


Abstract
DB289 [2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime] is biotransformed to the potent antiparasitic diamidine DB75 [2,5-bis(4-amidinophenyl) furan] by sequential oxidative O-demethylation and reductive N-dehydroxylation reactions. Previous work demonstrated that the N-dehydroxylation reactions are catalyzed by cytochrome b5/NADH-cytochrome b5 reductase. Enzymes responsible for catalyzing the DB289 O-demethylation pathway have not been identified. We report an in vitro metabolism study to characterize enzymes in human liver microsomes (HLMs) that catalyze the initial O-demethylation of DB289 (M1 formation). Potent inhibition by 1-aminobenzotriazole confirmed that M1 formation is catalyzed by P450 enzymes. M1 formation by HLMs was NADPH-dependent, with a Km and Vmax of 0.5 μM and 3.8 nmol/min/mg protein, respectively. Initial screening showed that recombinant CYP1A1, CYP1A2, and CYP1B1 were efficient catalysts of M1 formation. However, none of these three enzymes was responsible for M1 formation by HLMs. Further screening showed that recombinant CYP2J2, CYP4F2, and CYP4F3B could also catalyze M1 formation. An antibody against CYP4F2, which inhibited both CYP4F2 and CYP4F3B, inhibited 91% of M1 formation by HLMs. Two inhibitors of P450-mediated arachidonic acid metabolism, HET0016 (N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine) and 17-octadecynoic acid, effectively inhibited M1 formation by HLMs. Inhibition studies with ebastine and antibodies against CYP2J2 suggested that CYP2J2 was not involved in M1 formation by HLMs. Additionally, ketoconazole preferentially inhibited CYP4F2, but not CYP4F3B, and partially inhibited M1 formation by HLMs. We conclude that CYP4F enzymes (e.g., CYP4F2, CYP4F3B) are the major enzymes responsible for M1 formation by HLMs. These findings indicate that, in human liver, members of the CYP4F subfamily biotransform not only endogenous compounds but also xenobiotics.
As part of our search for new lead compounds for the treatment of African trypanosomiasis (African sleeping sickness) and other parasitic infections, aromatic dicationic compounds, such as DB75 [2,5-bis(4-amidinophenyl) furan], have been evaluated for efficacy in multiple models of infection. These diamidine-type compounds are effective against a broad range of pathogens in vitro, including Trypanosoma brucei, Leishmania spp., Pneumocystis carinii, and Plasmodium falciparum (Das and Boykin, 1977; Bell et al., 1990, 1993; Tidwell et al., 1990). However, antiparasitic activity after oral administration of these compounds in rodents is poor (Steck et al., 1981; Boykin et al., 1996; Hall et al., 1998); this is believed to be largely due to the positively charged cationic moieties that limit transcellular transport across the intestinal epithelium (Zhou et al., 2002). A prodrug of DB75, 2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime (DB289), exhibits enhanced oral efficacy and reduced acute toxicity in animal models of Pneumocystis pneumonia and African trypanosomiasis (Boykin et al., 1996; Fitzpatrick et al., 2004). In clinical trials involving patients with primary stage African trypanosomiasis, treatment with oral DB289 (100 mg, twice daily for 10 days) achieved cure rates of approximately 95% (Dr. Carol Olson, Immtech Pharmaceuticals Inc., personal communication). In clinical trials involving patients with Plasmodium vivax or acute, uncomplicated P. falciparum, treatment with oral DB289 (100 mg, twice daily for 5 days) achieved cure rates of 96% (Yeramian et al., 2005). Oral DB289 was also well tolerated, with no serious side effects (Yeates, 2003). DB289 has completed phase II clinical trials for African trypanosomiasis in the Democratic Republic of Congo and Angola, for malaria in Thailand, and for Pneumocystis pneumonia in Peru.

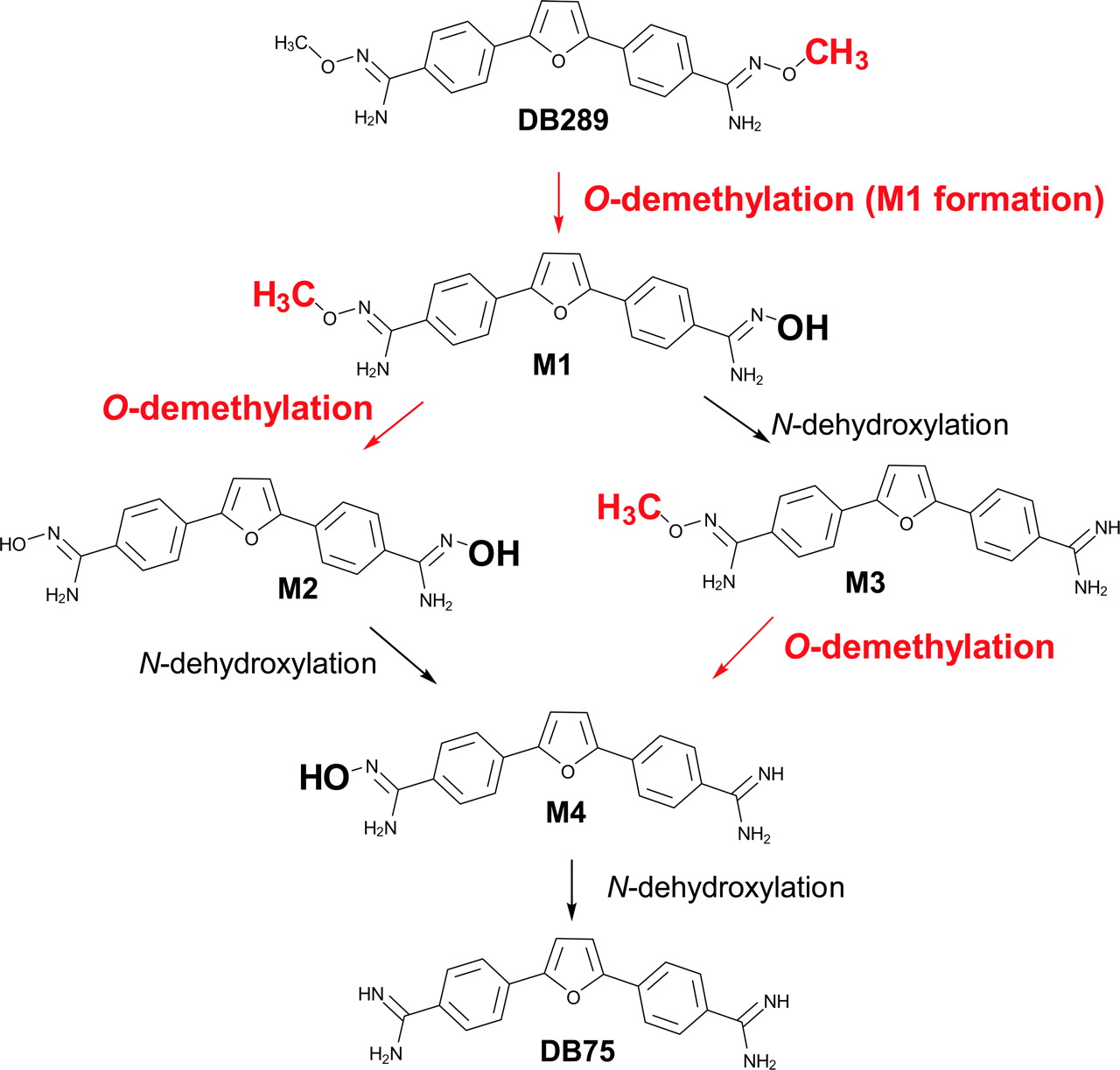
Metabolic pathway of DB289 biotransformation to DB75.
