


Abstract
Predictive in vitro methods to investigate drug metabolism in the human intestine using intact tissue are of high importance. Therefore, we studied the metabolic activity of human small intestinal and colon slices and compared it with the metabolic activity of the same human intestinal segments using the Ussing chamber technique. The metabolic activity was evaluated using substrates to both phase I and phase II reactions: testosterone, 7-hydroxycoumarin (7HC), and a mixture of cytochrome P450 (P450) substrates (midazolam, diclofenac, coumarin, and bufuralol). In slices of human proximal jejunum, the metabolic activity of several P450-mediated and conjugation reactions remained constant up to4hof incubation. In the colon slices, conjugation rates were virtually equal to those in small intestine, whereas P450-mediated conversions occurred much slower. In both organs, morphological evaluation and ATP content implied tissue integrity within this period. P450 conversions using the Ussing chamber technique showed that the metabolic rate (sum of metabolites measured in apical, basolateral, and tissue compartments) was constant up to 3 h. For 7HC conjugations, the metabolic rate remained constant up to 4 h. The distribution of the metabolites in the compartments differed between the substrates. Overall, metabolic rates were surprisingly similar in both techniques and appear similar to or even higher than in liver. In conclusion, this study shows that both human intestinal precision-cut slices and Ussing chamber preparations provide useful tools for in vitro biotransformation studies.
Although the liver has long been thought to play the major role in drug metabolism, the metabolic capacity of the intestine has been increasingly recognized (von Richter et al., 2001; Glaeser et al., 2004).
In the 1970s, intestinal metabolism was already reported (Stohs et al., 1976). Nonetheless, until recently the clinical significance of intestinal drug metabolism remained under debate (Lin et al., 1999; Doherty and Charman, 2002). Although basic knowledge concerning human intestinal drug enzyme and transporter expression has been collected over the past decade (Obach et al., 2001; Thorn et al., 2005), the ultimate proof for their significance has been given by in vivo studies. For several compounds, such as cyclosporin, verapamil, and midazolam, in vivo studies have shown significant first-pass metabolism by the intestinal wall (Kolars et al., 1991; von Richter et al., 2001; Glaeser et al., 2004). The significance of the intestine in determining the faith of drugs within the body is not only expressed by its high capacity to metabolize drugs but also by its sensitivity to induction and inhibition of drug-metabolizing enzymes (Pelkonen et al., 2001), in addition to the interplay between transporters and metabolic activity (Suzuki and Sugiyama, 2000; Benet et al., 2004; Jeong et al., 2005).
The challenge in studying human intestinal metabolism nowadays is to develop in vitro systems in which intact cells remain functioning because quantitative in vivo studies are difficult to perform. Subcellular human intestinal fractions, such as microsomal preparations and S-9 fractions, have been used up to 30 min of incubation (Wynalda et al., 2003). However, in addition to their limited lifespan, they lack the operable cell membrane transporters and are often incubated with unphysiological concentrations of cofactors. Furthermore, Caco-2 cells have been proven to be useful for absorption studies (Artursson and Karlsson, 1991; Ungell and Karlsson, 2003). However, interlaboratory reproducibility of Caco-2 cells is problematic, and they are lacking normal levels of important metabolic enzymes such as CYP3A4 (Sun et al., 2002).
Until now, several intact cell systems exist to study intestinal metabolism. Recently, the precision-cut slice technique was applied to rat intestinal tissue, and these studies showed that intestinal precision-cut slices are superior to so-called intestinal tissue biopsy punches (de Kanter et al., 2005). In addition, they exhibit drug metabolism at a constant rate for at least 3 h of incubation (van de Kerkhof et al., 2005). With the very efficient and rapid slice technique, many slices can be produced from a small tissue sample maintaining the cells in their physiological context with respect to metabolic enzymes, membrane transporters, and cofactors. The applicability of this system for human tissue, however, has not been validated to date. The Ussing chamber technique, in which a small piece of mucosal tissue is mounted such that mucosal and serosal sides are separately exposed to incubation medium, has been proven to be useful in absorption studies (Ungell et al., 1998) and successfully predicts human intestinal permeability in vivo for several compounds (Lennernas et al., 1997). However, to our knowledge, studies using Ussing chamber preparations were mainly focused on the prediction of the fraction absorbed, whereas metabolic capacity and disposition of the formed metabolites have not been reported. Nonetheless, indications exist that when only intestinal transport is measured, the metabolic extraction of the tissue may lead to false predictions of fraction absorbed (Ungell, 2005).
In this study, we adapted the precision-cut slice method to human tissue previously developed for rat intestinal tissue to study drug metabolism. The viability of slices was evaluated by measuring intracellular ATP levels, morphology, and metabolic stability during 4 h of incubation. Slices of proximal jejunum and colon tissue were incubated with high concentrations of testosterone (TT) or 7-hydroxycoumarin (7HC) to assess phase I and phase II, respectively, capacity at Vmax. In addition, slices were incubated with a mixture of cytochrome P450 (P450) substrates (midazolam, coumarin, diclofenac, and bufuralol) at concentrations below Km. These compounds are probes for CYP3A4/5, CYP2A6, CYP2C9, and CYP2D6. Second, from the same proximal jejunum tissue samples we prepared mucosal sheets for the Ussing chamber setup. These sheets were incubated up to 4 h with the mixture or 7HC while the viability was monitored by electrical parameters (Polentarutti et al., 1999). This enabled a direct comparison between the metabolic activity of human intestinal tissue either mounted in Ussing chambers or in the form of precision-cut slices.
