotransformation reactions that convert a parent drug to significantly smaller molecules is needed (Table 3). An improved MDF algorithm that automatically processes LC/MS data based on possible core substructures of the parent drugs would be helpful.

Mass spectra of omeprazole metabolite M3 in plasma obtained by Q-ToF LC/MS without (A) and with (B) MDF processing.
In summary, a simple MDF method using both drug and core structure filter templates has been applied to detect and identify oxidative metabolites that have mass defects similar to or significantly different from that of the parent drug. The effectiveness of this method was demonstrated for the model compounds nefazodone and omeprazole. The comparison with the PI scan and the NL scan clearly showed that the MDF-based high resolution LC/MS approach was more comprehensive in identifying metabolites. The metabolite search capability of the MDF technique, together with the empirical formula information associated with the data, makes high resolution LC/MS instruments a useful platform for the screening and identification of both common and uncommon metabolites.
Acknowledgments
We thank Dr. Vinod Arora and Yue-Zhong Shu for their contribution to the preparation of the common biotransformation reactions summary.
Footnotes
-
Part of this work was presented at the 7th International ISSX Meeting, Vancouver, Canada, 29 Aug–2 Sep, 2004 (Zhu et al., 2004).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009241.
-
ABBREVIATIONS: LC/MS, liquid chromatography/mass spectrometry; BPC, base peak chromatogram; FT, Fourier transform; MS/MS, tandem mass spectrometry; HLM, human liver microsome; NEF, nefazodone; m-CPP, meta-chlorophenylpiperazine; MDF, mass defect filter; MH+, protonated molecule; NL, neutral loss; PI, precursor ion; TIC, total ion chromatogram; ToF, time-of-flight; Q-ToF, quadrupole ToF.
-
↵1 Current affiliation: Novatia, LLC, Monmouth Junction, New Jersey.
-
↵2 Current affiliation: Amgen, Thousand Oaks, California.
- Received January 12, 2006.
- Accepted June 28, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}