As a prodrug, DB289 must be biotransformed to the active compound DB75 in sufficient quantities for effective treatment. The phase I metabolic pathway for the conversion of DB289 to DB75 has been studied in vitro with freshly isolated rat hepatocytes (Zhou et al., 2004). This pathway consisted of sequential oxidative O-demethylation and reductive N-dehydroxylation reactions with four intermediate metabolites formed (Fig. 1). Clinical trials in humans demonstrated oral DB289 to be well absorbed and efficiently converted to DB75. However, pharmacokinetic studies have indicated that DB75 plasma concentrations are highly variable among patients (Yeramian et al., 2005). Such variability could be due in part to interindividual differences in the extent of metabolic conversion of DB289 to DB75. Therefore, to provide a more comprehensive understanding of DB289 in vivo, an in-depth in vitro study was undertaken to identify the enzymes responsible for the biotransformation of DB289 to DB75.
The reductive N-dehydroxylation reactions (conversion of M1 to M3, conversion of M2 to M4, and M4 conversion to DB75; Fig. 1) were recently shown to be catalyzed by cytochrome b5/NADH-cytochrome b5 reductase, with no requirement for P450s (Saulter et al., 2005). However, little is known about the oxidative O-demethylation pathways of DB289. Accordingly, we have performed a reaction phenotyping study to identify the enzymes responsible for the first step in the conversion of DB289 to DB75, i.e., oxidative O-demethylation (M1 formation).
Our results suggest that the initial oxidative O-demethylation of DB289 is predominantly catalyzed by CYP4F enzymes in human liver microsomes. These findings not only advance our understanding of the metabolism of this new drug candidate but, by implicating CYP4F enzymes in the metabolism of a xenobiotic, have important implications for reaction phenotyping in practice and human drug metabolism in general.
Materials and Methods
Chemicals. DB289 was synthesized by Medichem (Woodlake, IL) using previously described methods (Das and Boykin, 1977; Boykin et al., 1996). The intermediate phase I metabolites (M1, M2, and M3), the active diamidine DB75, and deuterium-labeled DB289 (DB289-d8) (internal standard) were synthesized as described previously (Stephens et al., 2001; Anbazhagan et al., 2003). HPLC-grade water and acetonitrile were obtained from Fisher Scientific (Pittsburgh, PA). Analytical-grade ammonium formate, formic acid, magnesium chloride, dimethyl sulfoxide, potassium dihydrogen phosphate, disodium hydrogen phosphate anhydrous, NADPH, α-naphthoflavone, fluvoxamine, furafylline, coumarin, thio-TEPA, sulfaphenazole, trimethoprim, omeprazole, quinidine, diethyldithiocarbamate, ketoconazole, troleandomycin (TAO), lauric acid, and 1-aminobenzotriazole (ABT) were purchased from Sigma-Aldrich (St. Louis, MO). 17-Octadecynoic acid (17-ODYA), HET0016 (N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine), and arachidonic acid were purchased from Cayman Chemical Co. (Ann Arbor, MI). Ebastine was purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). All chemicals were of the highest purity available and used without further purification.
Human Liver Microsomes, Immunoinhibitory Antibodies, and Recombinant Human P450s. Pooled human liver microsomes (n = 50; mixed gender), polyclonal antibody against CYP3A4/5, and preimmune immunoglobulin (IgG) from rabbit were prepared by XenoTech, LLC (Lenexa, KS). Polyclonal antibody against CYP4F2, raised in rabbits, was purchased from Research Diagnostics, Inc. (Concord, MA) (source A; 1 mg IgG/ml) or was kindly provided by Dr. Yoshihiko Funae (Osaka City University Medical School, Osaka, Japan) (source B; 40 mg IgG/ml). Polyclonal antibody against CYP2J2 (40 mg IgG/ml) was also a gift from Dr. Funae (Hashizume et al., 2001, 2002). Monoclonal antibody against CYP2J2 (1.1 mg IgG/ml) and a control monoclonal antibody against egg lysozyme were generated in mouse hybridoma cells as described previously (Gelboin et al., 1998; Krausz et al., 2000; Xiao et al., 2004). Supersomes prepared from baculovirus-infected insect cells expressing human P450 enzymes and NADPH-cytochrome P450 reductase were purchased from BD Gentest (Woburn, MA). In the case of recombinant CYP2E1, CYP2J2, CYP3A7, CYP4F2, CYP4F3A, CYP4F3B, and CYP4F12, the enzymes were coexpressed with both NADPH-cytochrome P450 reductase and cytochrome b5. Control Supersomes were obtained from insect cells infected with wild-type baculovirus or insect cells infected with baculoviruses containing cDNA-expressed human NADPH-cytochrome P450 reductase and cytochrome b5.
Standard Incubation Conditions. Incubation mixtures contained 100 mM phosphate buffer (pH 7.4), 3.3 mM MgCl2, and 1 mM NADPH, if not indicated otherwise. Reactions were initiated with the addition of NADPH (or substrate for incubations with mechanism-based inhibitors) and were carried out at 37°C. DB289, M1, M2, and M3 were dissolved in dimethyl sulfoxide. DB75 was dissolved in HPLC-grade water. With the exception of ebastine, all chemical inhibitors were dissolved in methanol. Ebastine was initially dissolved in chloroform at a concentration of 25 mM, which was serially diluted with methanol to concentrations of 2.5 mM, 0.25 mM, and 0.025 mM. Final incubation mixtures contained less than 0.9% (v/v) organic solvent. Reactions were stopped with 2 volumes of ice-cold acetonitrile containing 0.1% formic acid (v/v) and 15 or 30 nM DB289-d8 as internal standard, if not indicated otherwise. The mixtures were vortex-mixed, and precipitated protein was removed by centrifugation (∼1400g) for 15 min. The supernatant fractions were analyzed by LC/MS/MS (described below). For all the experiments, the amount of product formed (or substrate consumed) was linear with respect to the incubation time and the amount of enzyme (HLMs or recombinant P450 enzyme).
Incubations with Pooled Human Liver Microsomes. Incubation mixtures (1 ml) contained 15 μM DB289 and 0.2 mg/ml HLMs. After a 5-min equilibration period at 37°C, reactions were initiated with the addition of NADPH. Aliquots (50 μl) of the reaction mixtures were removed at 0, 5, 15, 30, and 120 min and mixed with 25 μl of ice-cold acetonitrile. The mixtures were processed (described above) and analyzed immediately by LC/UV (described below, single determination). Metabolite identification was performed by comparing retention times to those of synthetic standards. Control incubations were run as described above, except that NADPH, microsomal protein, or prodrug was absent.
Incubations with Recombinant Human P450 Enzymes. The P450 enzymes, namely, 1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5 were incubated individually (500 μl) with DB289 (3.0 μM) in the presence of NADPH for 15 min to assess the percentage of substrate consumed. The final P450 enzyme concentration was 50 pmol/ml for all except CYP2C8 and CYP2C9 (100 pmol/ml). Aliquots (50 μl) of the reaction mixtures were removed and processed as described under Standard Incubation Conditions. Control incubations were run with control Supersomes.
In a subsequent experiment, recombinant human CYP2C18, 2J2, 3A7, 4A11, 4F2, 4F3A, 4F3B, and 4F12 were incubated individually (500 μl) with DB289 (3.0 μM) in the presence of NADPH for 15 min to assess the percentage of substrate consumed. Aliquots (50 μl) of the reaction mixtures were removed and processed as described under Standard Incubation Conditions. Control incubations were run with control Supersomes.
Enzyme Kinetic Studies. Under initial rate conditions (<20% substrate consumption), primary metabolite formation (M1 formation) was evaluated at concentrations of DB289 ranging from 0.05 μM to 25 μM with pooled HLM (0.02 mg/ml) or recombinant P450 enzymes (10–50 pmol/ml). After a 5-min equilibration period at 37°C, reactions (250 μl, final) were initiated with the addition of NADPH. The incubation time was 3 min for HLMs, 2 min for CYP1A1 and CYP1A2, 5 min for CYP1B1, CYP2J2, and CYP4F2, and 15 min for CYP2C8, CYP2D6, CYP2E1, CYP3A4, CYP3A5, CYP4F3B, and CYP4F12 (see Tables 1 and 2 for complete details). Reactions were stopped and processed as described under Standard Incubation Conditions. M1 formation rates were determined by LC/MS/MS analysis.