Materials and Methods
Chemicals. Dimethyl sulfoxide, trifluoroacetic acid (TFA), low gelling temperature agarose (type VII-A), amphotericin B solution (250 μg/ml), d-glucose, 7HC, 7-hydroxycoumarin-glucuronide (7HC-GLUC), HEPES, sodium bicarbonate, β-glucuronidase, sulfatase, TT, 11β-hydroxytestosterone (11β-TOH), 6β-TOH, and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). Williams medium E with Glutamax-I (WME) and gentamicin (50 mg/ml) solution were obtained from Invitrogen (Paisley, UK). Formaldehyde solution (3.8%) was purchased from Mallinckrodt Baker B.V. (Deventer, The Netherlands). Hydroxy-bufuralol maleate salt, bufuralol hydrochloride salt, 1-hydroxy midazolam, and 7-hydroxycoumarin-sulfate (7HC-SULF) were purchased from Ultrafine (Manchester, UK). 4-Hydroxy-diclofenac was obtained from Gentest (Woburn, MA). Midazolam was purchased from Lipomed AG (Arlesheim, Switzerland). Sodium azide was obtained from Kebo lab (Spånga, Sweden). Sodium chloride, EDTA, formic acid, Tris-HCl, dichloromethane, and sodium acetate were obtained from Merck (Darmstadt, Germany). Acetonitrile (ACN) and methanol (MeOH) were purchased from Rathburn (Walkerburn, Scotland). Ethanol was obtained from Kemetyl (Haninge, Sweden). 2α-, 16α-, and 11α-TOH were obtained from Steraloids (Newport, RI).
Human Tissue. Human tissue originated from surgical resections with approval from the regional ethical committee in Gothenburg (Sweden) and from each of the individual patients. Proximal jejunum was obtained from obese patients from Sahlgren's University Hospital (Sweden), and the colon resections were from patients with colon carcinomas at East Hospital (Gothenburg, Sweden). Donor characteristics are listed in Table 1.
Donor characteristics
After surgical removal, tissue was directly stored in ice-cold constantly oxygenated (with carbogen, 95% O2 and 5% CO2) Krebs-bicarbonate Ringer (KBR) solution (pH 7.4) with the composition reported earlier (Polentarutti et al., 1999).
Preparation and Incubation of Human Tissue. Directly after tissue arrival in the laboratory, the tissue was put in fresh oxygenated ice-cold KBR solution, and the muscle layers and serosa were carefully removed. In all the cases, preparation of mucosal tissue was finished within 1 or 2 h calculated from excision. The end of mucosal preparation is referred to as t = 0 in experiments.
Preparation of precision-cut slices. Mucosal tissue was transferred to ice-cold oxygenated Krebs-Henseleit buffer (containing 10 mM HEPES and 25 mM d-glucose, pH 7.4). Tissue was cut in pieces of 7 to 9 mm width and variable length, and segments were subsequently vertically embedded in 3% low-gelling temperature agarose in 0.9% NaCl solution (37°C) to ensure that every slice consisted of all the cell layers present in the mucosa. Then, the agarose solution was allowed to gel using a precooled (0°C) tissue-embedding unit (Alabama Research and Development, Munford, AL). After the agarose solution had gelled, precision-cut slices (thickness about 400 μM) were cut using a Krumdieck tissue slicer as described previously (de Kanter et al., 2005).
Incubation of precision-cut slices. Slices were incubated individually in 12-well culture plates (Costar no. 3512, Corning Incorporated, NY) in 1.3 ml of WME with Glutamax-I, supplemented with d-glucose (final concentration 25 mM), gentamicin (final concentration 50 μg/ml), and amphotericin B (final concentration 2.5 μg/ml). The culture plates were placed in a prewarmed cabinet (37°C) in plastic boxes. Slices were incubated under humidified carbogen (95% O2/5% CO2) and shaken using an orbital shaker at approximately 45 rpm.
Preparation and incubation for Ussing chambers. Mucosal tissue was mounted onto the Ussing chamber segment holder as flat sheets with a surface area of 1 cm2. Each of the two-chamber compartments on both sides of the segment was subsequently filled with 10 ml of oxygenated KBR (room temperature) continuously gassed with carbogen (95% O2/5% CO2), stirred with approximately 300 rpm, and slowly warmed until 37°C by water jackets. The tissue was mounted and placed in the Ussing chamber setup, after which the tissue was allowed to stabilize for 30 min to recover from the preparation and to equilibrate in the chambers.
Viability Testing of Precision-Cut Slices.Morphology. The condition of precision-cut slices was evaluated after 4 h of incubation by morphology. After incubation in triplicate per experiment, slices were stored in 3.8% formaldehyde solution at 4°C. In addition, directly after tissue arrival in the laboratory, a piece of tissue was put in ice-cold 3.8% formaldehyde solution as a control. Samples were sent to HistoCenter AB (Frölunda, Sweden) for further processing and H&E staining. Colon slice incubations for morphology were performed in two experiments with tissue from two subjects in triplicate; small intestinal slice incubations were performed in five experiments with tissue from five subjects in triplicate.
Intracellular ATP levels. Intracellular ATP levels were evaluated to judge the overall metabolic condition of the tissue after 4 h of incubation. Directly after tissue preparation, three pieces of tissue were snap-frozen as controls. Intracellular ATP levels were determined according to the method described by De Kanter et al. (2002). Directly after incubation, slices were put in 1 ml of ice-cold 70% ethanol containing 2 mM EDTA (pH 10.9) to precipitate the proteins, and directly snap-frozen in N2(l) and stored at –80°C until further use. Subsequently, samples were thawed on ice and homogenized by sonication. Samples were centrifuged (3 min, 4°C, 10,000g) to spin down the proteins, after which the supernatant was transferred to a new cup, and the pellet was used for protein determination. The supernatant was diluted with 100 mM Tris-HCl and 2 mM EDTA solution (pH 7.8) and analyzed using the ATP Bioluminescence Assay Kit CLS II (Roche Applied Sciences, Mannheim, Germany) measuring luminescence using a plate reader (Spectramax microplate reader, Molecular Devices, Sunnyvale, CA). ATP was determined in four experiments using tissue from four different donors in triplicate.
Stability of drug metabolism rate. The stability of metabolic rate was determined between 0 and 4 h of incubation. To evaluate phase I metabolism, slices were incubated with TT (final concentration 250 μM, 1% MeOH) or a mixture of 4 compounds: midazolam (3 μM), diclofenac (5 μM), bufuralol (10 μM), and coumarin (3 μM), with 0.35% MeOH as a final solvent concentration. As control, medium was incubated with substrate but without slice. After 1, 2, 3, and 4 h of incubation with TT, slice and medium were harvested together and stored at –20°C until further use. In earlier rat studies, we found that significant amounts of TT metabolites were retained within the slice (de Kanter et al., 2005). Therefore, slice and medium were analyzed together.