Kinetic analysis of M1 formation rates in incubations of DB289 with pooled HLMs and recombinant human P450 enzymes
Incubation mixtures contained 0.02 mg/ml HLMs or 10 to 50 pmol/ml recombinant cytochrome P450, and DB289 ranging from 0.05 to 25 μM. Reactions were carried out at 37°C for the period of time indicated. M1 formation rates were determined by the LC/MS/MS method as described under Materials and Methods. Apparent kinetic parameters were derived from the Michaelis-Menten kinetic analysis of the mean of duplicate determinations. Results for recombinant P450 enzymes are arranged according to their intrinsic clearance values (CLint).
Kinetic analysis of M1 formation rates in incubations of DB289 with recombinant human P450 enzymes
Incubation mixtures contained 20 to 50 pmol/ml recombinant cytochrome P450, and DB289 ranging from 0.05 to 25 μM. Reactions were carried out at 37°C for the period of time indicated. M1 formation rates (mean of duplicate determinations) were determined by the LC/MS/MS method as described under Materials and Methods. Kinetic parameters were derived from the Hill equation. Results are arranged according to their maximum clearance values (CLmax).
Chemical Inhibition Assays. Cytochrome P450-selective chemical inhibitors were added to standard incubation mixtures (250 μl) containing 0.2 mg/ml HLMs or 10 pmol/ml recombinant P450 enzymes and 0.3 μM or 3 μM DB289. Reactions were carried out for 5 min with HLMs, recombinant CYP2J2, or CYP4F2, 2 min with recombinant CYP1A1 or CYP1A2, or 15 min with recombinant CYP4F3B. The chemical inhibitors used in the initial study were 1 μM α-naphthoflavone (CYP1A inhibitor), 3 μM fluvoxamine (CYP1A2 inhibitor), 10 μM furafylline (mechanism-based CYP1A2 inhibitor), 100 μM coumarin (CYP2A6 inhibitor), 50 μM thio-TEPA (CYP2B6 inhibitor), 10 μM sulfaphenazole (CYP2C9 inhibitor), 60 μM trimethoprim (CYP2C8 inhibitor), 10 μM omeprazole (CYP2C19 inhibitor), 1 and 10 μM quinidine (CYP2D6 inhibitor), 5 and 50 μM diethyldithiocarbamate (mechanism-based CYP2E1 inhibitor), 3 μM ketoconazole (CYP3A inhibitor), 50 and 100 μM troleandomycin (mechanism-based CYP3A4 inhibitor), 100 μM lauric acid (CYP4A substrate), and 100 μM 1-aminobenzotriazole (mechanism-based, general P450 inhibitor). The selection of chemical inhibitors and concentrations was based on previous literature values or concentrations corresponding to 10 times the Ki value for each P450-specific probe reaction. Mechanism-based inhibitors were incubated with HLMs or P450 enzymes for 15 min in the presence of NADPH, before the addition of the substrate. At the end of the incubation, aliquots (100 μl) were removed and processed as described under Standard Incubation Conditions. Control incubations contained methanol in place of chemical inhibitors.
In a subsequent experiment, ebastine (a CYP2J2, CYP4F12, and CYP3A4 substrate), arachidonic acid (a CYP4F2, CYP4F3B, and CYP2J2 substrate), HET0016 (an arachidonic acid ω-hydroxylase inhibitor), and 17-ODYA (a nonselective mechanism-based arachidonic acid ω-hydroxylase and epoxygenase inhibitor) were evaluated for their ability to inhibit the conversion of DB289 to M1 by HLMs and selected recombinant human P450 enzymes essentially as described above. Varying concentrations of these inhibitors were used to assess concentration-dependent inhibition, generally covering 0.1 to 100 times their reported Km or IC50 values.
Inhibition of P450 Activities by DB289. DB289 (3.0 μM, final) was incubated with pooled HLMs (0.25 mg/ml, final) and NADPH (2 mM, final) for both 0 and 15 min before the addition of one of a range of marker substrates, each selective for a given P450 enzyme (7-ethoxyresorufin O-deethylation for CYP1A2, coumarin 7-hydroxylation for CYP2A6, diclofenac 4′-hydroxylation for CYP2C9, S-mephenytoin 4′-hydroxylation for CYP2C19, debrisoquine 4-hydroxylation for CYP2D6, lauric acid 11-hydroxylation for CYP2E1, testosterone 6β-hydroxylation for CYP3A4/5, and lauric acid 12-hydroxylation for CYP4A11). The effects of DB289 on these activities were compared with those elicited by a range of P450 enzyme-selective chemical inhibitors (30 μM furafylline for CYP1A2, 1 μM 8-methoxypsoralen for CYP2A6, 20 μM sulfaphenazole for CYP2C9, 100 μM tranylcypromine for CYP2C19, 10 μM quinidine for CYP2D6, 300 μM diethyldithiocarbamate for CYP2E1, and 100 μM troleandomycin for CYP3A4/5). Similar comparison was not performed for CYP4A11 because no selective chemical inhibitor of CYP4A11 has been established. Control incubations were run using the appropriate solvent in place of inhibitor.
Immunoinhibition Assays. Polyclonal antibody (1, 5, 12.5, and 25 μl) was preincubated with HLMs (0.2 mg/ml) or recombinant P450 enzymes (10 pmol/ml) at 37°C for 15 min before the addition of DB289 (3 μM, final). Reactions (250 μl) were initiated by addition of NADPH and were carried out for 5 min with HLMs, CYP2J2, and CYP4F2, or for 15 min with CYP4F3B. At the end of the incubation, aliquots (100 μl) were removed and processed as described under Standard Incubation Conditions. Control incubations contained the same volume of preimmune rabbit IgG to account for nonspecific protein binding.
Correlation Analysis and Evaluation of FMO Involvement. DB289 consumption activity was determined in standard incubations with 10 individual samples of human liver microsomes, characterized by the manufacturer for individual P450 enzyme activities. The initial concentration of DB289 was 0.3 μM. Pearson correlation coefficients (r) were determined by plotting the rate of DB289 disappearance versus the marker activity for each P450 enzyme, and a p value less than 0.05 was considered significant. Additional experiments with flavin monooxygenase 3 (FMO3) were conducted to evaluate whether this enzyme could catalyze the oxidative O-demethylation reaction of DB289. DB289 was incubated with baculovirus insect cell-expressed FMO3 (0.5 mg/ml) under the same experimental conditions as described above. Experiments with heat-inactivated human liver microsomes (incubations conducted at 60°C for 90 s) were performed to further assess the role of FMO in DB289 metabolism.
HPLC/UV Assays. Quantitative analyses of DB289 and its metabolites were performed on an Agilent 1100 Series HPLC system (Palo Alto, CA) equipped with a multiwavelength detector. All samples were monitored at 359 nm. Samples were separated on an Agilent ZORBAX Bonus-RP column (2.1 × 50 mm, 3.5 μM) with a 0.35 ml/min flow rate. Mobile phase composition (A, 35 mM formic acid and 15 mM ammonium formate in HPLC-grade water; B, 35 mM formic acid and 15 mM ammonium formate in acetonitrile/HPLC-grade water 80:20 v/v) began with 5% B and was increased to 80% B over 15 min. Then it was maintained at 100% B for 3 min to wash the column. Postrun time was set to 6 to 7 min for column reconditioning before the next injection. Separation was carried out at room temperature. Typically, 8 μlof sample was injected. An external calibration curve (0.1–15 μM) was used to quantify the levels of DB289, M1, M2, and M3. Metabolite identities were confirmed by comparing retention times to those of synthetic standards.