For mixture incubations, no metabolic interactions were detected between substrates at these concentrations (T. Andersson, AstraZeneca, Mölndal, Sweden, personal communication). Furthermore, pilot experiments showed that only minor amounts of the mixture metabolites were retained in the slice and were barely detectable. Therefore, only the medium samples were analyzed. During 0 to 4 h of incubation, medium samples (100 μl) were taken every hour. The last sample contained slice tissue and the remaining medium. Samples were stored at –20°C until further use.
To evaluate phase II metabolism, slices were incubated with 7HC (500 μM, 1% MeOH), and as a control, medium was incubated with substrate. Medium samples (200 μl) were taken every hour between 0 and 4 h and stored at –20°C. After incubation, slices were stored at –20°C for protein determination.
Metabolism experiments with proximal jejunum were performed on tissues from five to eight donors in triplicate. Metabolism experiments in colon were performed on tissue from five donors in triplicate.
Viability Testing Ussing Chambers.Electrical parameters. Viability was monitored by measuring electrical parameters as described previously (Polentarutti et al., 1999): the potential difference (PD) reflects the voltage gradient generated by the tissue; the short-circuit current (SCC) reflects the ionic fluxes across the epithelium; and the resistance (R) reflects tissue integrity.
In brief, a four-electrode system recorded electrical parameters during the experiment. The PD was recorded using calomel electrodes connected by agar/NaCl bridges. Every 5 min, five current (I) pulses (0 ± 15 ± 30 μA) were sent via AgCl electrodes through the chamber, after which the voltage response (U) was measured. The current (I) – voltage (U) pairs form a linear plot from which the PD can be deduced. PD was obtained from the intersection with the voltage axes, when I = 0, and the R of the tissue segment was the slope of this plot. R was corrected for external, nontissue resistance by the recorder system. The SCC was calculated from Ohm's law: I = U/R.
Stability of drug metabolism rate. Tissue from four individual donors was used to prepare both precision-cut slices and Ussing chamber preparations to enable a direct comparison between the metabolic activity in the Ussing chamber preparations and precision-cut slices. In Table 1 it is indicated which tissue was used for both experiments.
For a set of eight chambers per subject and per day, two experimental setups were chosen: 1) Four chambers were incubated with the mixture of the four P450 substrates: midazolam (3 μM), diclofenac (5 μM), bufuralol (10 μM), and coumarin (3 μM); and 2) Four chambers were incubated with 7HC (500 μM, 1% MeOH) to assess phase II metabolism. The metabolic reactions were monitored during 4 h of incubation. At 1, 2, 3, and 4 h of incubation with the model substrates from one of the chambers, tissue and medium [both apical (A) and basolateral (B) individually] were harvested. This terminated the incubation of one chamber at that specific time point. Samples were stored at 4°C until the end of the experiment and subsequently stored at –20°C until further use.
Metabolite Analyses.TT sample analysis. After thawing (at 4°C), samples were homogenized using a sonicator. Then, 10 μl of 11β-TOH (internal standard, 0.2 μg/ml) and 6 ml of dichloromethane were added. The total mixture was vortexed and centrifuged using a Beckman-Coulter CS-6KR centrifuge (Beckman Coulter, Fullerton, CA) (10 min, 4°C, at 800g). The water layer was removed, and the dichloromethane evaporated overnight. The residue was dissolved in 130 μl of 50% MeOH and then analyzed by high-performance liquid chromatography as described previously (van't Klooster et al., 1993). The concentration range of the standard curve was 2 to 10 μM. The lower limit of quantitation (LLOQ) was 0.1 μM. The intraday and interday variability (2 μM) were 6.1 and 8.8% CV, respectively.
Mixture sample analysis. Slice samples containing medium and tissue were homogenized by sonication. Ussing chamber tissue samples were homogenized in 2 ml of fresh WME using a Potter-Elvehjen homogenizer. Subsequently, two volumes of ACN were added to precipitate the proteins. Then, samples were centrifuged (10 min, 20°C, 4000g). The supernatant, used for metabolite analysis, was diluted with two volumes of water (final concentration of ACN: 30%). The precipitate was stored at –20°C for protein determination. Medium samples were directly pipetted into a 96-well plate together with standards (1′OH-midazolam, 4′OH-diclofenac, OH-bufuralol, and 7′OH-coumarin) dissolved in both water and 30% ACN. The plate was centrifuged for 20 min (4000g, 4°C) before injection into a liquid chromatography/mass spectrometry (LC/MS). For OH-bufuralol, the concentration range of the standard curve used was 0.05 to 0.25 μM, and the interday variability (0.25 μM) was 5.6% CV. For 4′OH-diclofenac and 1′OH-midazolam, the concentration range of the standard curves was 0.25 to 1 μM, and the interday variability (1 μM) was 3.4 and 1.3% CV, respectively. The LLOQ was lower than 5 nM for OH-bufuralol, 1′OH-midazolam, and 4′OH-diclofenac.
LC separations were performed on a reversed-phase HyPURITY C18 analytical column (50 × 2.1 mm i.d., 5 μm, ThermoQuest, Runcorn, UK) with a HyPURITY C18 guard column (2.1 × 10 mm, 5 μm) at 40°C. The mobile phase consisted of a mixture of (A) 0.1% formic acid in water and (B) 0.1% formic acid in ACN. A gradient elution containing solvents A and B was performed: solvent A was linearly decreased from 95% to 20% during 3 min, where it remained isocratic for 0.2 min. Then A was increased to 95% during 0.1 min and re-equilibrated at 95% A for 2.5 min. A flow rate of 0.75 ml/min was used, and an aliquot of 15 μl was injected for analysis. The MS analyses were performed with a triple quadrupole mass spectrometer, API4000, equipped with electrospray interface (Applied Biosystems/MDS Sciex, Concord, Canada). The instrument settings were optimized using the individual analytes. The MRM transitions of the precursor ions to the corresponding product ions were monitored for the analytes. Instrument control, data acquisition, and data evaluation were performed using Applied Biosystems/MDS Sciex Analyst 1.4 software.