LC/MS/MS Assays. The quantification of DB289 and M1 by LC/MS/MS was performed on an Applied Biosystems (Foster City, CA) API 4000 triple quadrupole mass spectrometer equipped with a Turbo IonSpray interface (MDS Sciex, San Francisco, CA). Samples were introduced to the mass spectrometer using a thermostatted (6°C) CTC PAL LEAP autosampler (LEAP Technologies, Carrboro, NC), a Shimadzu pumping system (Shimadzu, Kyoto, Japan), and a Valco solvent divert valve (Valco, Houston, TX). All equipment was controlled using Analyst software (Version 1.3; Applied Biosystems). DB289 and M1 were separated on an Aquasil C18 HPLC column (2.1 mm × 50 mm, 5 μm) (Thermo Electron, Waltham, MA). HPLC mobile phases that were optimized for mass spectrometry were HPLC-grade water containing 0.1% formic acid (C) and methanol containing 0.1% formic acid (D). After a 0.4-min initial hold at 30% D, mobile phase composition started with 30% D and was increased to 80% D over 2.6 min with a flow rate of 0.5 ml/min. Then the column was washed with 90% D for 1.5 min with a flow rate of 1.5 ml/min and was reequilibrated with 30% D with a flow rate of 0.5 ml/min for 0.5 min before injecting the next sample. The characteristic multiple reaction monitoring transitions for DB289, M1, and DB289-d8 were m/z 365.10 → 334.10, m/z 351.10 → 320.10, and m/z 373.00 → 342.00, respectively. Typically, 4 μlof sample was injected. The range of the calibration curve was 25 to 5000 nM for DB289 and 1 to 2500 nM for M1.
Data Analysis. Data are presented as the means of duplicate determinations unless indicated otherwise. Percentage of substrate consumed for DB289 incubations with HLMs or recombinant P450 enzymes was determined by normalizing DB289 concentrations at 15 min to the DB289 concentration at 0 min (expressed as 100%). For inhibition studies, the amount of M1 formed in control incubations was set to 100%, to which the amount of M1 formed in the presence of chemical inhibitors or inhibitory antibodies was normalized. Eadie-Hofstee plots were generated from V0 versus V0/[DB289] data. Apparent Km (or S50) and Vmax values were obtained by fitting the Michaelis-Menten equation or the Hill equation (Houston and Kenworthy, 2000) with the initial rate (V0) of M1 formation versus substrate concentration data by nonlinear regression analysis (Prism 4.0; GraphPad Software Inc., San Diego, CA). In vitro intrinsic clearance (CLint) was determined by dividing Vmax by Km. The maximal clearance (CLmax) was determined with the equation CLmax = (Vmax/S50) × [(n–1)/n(n–1)1/n] (Houston and Kenworthy, 2000).

A, LC/UV chromatograms of DB289 (15 μM) incubated with pooled HLMs (0.2 mg/ml) for 0, 5, 15, 30, and 120 min. B, plot of concentrations of DB289 and its metabolites (M1, M2, and M3) versus incubation time. Concentrations of M4 and DB75 were below the lower level of quantification.
Results
Phase I Biotransformation of DB289 in Human Liver Microsomes. DB289 was incubated with pooled HLMs in the presence of NADPH, and the phase I metabolites were identified by LC/UV analysis with comparison to synthetic standards. Representative chromatograms at 0, 5, 15, 30, and 120 min of incubation are shown in Fig. 2A. DB289 had a half-life of 12 min in this pooled HLM and was completely consumed by 120 min. M1 appeared to increase over the first 30 min before decreasing over the next 90 min, presumably because of the further conversion of M1 to subsequent metabolites, i.e., M2 and M3 (Fig. 2B). The major metabolites of DB289 at 120 min were the two oxidative O-demethylation products, M1 and M2. Neither DB289 consumption nor metabolite formation was observed in control incubations without NADPH or HLMs (data not shown).
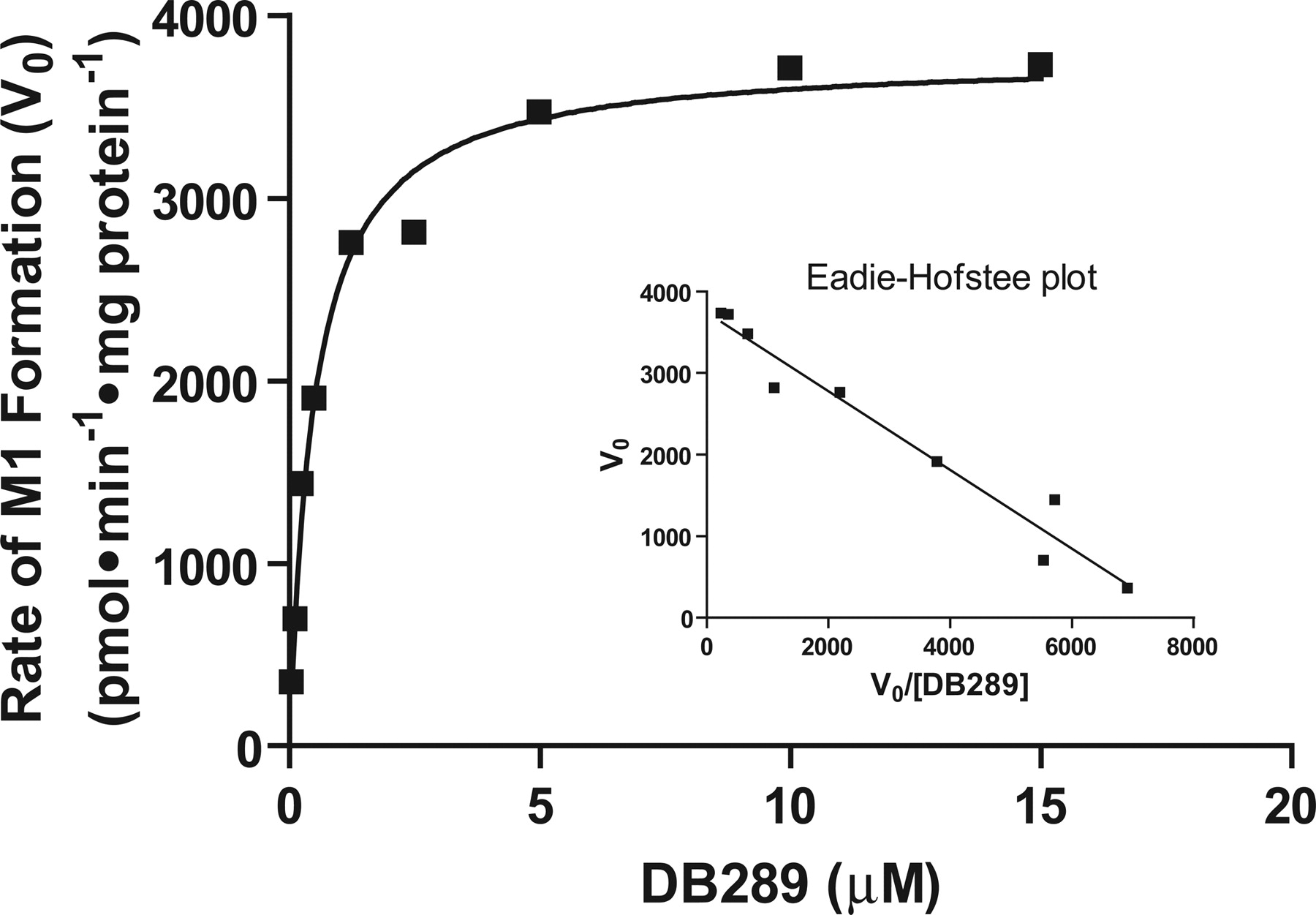
Oxidative O-demethylation of DB289 (M1 formation) is the first step in the biotransformation of DB289 to its active diamidine DB75 (Fig. 1). An Eadie-Hofstee plot of the M1 formation rate-substrate concentration data displayed monophasic kinetics over the DB289 concentration range 0.05 to 15 μM (Fig. 3, Table 1).
Initial Reaction Phenotyping Studies for M1 Formation in Human Liver Microsomes.Inhibition with cytochrome P450-selective chemical inhibitors. The effects of cytochrome P450-selective chemical inhibitors on M1 formation by HLMs are shown in Fig. 4. The pattern of inhibition was the same whether the concentration of DB289 was 0.3 μM or 3 μM. Therefore, only the results with 3 μM DB289 are shown. The nonselective cytochrome P450 inhibitor, ABT, markedly inhibited M1 formation by HLMs (86% relative to control), indicating that this O-demethylation reaction is mediated by cytochrome P450. The CYP1A inhibitor, α-naphthoflavone, showed negligible inhibition of M1 formation. The CYP3A4 inhibitor, ketoconazole, partially inhibited (61%), whereas troleandomycin did not inhibit M1 formation at either 50 or 100 μM. Chemical inhibitors of the remaining P450 enzymes examined (CYP1A2, 2A6, 2B6, 2C, 2D6, 2E1, and 4A) caused little or no inhibition.

Kinetic analysis of M1 formation rates with pooled HLMs. Reaction mixtures contained 0.02 mg/ml HLM, and 0.05 μM to 15 μM DB289 as described under Materials and Methods. Reactions were carried out for 3 min at 37°C. Symbols denote the mean of duplicate determinations. The smoothed curve represents the best fit by nonlinear regression analysis using the Michaelis-Menten equation for a unienzyme system. The Eadie-Hofstee plot (inset) was generated as described under Materials and Methods.
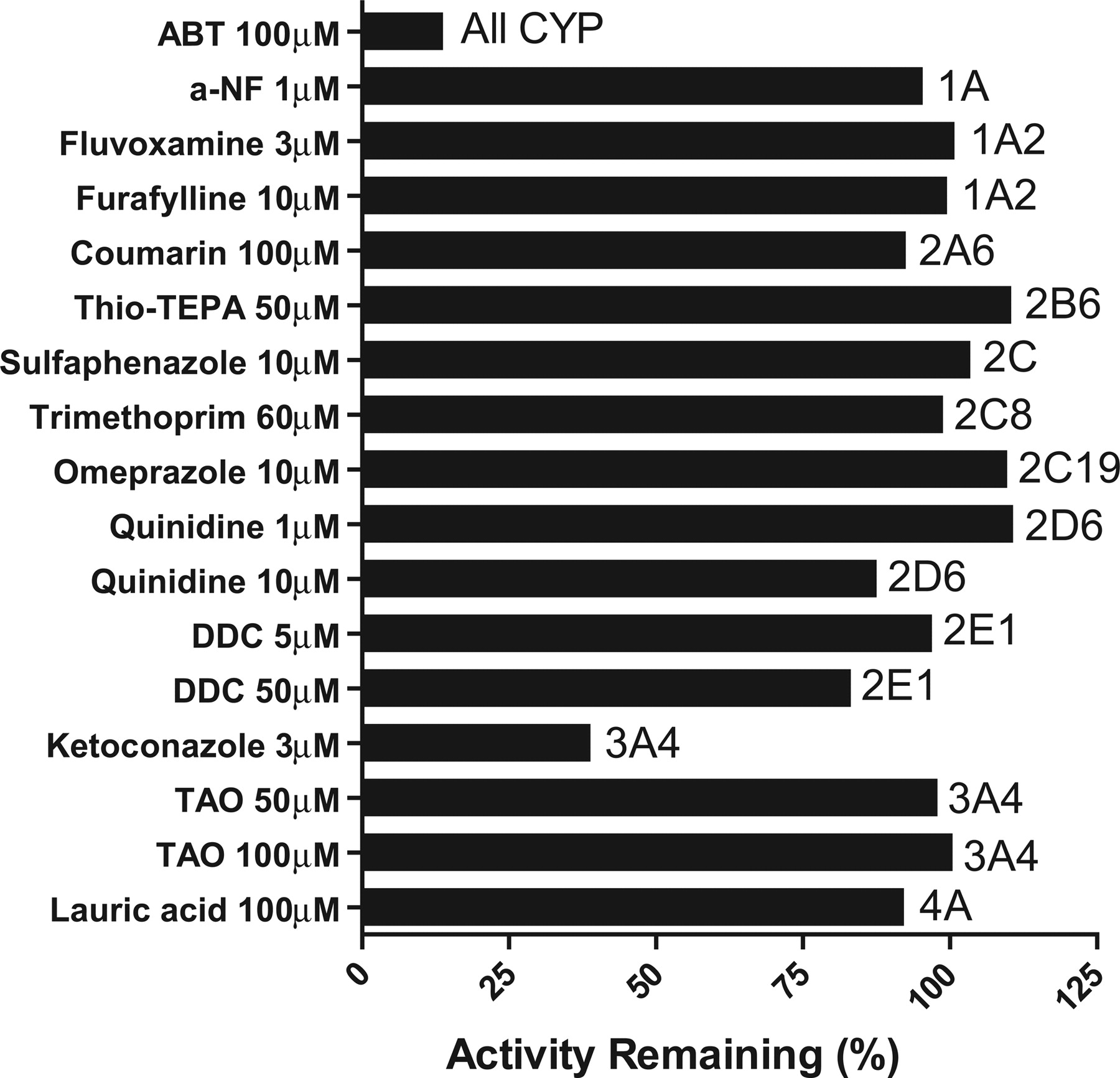
Inhibition of M1 formation by cytochrome P450 (CYP)-selective chemical inhibitors. Incubation mixtures contained 0.2 mg/ml HLM, 3.0 μM DB289, and a chemical inhibitor at the indicated concentration. Reactions were carried out for 5 min at 37°C. Controls were run with methanol in place of chemical inhibitors. Bars denote the average of duplicate determinations and are expressed as percentage activity remaining relative to the control. a-NF, α-naphthoflavone; DDC, diethyldithiocarbamate.
Metabolism of DB289 to M1 by recombinant human P450 enzymes. Although the chemical inhibition studies suggested that none of the P450 enzymes examined above is responsible for M1 formation by HLMs, a combination of multiple approaches is generally required for accurate determination (Bjornsson et al., 2003). Therefore, a panel of recombinant human P450 enzymes was evaluated for their ability to metabolize DB289 (3.0 μM) based on substrate consumption (Fig. 5A). The percentage of substrate consumed after a 15-min incubation was 82% for CYP1A1, 61% for CYP1A2, 46% for CYP1B1, and 18% for CYP3A4. In addition, CYP2C8, 2D6, 2E1, and 3A5 showed detectable activity toward DB289 (5, 9, 7, and 8% consumption, respectively). CYP2A6, 2B6, 2C9, and 2C19 only showed marginal activity (less than 5% consumption). Neither control microsomes obtained from insect cells infected with wild-type baculovirus nor those infected with baculovirus containing human NADPH-cytochrome P450 reductase and cytochrome b5 metabolized DB289 (data not shown).

Percentage of DB289 consumed after a 15-min incubation with recombinant human P450 enzymes at 37°C. Incubation mixtures contained 50 pmol/ml recombinant P450 enzyme (100 pmol/ml for CYP2C8 and CYP2C9) and 3.0 μM DB289. Negative control was run with microsomes not transfected with human P450 enzymes. Bars denote the average of duplicate determinations and are expressed as percentage of substrate consumed as described under Materials and Methods.
To evaluate the relative efficiency of P450 enzymes in DB289 metabolism, we determined the kinetics of M1 formation by these recombinant P450 enzymes (Tables 1 and 2). Based on in vitro intrinsic or maximal clearance, CYP1A1 was the most efficient at converting DB289 to M1, followed by CYP1A2. CYP1A2 was at least ∼300-fold more efficient than CYP3A4, 2D6, 2E1, or 3A5.
Correlation analysis. Correlation studies were carried out by measuring DB289 (0.3 μM) consumption in incubations with 10 individual samples of HLMs. The rate of DB289 consumption varied only 4-fold. When DB289 consumption rates were compared with marker activities for individual P450 enzymes (namely, CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4), none showed a statistically significant correlation (data not shown). However, there was an apparent correlation between DB289 consumption rate and FMO activity (r = 0.72, p = 0.01). Therefore, further experiments were conducted to ascertain a role for FMO in DB289 metabolism.
Incubations of DB289 with recombinant human FMO3 did not show any detectable metabolism of DB289. Additionally, experiments with heat-inactivated microsomes, which render FMO inactive but have no effect on P450 enzymes, did not impede DB289 metabolism (data not shown). Therefore, the statistically significant relationship between DB289 metabolism and FMO activity was considered a spurious correlation.