Deconjugation of mixture samples. To test whether further conjugation of the hydroxy-metabolites of the mixture compounds had occurred, Ussing chamber medium samples after 2 h of incubation were deconjugated. For this, 40 μl of medium was added to 65 μl of 1 N sodium acetate buffer (pH 4.5) containing 5480 U/ml β-glucuronidase and 59 U/ml sulfatase and was incubated for 2 h at 37°C. The reaction was terminated by precipitation of the proteins adding two volumes of ACN. After vortexing, the samples were centrifuged and diluted twice with water before analysis. Analysis occurred as described in the section mixture sample analysis.
7HC sample analysis. After thawing, sodium azide (final concentration 0.1 mg/ml) was added to the samples to prevent bacterial growth. Subsequently, the samples were centrifuged (3 min, 4°C, 10,000g), and metabolites were determined by high-performance liquid chromatography. 7HC, 7HC-GLUC, and 7HC-SULF were used as references. Chromatography was performed on a Zorbax Column SB-C18 (4.6 mm × 15 cm, 5 μm, Hewlett Packard, ChromTech AB, Hägersten, Sweden). The mobile phase consisted of (A) 0.1% TFA and (B) 0.1% TFA in 100% ACN. The separation run lasted 16.0 min, including column equilibration. The gradient was as follows: 0% B for 1 min, followed by linear increase to 20% B over 4 min, followed by a step gradient to 85% B for 6 min, remained at 85% B for 3.5 min, and returned to 0% B for 1.5 min. Per sample, 50 μl was injected, and the flow rate was 0.8 ml/min. The concentration range of the standard curves of 7HC-GLUC and 7HC-SULF was 0.25 to 1 μM, and the interday variability (1 μM) was 0.5 and 3.4% CV, respectively. The LLOQ was lower than 0.25 μM.

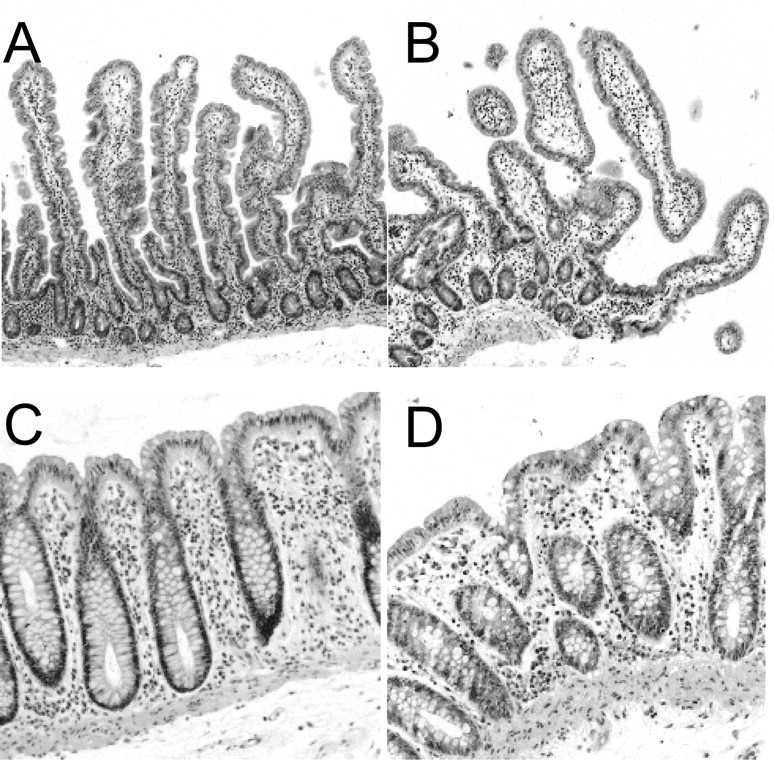
Micrographs of small intestinal tissue as control (A) and slices after 4 h of incubation (B). In addition, micrographs of colon tissue as control (C) and colon slices after 4 h of incubation (D). Staining: H&E (100× for small intestine; 200× for colon). Representative micrographs from five experiments (small intestine) and two experiments (colon) using three slices per incubation.
Protein Determination. Because slices had somewhat different sizes, the protein content was determined on all the slice samples after ATP analysis, 7HC incubations, or mixture incubations to enable expression on protein basis. Protein values were determined on all the Ussing chamber tissue samples as well. After incubation, samples were stored at –20°C until further use. Subsequently, samples were thawed, and 5 N NaOH was added: 20 μl of 5 N NaOH for slice samples was used for ATP determination and 7HC metabolism in Eppendorf cups, and 200 μl of 5 N NaOH was used for mixture slice samples and Ussing chamber tissue samples in centrifuge tubes. Forty minutes of incubation at 37°C followed to dissolve the tissue. Then, water was added to obtain a final concentration of 0.1 N NaOH needed for the analysis. Tissue was homogenized using sonication and further diluted to determine the protein content using Bio-Rad protein assay dye reagent (Bio-Rad, Munich, Germany) with bovine serum albumin as standard. For TT experiments, the protein content could not be determined because the proteins were removed together with the water layer during extraction. Therefore, the average of the protein values of 7HC incubations was used. Per experiment, 79 to 97% of the slices had a protein content ranging from 50 to 150% of the average protein content.
Statistics. Statistical significance of the difference between Ussing chamber preparations and precision-cut slices was determined using a paired Student's t test. The linearity of metabolic rates in time was determined using linear regression analyses.
Results
Viability Assessment of Small Intestinal and Colon Slices. Three different parameters were assessed to evaluate the viability of small intestinal and colon slices after 4 h of incubation: morphology, ATP levels, and metabolic stability. Small pieces of nonincubated tissue served as controls for morphology and ATP levels.