Inhibition of human P450 enzyme activities by DB289. The inhibitory effects of DB289 on hepatic P450 enzymes were investigated using selective marker substrates and pooled HLMs. DB289 (3.0 μM) exhibited modest inhibition (∼20–25%) toward CYP1A2 and CYP2C9 and minimal to no inhibition toward the remaining P450 enzymes (CYP2A6, 2C19, 2D6, 2E1, 3A, and 4A11) (data not shown). In contrast, cytochrome P450-selective chemical inhibitors exhibited potent inhibition (≥60%) of the corresponding P450 enzyme activity (data not shown).
Identification of Other P450 Enzymes That Catalyze M1 Formation. Initial reaction phenotyping experiments using routine methodology indicated that one or more P450 enzymes other than those examined above are responsible for the oxidative O-demethylation of DB289 to M1. To identify potential P450 enzymes, DB289 was incubated with eight additional recombinant human P450s, namely, CYP2C18, 2J2, 3A7, 4A11, 4F2, 4F3A, 4F3B, and 4F12. The percentage of substrates consumed after a 15-min incubation is shown in Fig. 5B. Like CYP1A1 (Fig. 5A), CYP2J2 and CYP4F2 nearly completely consumed DB289 in 15 min (85% and 98%, respectively). CYP4F3B and CYP4F12 exhibited modest and minimal activity toward DB289, causing 40% and 10% consumption, respectively. CYP4F3A, an alternative splicing variant of CYP4F3B (Christmas et al., 2001), caused little DB289 consumption up to 120 min. CYP2C18, CYP3A7, and CYP4A11 caused only marginal consumption (less than 5%). It should be noted that the DB289 consumption by recombinant CYP4F2, CYP2J2, and CYP4F3B was NADPH-dependent, and the N-dehydroxylation product, M3 (Fig. 1), was also detected in the LC/UV analyses. The mechanism for the formation of the N-dehydroxylation product by these recombinant P450 enzymes, which were coexpressed with cytochrome b5, remains unclear.
The kinetic parameters of M1 formation were also determined for recombinant CYP2J2, CYP4F2, CYP4F3B, and CYP4F12 (Table 1). The apparent Km and Vmax values for CYP4F2 and CYP2J2 were comparable to those observed with HLM. The intrinsic clearance value for CYP4F2 was 1.3-, 3.5-, and 92-fold greater than that for CYP2J2, CYP4F3B, and CYP4F12, respectively.
Immunoinhibition of M1 Formation. To clarify a potential role of CYP2J2 and CYP3A4 in M1 formation by HLMs, immunoinhibition experiments were performed with polyclonal antibodies against CYP2J2 or CYP3A4/5. As shown in Fig. 6A, no inhibition was observed at any antibody concentration tested. The same experiment was repeated with a monoclonal antibody against CYP2J2 at a single concentration (25 μl antibody/0.05 mg HLMs) and, again, inhibition was not observed (data not shown).
To ascertain the potential role of CYP4Fs in the metabolism of DB289 by HLMs, immunoinhibition studies were carried out with polyclonal antibodies raised against CYP4F2 from two independent sources (source A and source B as described under Materials and Methods). As shown in Fig. 6A, both antibodies against CYP4F2 inhibited M1 formation by HLMs in a concentration-dependent manner, with 91% inhibition at the highest concentration tested. Given the extensive amino acid sequence homology (∼93%) between human CYP4F2 and CYP4F3B (Christmas et al., 2001), it is reasonable to expect that the polyclonal antibodies against CYP4F2 cross-react with CYP4F3B. Hence, the specificity of the antibody from source A was examined against recombinant CYP4F2, CYP4F3B, and CYP2J2 (Fig. 6B). As expected, this antibody did not inhibit M1 formation by recombinant CYP2J2. However, the same antibody inhibited M1 formation by both recombinant CYP4F2 and CYP4F3B in a concentration-dependent manner, although it appeared to be less inhibitory for CYP4F3B than for CYP4F2. This antibody exhibited less potent inhibition of M1 formation by recombinant CYP4F2 and CYP4F3B than by HLMs, possibly because of the higher amount of recombinant enzymes used relative to the amount of the enzymes contained in the 0.05 mg of HLMs tested (incubation volume and HLM concentration were 0.25 ml and 0.2 mg/ml, respectively). A recent study by Ito et al. (2006) showed that their polyclonal antibody raised against CYP4F2 also cross-reacted with these same CYP4F enzymes.
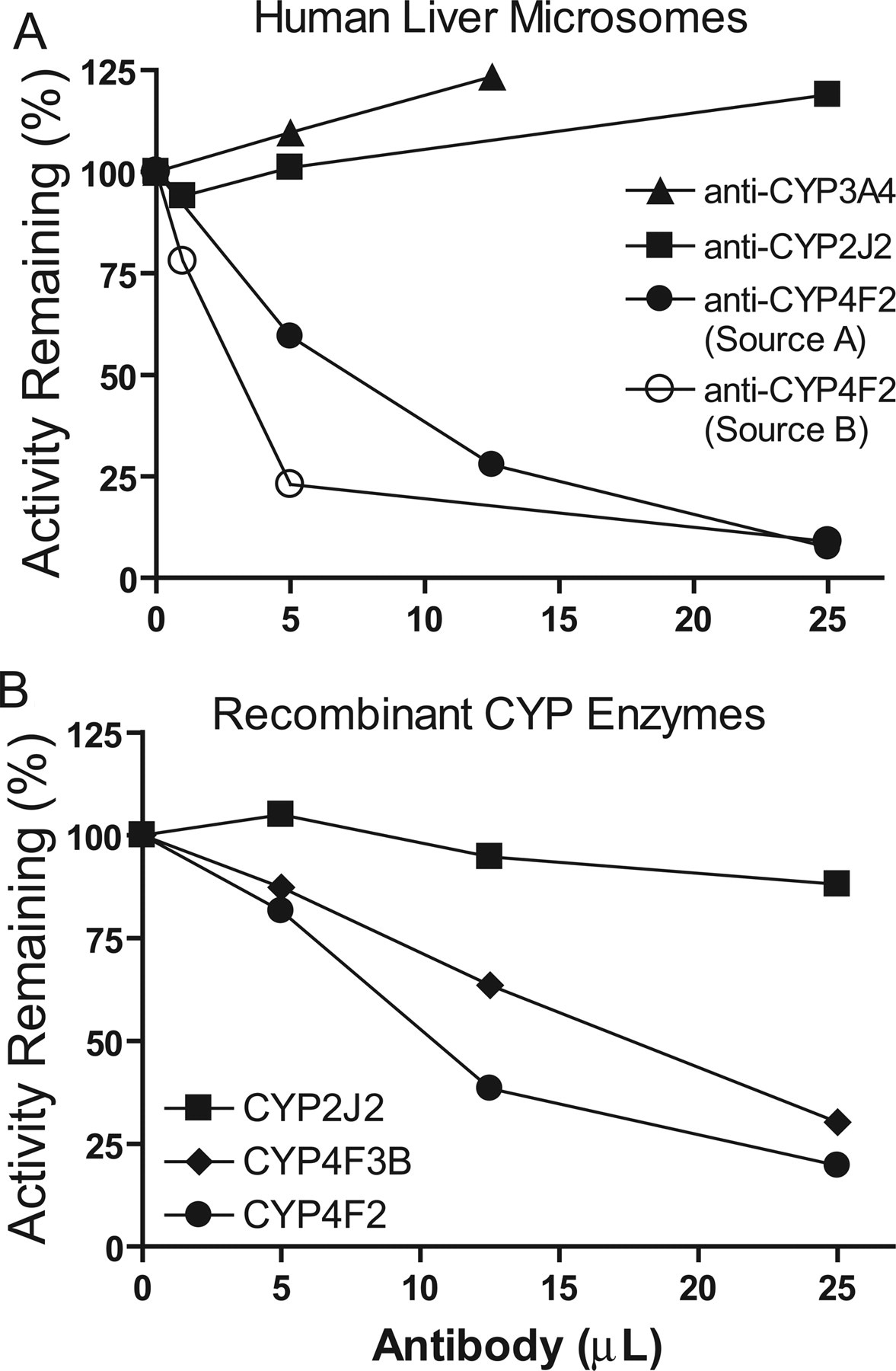
Immunoinhibition of M1 formation in incubations of DB289 with pooled HLMs (A) and recombinant P450 enzymes (CYP2J2, CYP4F2, and CYP4F3B) (B). Only antibody against CYP4F2 from source A was used for immunoinhibition with recombinant P450 enzymes (B). Incubation mixtures (250 μl) contained 0.2 mg/ml HLMs or 10 pmol/ml recombinant P450 enzyme, 3 μM DB289, and various amounts of antibody against CYP4F2, CYP3A4/5, or CYP2J2. Reactions were carried out at 37°C for 5 min with HLMs and recombinant CYP2J2 and CYP4F2, and for 15 min with CYP4F3B. Controls were run by substituting antibodies with the corresponding volumes of preimmune IgG proteins. Symbols denote the mean of duplicate determinations and are expressed as percentage activity remaining relative to the control.