Morphology. Although the villi showed some flattening, the overall tissue morphology and the morphological appearance of the colonocytes/enterocytes of colon and proximal jejunal slices remained intact during 4 h of incubation compared with nonincubated control tissue. No cell swelling or necrosis was observed. The morphological evaluation indicated that the number of intact epithelial cells was not reduced during this incubation period (Fig. 1).
Intracellular ATP. General viability of small intestinal slices was assessed in four experiments by measurement of intracellular ATP levels. The level of ATP in tissue pieces harvested directly after preparation of the mucosal sheets was 1.2 nmol/mg protein and increased significantly during the first 4 h of incubation to 3.3 nmol/mg protein (p < 0.02).
Viability in Ussing Chamber Preparations. To evaluate the tissue integrity of Ussing chamber preparations, electrical parameters were monitored.
Electrical parameters. In Fig. 2, the electrical parameters of Ussing chamber preparations are shown during 4 h of incubation with substrates. The PD, reflecting the voltage gradient generated by the tissue, was 7.9 mV and decreased significantly (p < 0.003) to 4.5 mV after 4 h of incubation. The SCC values (reflecting the ionic fluxes across the epithelium) were 2 μA/mm2 at the beginning of the experiment and, after a small increase to 2.3 μA/mm2, decreased nonsignificantly to 1.2 μA/mm2 after 4 h of incubation. The R, reflecting tissue integrity, increased nonsignificantly from 42 to 48 Ω× cm2 after 4 h of incubation.

Electrical parameters of human intestinal segments in the Ussing chamber technique during 4 h of incubation: PD (A), SCC (B), and R (C). Results show mean ± S.E.M. of four donors; each time point represents 6 to 32 chambers.

Metabolic activities as detected in precision-cut slices toward TT prepared from small intestine: A, androstenedione; and B, 6β-TOH, 16α-TOH, and 2β-TOH. Results show mean ± S.E.M. of five to seven donors; in each experiment three slices were incubated per time point.
Stability of Metabolic Rate in Precision-Cut Slices.TT metabolism. The rates of TT metabolite formation in human proximal jejunum are shown in Fig. 3. Four different metabolites could be quantified: androstenedione, 16α-, 6β-, and 2β-hydroxytestosterone. Androstenedione and 16α-TOH were formed at a constant rate up to 4 h of incubation (R2 > 0.93). For 6β- and 2β-TOH, the highest formation rate was detected between 0 and 1 h of incubation. In order from highest to lowest formation rate: androstenedione (316 pmol/min/mg protein), followed by 6β-TOH (107 pmol/min/mg protein in first hour), 2β-TOH (25 pmol/min/mg protein in first hour), and 16α-TOH (9 pmol/min/mg protein).
Hydroxylation of midazolam, diclofenac, bufuralol and coumarin. Slices were incubated with a mixture of drugs (midazolam, diclofenac, bufuralol, and coumarin) to further evaluate the stability of P450-mediated reactions. The metabolite formation in time is depicted in Fig. 4, A through C. No 7′OH-coumarin metabolite formation could be detected in the slices. 1′OH-midazolam (R2 > 0.92), 4′OH-diclofenac (R2 > 0.97), and OH-bufuralol (R2 > 0.93) formation were time-dependent up to 4 h of incubation. The fastest metabolic hydroxylation rate was detected using midazolam as a substrate (11.6 pmol/min/mg protein), followed by diclofenac (7.7 pmol/min/mg protein) and bufuralol (1.3 pmol/min/mg protein). In Fig. 4, the metabolite values in the slices are depicted as mean values of four experiments using tissue from four different donors. These were performed simultaneously with Ussing chamber experiments. Two additional slice experiments were performed with the mixture. The obtained results were in line with the results depicted in Fig. 4 (data not shown). The range of metabolite formation rates in slices in the six experiments were 8.3 to 17 (1′OH-midazolam), 4.4 to 9.4 (4′OH-diclofenac), and 0.2 to 2.2 pmol/min/mg protein (OH-bufuralol). In colon slices, the hydroxy-metabolite formation for all the substrates was below the detection limit.
7HC conjugation.Figure 5 shows that for both glucuronidation and sulfation, the metabolic rates were constant during the first 4 h of incubation in both small intestine (open symbols, R2 > 0.98) and colon (closed symbols, R2 > 0.98).

Metabolic activities detected using precision-cut slices and Ussing chamber preparations prepared from proximal jejunum. Phase I reaction rates were assessed with a mixture of four compounds: midazolam toward 1′OH-midazolam (A), diclofenac toward 4′OH-diclofenac (B), bufuralol toward OH-bufuralol (C), and coumarin toward 7′OH-coumarin (not detectable). Phase II reactions were monitored using 7HC toward 7HC-GLUC (D) and 7HC-SULF (E). Results are mean ± S.E.M. of four donors; in each experiment three slices and one Ussing chamber preparation were incubated per time point.
In small intestinal and colon slices, the glucuronidation rate is higher than the sulfation rate. Comparing colon and small intestinal slices, comparable rates were found for both glucuronidation (small intestine: 315 pmol/min/mg protein, colon: 377 pmol/min/mg protein) and sulfation (small intestine: 52 pmol/min/mg protein, colon: 62 pmol/min/mg protein).
Stability of Metabolic Rate in Ussing Chamber Preparations.Hydroxylation of midazolam, diclofenac, bufuralol, and coumarin. In the Ussing chamber incubations, metabolites that were detected in the apical chamber (A), basolateral chamber (B), and in the tissue (T) are indicated in Fig. 6. The sums of the metabolites in these three compartments correspond to the total metabolite formation in the Ussing chamber incubations and are depicted in Fig. 4.
As in the slices, 7′OH coumarin could not be detected in Ussing chamber experiments after 4 h of incubation in either compartment A or B. However, low amounts of the metabolite could be detected in tissue, indicating a low activity of the CYP2A6 enzyme.