Inhibition of M1 Formation by Arachidonic Acid P450-Mediated Metabolism Inhibitors and Ebastine. To help determine the contribution of CYP4F2, CYP4F3B, and CYP2J2 to M1 formation by HLMs, we examined the inhibitory effects of arachidonic acid (a CYP4F2, CYP4F3B, and CYP2J2 substrate) and ebastine (a CYP2J2, CYP4F12, and CYP3A4 substrate). Similarly, the effects of 17-ODYA (a nonselective mechanism-based arachidonic acid ω-hydroxylase and epoxygenase inhibitor) and HET0016 (an arachidonic acid ω-hydroxylase inhibitor) (Zou et al., 1994; Wang et al., 1998; Miyata et al., 2001) were examined (Fig. 7). The ranges of the concentrations of the inhibitors encompassed the previously reported Ki or Km values. In HLMs (Fig. 7A), HET0016 potently inhibited M1 formation by 78% at 0.1 μM and by 95% at 0.5 μM. 17-ODYA inhibited M1 formation by 39% at 1 μM and by 84% at 10 μM. In contrast, as a competitive inhibitor of CYP2J2, ebastine marginally inhibited M1 formation at concentrations up to 10 μM, which is about 8-fold higher than the Km value (1.3 μM) reported by Hashizume et al. (2002). Arachidonic acid at 500 μM inhibited M1 formation by 60% but showed no effect up to 100 μM.

Inhibition of M1 formation by HET0016, 17-ODYA, ebastine, and arachidonic acid. Incubations were carried out with pooled HLMs (A), recombinant CYP4F2 (B), recombinant CYP4F3B (C), and recombinant CYP2J2 (D). Incubation mixtures contained 0.2 mg/ml HLM or 10 pmol/ml recombinant P450 enzyme, 3 μM DB289, and a chemical inhibitor at the indicated concentration. Reactions were carried out at 37°C for 5 min with HLMs and recombinant CYP4F2 and CYP2J2, and for 15 min with CYP4F3B. The mechanism-based inhibitor, 17-ODYA, was preincubated with enzymes and NADPH for 15 min before adding the substrate. Controls were run with methanol in place of chemical inhibitors. Bars denote the average of duplicate determinations and are expressed as percentage activity remaining relative to the control.
In an attempt to use these inhibitors to determine the relative contribution of CYP4F2 and CYP4F3B to M1 formation in HLMs and to confirm that these inhibitors can indeed inhibit M1 formation by individual cytochrome P450s, we investigated the effects of these inhibitors on M1 formation catalyzed by recombinant CYP4F2, CYP4F3B, and CYP2J2. CYP4F2 (Fig. 7B) and CYP4F3B (Fig. 7C) exhibited similar inhibition profiles by all inhibitors except 17-ODYA, which appeared to activate CYP4F3B at low concentrations (0.2 μM and 1 μM). Such activation was not observed for either CYP4F2 or CYP2J2 (Fig. 7D). Unexpectedly, HET0016 inhibited recombinant CYP2J2 (by 53% at 0.1 μM), albeit to a much lesser extent than CYP4F2 and CYP4F3B (by 82% and 79% at 0.02 μM, respectively). Ebastine at 1 μM appeared to be a selective inhibitor of CYP2J2 (Fig. 7, B–D), but did not inhibit M1 formation by HLMs (Fig. 7A), indicating that CYP2J2 was not a major contributor to M1 formation by HLMs. At higher concentrations (>1 μM), ebastine became nonselective, partially inhibiting both CYP4F2 and CYP4F3B. At a concentration of 100 μM, arachidonic acid potently inhibited M1 formation by recombinant CYP4F2 and CYP4F3B. These results, although not conclusive, suggested that CYP4F2 and CYP4F3B, but not CYP2J2, contribute to M1 formation by HLMs.

Differential inhibition of M1 formation by ketoconazole (3 μM) in incubations of DB289 with pooled HLMs and recombinant CYP4F2, CYP4F3B, and CYP2J2. Incubation mixtures contained 0.2 mg/ml HLMs or 10 pmol/ml recombinant P450 enzyme, and 3 μM DB289. Reactions were carried out at 37°C for 5 min with HLMs and recombinant CYP2J2 and CYP4F2, and for 15 min with CYP4F3B. Controls were run with methanol in place of ketoconazole. Bars denote the average of duplicate determinations and are expressed as percentage activity remaining relative to the control.
Differential Inhibition of CYP4F2, CYP4F3B, and CYP2J2 by Ketoconazole. Due to the partial inhibition of M1 formation in pooled HLMs by 3 μM ketoconazole (Fig. 4), the effects of ketoconazole on recombinant CYP4F2, CYP4F3B, and CYP2J2 were investigated. As shown in Fig. 8, ketoconazole potently inhibited M1 formation by recombinant CYP4F2 and CYP2J2 (87% and 93%, respectively), but had no effect on M1 formation by recombinant CYP4F3B. The partial inhibition (∼43%) of M1 formation in pooled HLMs by 3 μM ketoconazole was consistent with the previous experiment (Fig. 4). The slight difference in the extent of inhibition was likely due to the lot difference in pooled HLMs used.
Discussion
Several lines of evidence from the current study indicate that in HLMs, M1 formation is predominantly and unexpectedly catalyzed by CYP4F enzymes. First, a polyclonal antibody raised against CYP4F2, which inhibited both recombinant CYP4F2 and CYP4F3B (Fig. 6B), inhibited M1 formation by HLMs in a concentration-dependent manner (up to 91%). Second, two arachidonic acid P450-mediated metabolism inhibitors, HET0016 and 17-ODYA, inhibited M1 formation by HLMs in a concentration-dependent manner (Fig. 7A). Third, with recombinant CYP4F2, the enzyme kinetics of M1 formation were comparable to those observed with HLMs (Table 1). It should be noted that, because of the high plasma protein-binding property of DB289 (Fitzpatrick et al., 2004), the apparent Km values are probably dependent on the protein concentrations used. In another study with a higher HLM concentration (0.5 mg/ml), the apparent Km value (uncorrected for protein binding) was reported as 2.1 μM.
In support of our conclusion, none of the cytochrome P450-selective chemical inhibitors evaluated (Fig. 4), with the exceptions of ketoconazole and ABT, substantially inhibited M1 formation by HLMs, arguing against roles for CYP1A, CYP2A6, CYP2B6, CYP2C, CYP2D6, CYP2E1, CYP3A, and CYP4A. This was further supported, with the exception of the CYP1 enzymes, by the minimal or modest activity of recombinant P450 enzymes toward DB289 consumption (Fig. 5A) and/or limited intrinsic or maximal clearance values from kinetic studies (Tables 1 and 2). Furthermore, CYP3A4/5 did not appear to contribute appreciably to M1 formation by HLMs as shown by the lack of inhibition by both TAO (Fig. 4) and a polyclonal antibody against CYP3A4/5 (Fig. 6A). However, our preliminary studies indicated that CYP3A4 may play a major role in the conversion of M3 to M4 in the DB289 metabolic activation pathway (Fig. 1).