1′OH-midazolam (R2 > 0.95) formation and 4′OH-diclofenac (R2 > 0.99) formation were linear up to 3 h, and OH-bufuralol formation was linear up to 4 h of incubation (R2 > 0.98) (Fig. 4, A–C). As for slices, the highest metabolite formation was 1′OH-midazolam (9.3 pmol/min/mg protein), followed by 4′OH-diclofenac (6.9 pmol/min/mg protein) and OH-bufuralol (1.3 pmol/min/mg protein). All the detectable hydroxy-metabolites were excreted to a higher extent in the apical compartment compared with the basolateral compartment [significant for 1′OH-midazolam (p > 0.005) and 4′OH-diclofenac (p > 0.007) but not for OH-bufuralol (p > 0.11)] (Fig. 6, A–C).
The total amount of metabolites that accumulated in the tissue remained constant during incubation: after 1 h of incubation, 22 to 34% of the formed metabolites were retained within the tissue and after 4 h of incubation, 10 to 13%.
Deconjugation of mixture metabolites. To evaluate possible conjugation of the hydroxy-metabolites formed, media of Ussing chamber preparations (after 2 h of incubation) from both A and B compartments were deconjugated and reanalyzed (Table 2). In the A and B compartments no relevant conjugation could be detected for 1′OH-midazolam, OH-bufuralol, or 7′OH-coumarin (remained below the detection limit). However, the amount of 4′OH-diclofenac increased to 133% in A and 249% in B after deconjugation, resulting in an increase in total metabolism of 145%.
Percentage increase in metabolite concentration after incubation with deconjugating enzymes
Deconjugated samples were Ussing chamber medium samples incubated with mixture of compounds for 2 h (n = 4, mean ± S.E.M.).

Conjugation activity toward 7HC was measured using precision-cut slices prepared from small intestine (open marks) and colon (closed marks). 7HC-GLUC (♦) and 7HC-SULF formation (▴) were measured during 0 to 4 h of incubation. Results are mean ± S.E.M. of five to seven donors; in each experiment three slices were incubated.
7HC conjugation. For 7HC, tissue glucuronidation and sulfation rates were constant during 4 h of incubation in the Ussing chambers (R2 > 0.95) (Fig. 4, D and E). As for slices, glucuronidation (415 pmol/min/mg protein) occurred faster than sulfation (66 pmol/min/mg protein). The excretion of both conjugates to the A and B compartments differed for the two conjugates. For 7HC-GLUC, a slightly higher excretion to B than to A was observed (nonsignificant: p > 0.08) (Fig. 6D). For 7HC-SULF, more metabolite (although nonsignificant: p > 0.08) was excreted toward A compared with B (Fig. 6E). After 1 h of incubation, 18% of 7HC-GLUC and 0% 7HC-SULF were retained within the tissue. However, after 4 h of incubation the total amount retained in the tissue was low in relation to the amount excreted (0% for 7HC-SULF, 2.5% for 7HC-GLUC).
Comparison of Metabolic Rates in Precision-Cut Slices and Ussing Chamber Preparations. Proximal jejunum tissue used in the Ussing chamber incubations and precision-cut slices were prepared at the same time from tissues of four donors. For 1′OH-midazolam formation, slices had a slightly, but significantly, higher metabolic rate (p < 0.03 tested with a paired two-tailed Student's t test) compared with Ussing chamber incubations (Fig. 4A). For 4′OH-diclofenac and OH-bufuralol formation (Fig. 4, B and C), the metabolic rates were similar for precision-cut slices and Ussing chamber preparations.
For 7HC conjugation (Fig. 4, D and E), the metabolic rates in Ussing chamber preparations were slightly, but significantly, higher (p < 0.02 tested with a paired two-tailed Student's t test) than in precision-cut slices.
Discussion
Recently, rat intestinal precision-cut slices were presented as a tool to study intestinal metabolism (Martignoni et al., 2004; de Kanter et al., 2005; van de Kerkhof et al., 2005). In addition, the rat and human intestinal Ussing chamber technique has been optimized as a tool to predict the absorption of the original compound (Ungell, 2005) and not for metabolism. Because predictive in vitro methods are of high importance to investigate human intestinal drug metabolism, we characterized the viability and metabolic activity of human intestinal slices and compared it with the metabolic activity in Ussing chamber preparations.
In slices, the intracellular ATP levels and morphology indicated that they were viable up to 4 h of incubation. The increase of intracellular ATP levels during 4 h of incubation can be explained by the temperature change from 4 (storage) to 37°C during incubation, allowing ATP levels to be re-established. This result is in line with earlier observations obtained with human liver, lung, and kidney slices (De Kanter et al., 2002). In Ussing chamber preparations, the electrical parameters indicate that the viability and integrity were maintained for at least 4 h because the tight junctions (reflected by the resistance) remained intact, which is a high-energy process. However, a small decrease of ion transport activity (PD and SCC) occurred, which is probably because of some loss of cells and surface area, as has been described by Polentarutti et al. (1999).
The metabolic stability was our major focus during these studies. Therefore, proximal jejunal slices were incubated with TT to test phase I activity. Formation rates of 6β-TOH and 2β-TOH, both mediated by CYP3A4 (Yamazaki and Shimada, 1997), tended to decline after 1 h. In contrast, androstenedione formation [mediated by 17β-HSD (Farthing et al., 1981) and CYP2C19 (Yamazaki and Shimada, 1997)] and 16α-TOH formation were constant up to 4 h. This may indicate different half-lives of the individual (iso)enzymes (Renwick et al., 2000) or product inhibition.
The decision to use a mixture of model substrates was made to increase the amount of information that could be obtained per donor tissue: midazolam 1′hydroxylation (CYP3A4/3A5), diclofenac 4′hydroxylation (CYP2C9), coumarin 7′hydroxylation (CYP2A6), and bufuralol hydroxylation (CYP2D6) were chosen based on substrate specificity (Bjornsson et al., 2003). Slice formation rates of 4′OH-diclofenac, 1′OH-midazolam, OH-bufuralol, 7HC-GLUC, and 7HC-SULF were constant up to 4 h of incubation. The difference in the stability of 1′OH-midazolam (nonsaturated conditions) and 2β-TOH and 6β-TOH formation (saturated conditions) could be explained by the exhaustion of cofactors and/or product inhibition of 2β-TOH and 6β-TOH.
A lag phase was observed only for 7HC-GLUC formation. Preincubation of slices for 1 h did not influence this lag phase (pilot experiment, data not shown). No lag phase was observed in Ussing chamber experiments with 7HC because both medium and tissue were harvested. In these tissues, a constant amount of metabolite was detected up to 4 h of incubation, indicating that accumulation of the metabolites in slices may explain the lag phase. Therefore, metabolic rates used for comparison are expressed as metabolite formation measured between 1 and 4 h of incubation.
Hydroxylation of mixture compounds in colon was undetectable and thus much slower compared with proximal jejunum. For 7HC-SULF and 7HC-GLUC, the metabolic rates expressed per milligram protein were similar in colon and proximal jejunum as was also found in rat (van de Kerkhof et al., 2005). In previous rat studies, intestinal slice incubations with 7HC showed higher glucuronidation rates [750 (colon) and 977 pmol/min/mg protein (proximal jejunum)] compared with human slices (377 and 315 pmol/min/mg protein, respectively). The glucuronidation/sulfation ratio was higher in rats (colon: 16 and proximal jejunum: 23) than in humans (colon and proximal jejunum: 6) (van de Kerkhof et al., 2005), suggesting a clear species difference. This is in contrast with liver, in which the glucuronidation/sulfation ratio is higher in humans (19) than in rats (15) (De Kanter et al., 2002). However, in future studies the test group should be expanded, and intestinal tissue from healthy subjects also should be taken into account to prove the existence of this species difference. To our knowledge, it is unknown whether obesity influences drug metabolism in the small intestine.
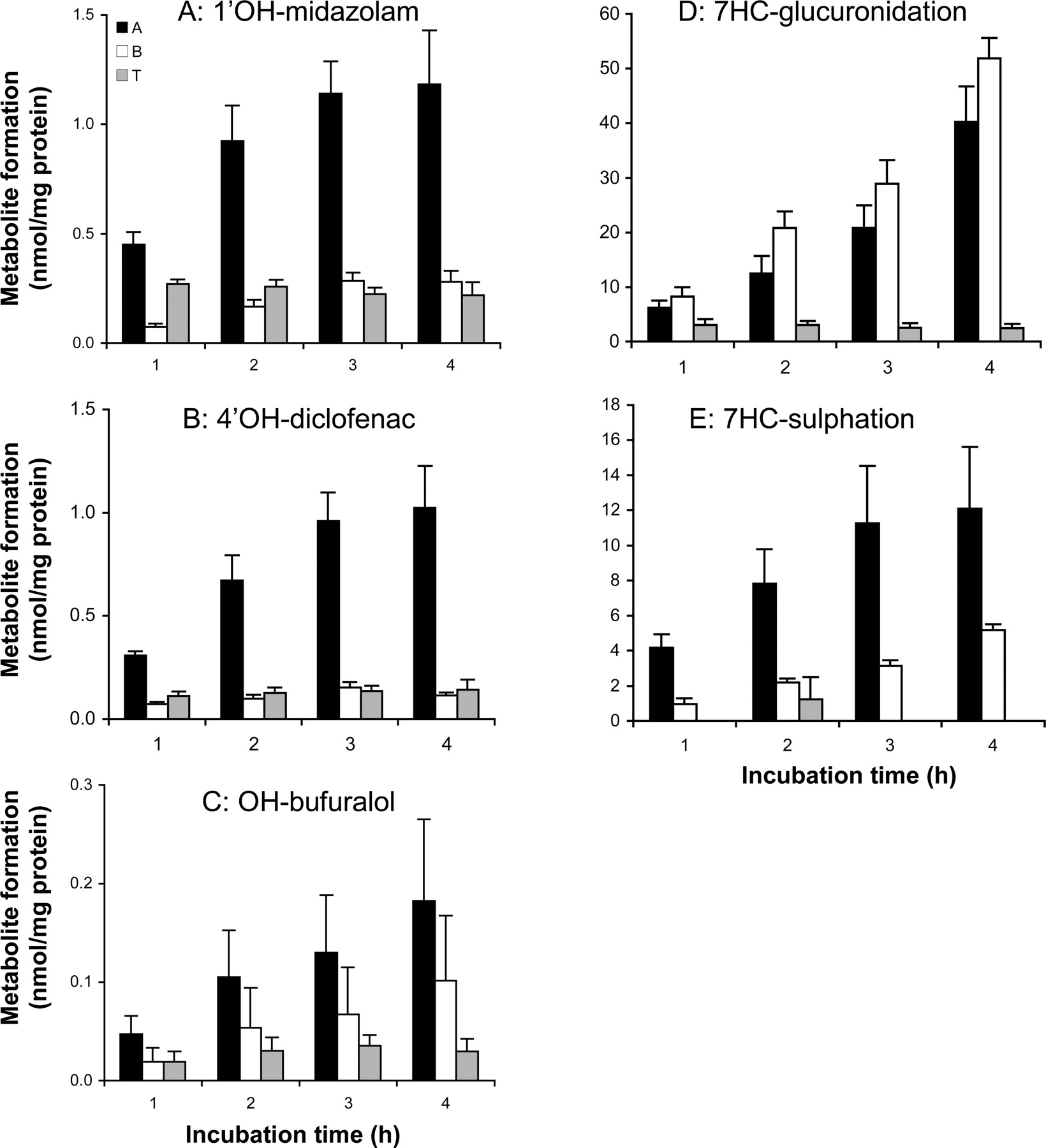
Distribution of formed metabolites over the apical (A), basolateral (B), and tissue (T) compartment was measured using human proximal jejunum in the Ussing chamber setup. Phase I reaction rates were assessed with a mixture of four compounds, formation of 1′OH-midazolam (A), 4′OH-diclofenac (B), OH-bufuralol (C), and 7′OH-coumarin (not detectable). Phase II reactions were monitored using 7HC toward 7HC-GLUC (D) and 7HC-SULF (E). Results are mean ± S.E.M. of four donors; in each experiment one Ussing chamber preparation was incubated per time point.