Although CYP1A1 and CYP1A2 had the highest intrinsic clearance values among all recombinant enzymes examined, neither appeared to contribute to M1 formation by HLMs (Fig. 4). This was confirmed by polyclonal antibodies against CYP1A1 or CYP1A2, which did not inhibit DB289 metabolism (data not shown). Moreover, because CYP1A1 (and CYP1B1) is predominantly an extrahepatic enzyme (Murray et al., 1997; Paine et al., 1999; Guengerich, 2004; Doshi et al., 2006), CYP1A1 is unlikely to play a role in M1 formation by HLMs.
It is unclear why CYP1A2, which had a high intrinsic clearance toward M1 formation (Table 1) and is considered a major hepatic drug-metabolizing enzyme, did not contribute to M1 formation by HLMs. DB289 (at 3.0 μM) had a negligible inhibitory effect toward CYP1A2 activity, as well as other major P450 enzymes, with or without a 15-min preincubation, using HLMs. These results argue against inhibition of CYP1A2 by DB289, or its metabolites, as a potential explanation of why CYP1A2 had no contribution to M1 formation by HLMs.
DB289 consumption rates did not correlate with marker activities for individual P450 enzymes, including CYP1A2 and CYP3A4. In these studies, individual CYP4F enzymes were not included because of the lack of marker activities. Although leukotriene B4 (LTB4) ω-hydroxylation could be used as a nonselective marker reaction for the CYP4F subfamily (Kikuta et al., 2002), the limited variation (4-fold) in DB289 consumption rates (current study), coupled with the limited variation (2- to 3-fold) in LTB4 ω-hydroxylation rates observed in a panel of HLMs (Drs. Chris Patten and David Stresser, BD Gentest, personal communication; and Jin et al., 1998), reduces or eliminates the power of such a correlation analysis.
Although recombinant CYP2J2 could catalyze M1 formation (Fig. 5B, Table 1), we concluded that CYP2J2 was not the major enzyme responsible for M1 formation by HLMs because of the lack of inhibition by antibodies against CYP2J2 (Fig. 6A) and by ebastine, a competitive inhibitor of CYP2J2 (Fig. 7A). Human CYP2J2 is predominantly expressed in extrahepatic tissues, such as the small intestine and heart (Wu et al., 1996; Scarborough et al., 1999). Therefore, like CYP1A1 and CYP1B1, CYP2J2 may be involved in the extrahepatic metabolism of DB289. Another study by Hashizume et al. (2002) demonstrated that CYP2J2 is the predominant ebastine hydroxylase in human intestinal microsomes. This raises the possibility that DB289 could undergo significant first-pass metabolism by CYP2J2 in human intestine after oral administration.
Over the last decade, a number of studies have shown that CYP4F2 and CYP4F3B are constitutively expressed at relatively high levels in human liver. Different methods of quantification were used, including immunoquantitation (Jin et al., 1998) and enzyme-specific quantitative reverse transcription-polymerase chain reaction (Christmas et al., 2001). Caution should be taken when considering the expression levels of CYP4F2 and CYP4F3B, because of the 93% amino acid sequence identity. If an antibody against CYP4F2 or CYP4F3B is used for the quantification of these enzymes, it should be presumed that final measurements are the sum of both enzymes, unless the antibody has been explicitly tested for its specificity toward CYP4F2 or CYP4F3B. Quantitative reverse transcription-polymerase chain reaction can distinguish different but closely related enzymes by measuring mRNA levels, which may or may not reflect the levels of the corresponding protein. According to the immunoquantitation by Jin et al. (1998), CYP4F2 and CYP4F3B should constitute an appreciable portion of total hepatic cytochrome P450. These authors' results from 10 individual HLMs also indicated that the variation in CYP4F2 and CYP4F3B protein content is limited (less than 5-fold), consistent with the limited variation in LTB4 20-hydroxylation rates (∼3-fold) observed in the same panel of HLMs. In the present study, we measured M1 formation rates by 10 individual HLMs and observed only a 4-fold variation. Overall, these findings support CYP4Fs as the major enzymes involved in DB289 O-demethylation in human liver.
Members of the CYP4F subfamily are important enzymes involved in the biotransformation of endogenous compounds (e.g., arachidonic acid, LTB4) and are involved in the regulation of numerous physiological functions, such as inflammation and vasoconstriction (Jin et al., 1998; Sarkis et al., 2004; Kroetz and Xu, 2005). Although the role of CYP4F enzymes in drug disposition is largely unknown, Zimmerlin and Patten (2000) reported that the promising immunomodulatory agent, FTY720 [2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol], is eliminated predominantly by CYP4F-mediated ω-hydroxylation of the aliphatic chain. FTY720 ω-hydroxylation kinetics in HLMs exhibited a Vmax value comparable to that obtained for DB289 (1850 versus 3770 pmol/min/mg protein). However, the apparent Km value for FTY720 was much higher than that for DB289 O-demethylation (180 μM versus 0.5 μM), presumably because of structural differences between the two drugs.
In our attempt to determine the relative contributions of CYP4F2 and CYP4F3B to M1 formation by HLMs, ketoconazole was shown to selectively inhibit M1 formation by recombinant CYP4F2, but had little effect on M1 formation by recombinant CYP4F3B (Fig. 8). This same concentration of ketoconazole only partially inhibited M1 formation by HLMs, suggesting that CYP4F2 and CYP4F3B both contribute to M1 formation. Ketoconazole may provide a tool to distinguish these two closely related enzymes if the contribution from CYP3A4/5 can be established by other means (e.g., TAO and immunoinhibition). Based on these results, CYP4F3B could be as important as CYP4F2 for M1 formation by HLMs. Another member of the CYP4F subfamily, CYP4F11, has also been shown to be expressed primarily in human liver (Cui et al., 2000). In addition, CYP4F8 was identified in human seminal vesicles, but limited mRNA expression was detected in human liver (Bylund et al., 1999). Potential contributions to M1 formation in HLMs by these P450s remain uncharacterized because of the paucity of tools available to evaluate these enzymes.
DB289 biotransformation to its active form, DB75, consists of three O-demethylation reactions, i.e., M1 and M2 formation, and conversion of M3 to M4 (Fig. 1). It is not known whether all of these reactions are mediated by the same enzyme(s) in HLMs. Preliminary data indicate that CYP4F enzymes are capable of catalyzing the O-demethylation of M1 to form M2, and CYP3A4 appears to efficiently catalyze the O-demethylation of M3 to form M4. Investigation to elucidate the complete pathway of DB289 biotransformation in HLMs is currently under way.
In conclusion, DB289 O-demethylation to M1 in HLMs appears to be mediated primarily by CYP4F enzymes, including CYP4F2 and CYP4F3B. Thus, in human liver, members of the CYP4F subfamily metabolize not only endogenous compounds, such as arachidonic acid and eicosanoids, but also xenobiotics. Their potential as drug-metabolizing enzymes remains to be fully understood.
Acknowledgments
We are grateful to Dr. Yoshihiko Funae (Osaka City University Medical School, Osaka, Japan) for providing polyclonal antibodies against CYP4F2 and CYP2J2.
We would like to dedicate this article to Jim Allen, colleague and friend.
Footnotes
-
This study was supported by grants from the Bill and Melinda Gates Foundation and the Medicines for Malaria Venture.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.010587.
-
ABBREVIATIONS: DB75, 2,5-bis(4-amidinophenyl) furan; DB289, 2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime; DB289-d8, deuterium-labeled DB289; 17-ODYA, 17-octadecynoic acid; ABT, 1-aminobenzotriazole; P450, cytochrome P450; FMO, flavin monooxygenase; HLM, human liver microsome; HPLC, high performance liquid chromatography; LTB4, leukotriene B4; TAO, troleandomycin; HET0016, N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine; LC/MS/MS, liquid chromatography-tandem mass spectrometry.
-
↵1 James L. Allen, deceased.
- Received April 19, 2006.
- Accepted September 21, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}