When Ussing chamber preparations were incubated with the mixture of P450 substrates or 7HC, the metabolite formation was linear up to 3 to 4 h of incubation. The distribution of metabolites in the A and B compartments, however, differed clearly per substrate. This may indicate a clear role of transporters. Possibly, metabolites appear at the apical site because of metabolism in sloughed-off cells. To exclude this, we sampled medium after incubation and incubated these for another 3 h. No further metabolism was detected in these incubations, confirming that the formation of the metabolites occurred in cells within the tissue and was then excreted into the apical compartment.
The role of transporters in uptake and excretion of compounds in the intestine has been extensively discussed (Benet et al., 2004; Ito et al., 2005). The colocalization of CYP3A4 and P-glycoprotein in the villi tips of human intestine has also been reported (Watkins, 1997), indicating a collaborative role between enzymes and efflux transporters in the gut. In Ussing chamber studies using pig intestine, extensive luminal efflux of the oxidative product hydroxy-sirolimus was reported (Lampen et al., 1998). For glucuronide and sulfate conjugates formed in rat enterocytes, a significant extrusion into the intestinal lumen has been reported as well (Adachi et al., 2005). Apical excretion was shown to be facilitated by MRP2 in Caco-2 cells (Suzuki and Sugiyama, 2000), which is in line with localization of MRP2 at the apical membranes of enterocytes (Borst and Elferink, 2002). The present study showed for the first time a clear luminal efflux of hydroxy-metabolites in human intestine. Elucidation of the identity of the transporters involved in the apical and basolateral excretion of the metabolites will be the focus of further research.
A direct comparison of metabolic activity between the methods could be made because tissues from the same donors were used to prepare both slices and Ussing chamber preparations. The two techniques offer different advantages and limitations. The slice technique requires only 4 mg of tissue per slice and is technically easier to use. Thus, many more experimental variations may be tested within one experiment. More intestinal tissue is needed to mount tissue in the Ussing chamber setup (around 160 mg mucosal tissue per setup). However, the latter has the major advantage that vectorial transport of drugs and metabolites can be studied. The incubation conditions for both systems were also slightly different. In fact, these were chosen based on the optimized conditions developed for the two systems separately. Surprisingly, despite these differences, similar metabolic activities were found for all the reactions (phase I and II) tested.
After intensive literature search, we believe that we are unable to compare the obtained results with earlier published microsomal data because no exact scaling factor is known. Although certain reports give the microsomal protein yield after isolation, the loss during isolation has not yet been reported. Furthermore, for intestinal preparations, the origin of the tissue (location along the tract, patient/donor background) and/or the storage and preparation techniques used will introduce differences in the metabolic rates. Metabolic rates of different methods can be compared only when studies are performed with the same donor material. This may be a subject for future studies.
To underline the intrinsic importance of the enterocytes in drug metabolism, we compared the intestinal metabolic rates with activities obtained using human liver. First, a remarkably low interindividual variation was seen in this study using gut tissues. For TT hydroxylation, only a 2-fold difference was detected between six donors. This is not in line with the observation of others that intestinal metabolism is highly variable (Lin and Lu, 2001), but it may be related to the relatively small number of obese donors used in our study. Liver slice values of human TT hydroxylation have been reported to be 65 (6β-TOH), 24 (2β-TOH), 223 (androstenedione), and 5 (16α-TOH) pmol/min/mg protein (De Kanter et al., 2002). The proximal jejunum rates detected in the present study were 1.6, 1, 1.4, and 1.8 times higher, respectively, compared with the liver activity. However, these activities cannot be directly ascribed to the activity of liver hepatocytes and intestinal enterocytes. In liver, hepatocytes represent more than 80% of wet weight, of which about 50% is contributing to the metabolic activity in slices of 250 μm thickness (de Graaf et al., 2006), indicating that hepatocyte activity is 2.5 times the tissue value. In intestinal mucosa, enterocytes account for approximately 25% of total wet weight (morphological observation), indicating that the enterocyte activity is 4 times the tissue value. From these observations, we come to the remarkable conclusion that the phase I metabolic rate in enterocytes is estimated to be 1.7 to 2.9 times higher than that in hepatocytes.
7HC-GLUC and 7HC-SULF rates in human liver slices have been reported as 880 and 33 pmol/min/mg protein for 7HC-GLUC and 7HC-SULF, respectively (de Kanter et al., 2002). If the aforementioned assumptions are taken into account, glucuronidation rates in enterocytes in the present study reach 60 to 70% of the hepatocyte values. Enterocyte sulfation rates are approximately 250 to 300% of the sulfation rate in hepatocytes. We conclude that sulfation rates in enterocytes of proximal jejunum and colon are higher than in hepatocytes, but glucuronidation rates are lower.
From the present study, it can be concluded that both precision-cut intestinal slices and Ussing chamber preparations are valuable tools to study human intestinal drug metabolism. Both systems remain viable up to 4 h and exhibit drug metabolism at similar rates. Interestingly, we estimate that human enterocytes exhibit at least comparable to even higher metabolic rates compared with hepatocytes.
Acknowledgments
We thank Sara Leandersson and Xueqing Li for assistance with the LC/MS analyses and Prof. D. K. F. Meijer for excellent advice and critical reading of the manuscript.
Footnotes
-
This study was supported by AstraZeneca, the Technology Foundation STW, Applied Science Division of NOW, and the Technology Programme of the Ministry of Economic Affairs, as well as Yamanouchi Europe.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011148.
-
ABBREVIATIONS: TT, testosterone; 7HC, 7-hydroxycoumarin; P450, cytochrome P450; TFA, trifluoroacetic acid; 7HC-GLUC, 7-hydroxycoumarin glucuronide; TOH, hydroxytestosterone; WME, Williams medium E; 7HC-SULF, 7-hydroxycoumarin sulphate; ACN, acetonitrile; MeOH, methanol; KBR, Krebs-bicarbonate Ringer solution; PD, potential difference; SCC, short-circuit current; R, resistance; I, current; U, voltage; LLOQ, lower limit of quantitation; LC/MS, liquid chromatography/mass spectrometry.
- Received May 23, 2006.
- Accepted August 10, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}