


Abstract
Variable interindividual expression of cytochrome P450 3A presents a challenge in dosing drugs. The use of potent inhibitors of CYP3A such as ketoconazole has been explored to reduce the clearance of CYP3A substrates, thereby resulting in smaller dose requirements; however, the impact of CYP3A inhibition on interindividual variability has not been well characterized. Our objective was to examine the effect of ketoconazole inhibition on CYP3A metabolic variability as measured by the CYP3A biomarker oral midazolam. A single dose of midazolam (0.075 mg/kg) was administered to 19 healthy Caucasian adults (38.7 ± 8.8 years, nine male/10 female) at baseline and concurrently with ketoconazole (400 mg daily for 10 days) on day 6 or 9 of ketoconazole. Plasma samples were collected over 6 to 30 h. A paired t test and percent coefficient of variation (CV%) were used to evaluate differences in midazolam clearance and interindividual variability during both phases. Monte Carlo simulation was performed to determine probability distribution of area under the concentration-time curves (AUCs). Midazolam apparent oral clearance decreased by 89% (p < 0.0001) during inhibition. Cmax increased from 23 ng/ml (95% CI 19–29 ng/ml) to 55 ng/ml (95% CI 46–66 ng/ml), p < 0.0001. CV% increased from 41 to 58% from baseline to ketoconazole inhibition. AUCs [median (range)] were 0.20 mg · min/ml (0.05–0.81 mg · min/ml) and 1.94 mg · min/ml (0.25–25.4 mg · min/ml) at baseline and inhibition phase, with CV% of 41 and 61%, respectively. Ketoconazole decreased CYP3A activity but did not reduce interindividual variability. Use of a CYP3A inhibitor to standardize dosing of CYP3A substrates may not be feasible in clinical practice.
The cytochrome P450 (P450) 3A subfamily comprises the most clinically important metabolic enzymes involved in drug metabolism. CYP3A exhibits broad substrate specificity, metabolizing nearly half of all marketed drugs (Gibbs and Hosea, 2003). This group is also the most abundant of the P450 isozymes, accounting for 29 and 70% of the P450s in the liver and intestinal mucosal, respectively (Guengerich, 1991; Watkins, 1992). Currently, four isoforms have been described in humans: CYP3A4, CYP3A5, CYP3A7, and CYP3A43, of which the latter two are believed to have only a minor role in CYP3A-mediated biotransformation (Daly, 2006).
The highly variable expression/function of CYP3A observed between individuals poses a therapeutic challenge. Intersubject variability in CYP3A-mediated activity has been reported to be large (approximately 10-fold difference). Some studies have reported variability of greater than 20-fold (Lin et al., 2001; Zhu et al., 2003). An obvious challenge is in optimizing drug therapy for maximization of efficacy while limiting toxicity. This is particularly a concern for drugs with narrow therapeutic indices, including many immunosuppressants and chemotherapeutic agents. Some studies have explored the use of potent CYP3A inhibitors such as the imidazole antifungal agent ketoconazole to increase exposure of drugs such as in cyclosporine at reduced doses. Usually, the intention of P450 inhibition in these studies has been to allow use of smaller doses and thereby decrease cost (Martin et al., 1999). However, investigation of the effect of inhibition on interindividual variability has not been well established.
Midazolam, a short-acting benzodiazepine, is selectively metabolized by CYP3A, specifically CYP3A4 and CYP3A5, to the major metabolite 1′-hydroxymidazolam (Tsunoda et al., 1999). It is widely used as a phenotyping probe for CYP3A activity. Apparent oral clearance of midazolam is the pharmacokinetic parameter that is recognized as the biomarker for hepatic and intestinal CYP3A activity. The purpose of this study is to examine whether the large intersubject variability in observed CYP3A activity can be significantly decreased with use of the potent CYP3A inhibitor ketoconazole.
Materials and Methods
The data for this analysis were obtained from a previously published study (Chung et al., 2006). Only the data from the midazolam phenotyping phases of baseline and inhibition were used. The study was approved by the Institutional Review Board of Bassett Healthcare. All subjects provided written informed consent prior to study procedures.

Midazolam plasma concentration (mean ± S.D.) versus time profile at baseline (▴) and ketoconazole inhibition (▪).
Subjects. Twenty healthy Caucasian adults between the ages of 18 to 55 years participated in the study. Subjects were required to have a body mass index between 18 to 30 kg/m2. They avoided tobacco or nicotine containing products for at least 3 months and were determined to be in good health by complete medical history, physical examination, electrocardiogram, and clinical laboratory tests. Women were required to be nonpregnant, non-nursing, premenopausal, and either surgically sterile or using acceptable nonhormonal methods of contraception at least 14 days before and until 30 days after completion of the study. Urine pregnancy tests were performed for all women of childbearing potential during the screening phase and before each dosing period. Use of any medication, herbal preparation, or nutritional supplement known to affect CYP3A activity within 7 days or five half-lives (whichever was longer) before the first dose of study medication was prohibited. Subjects were required to refrain from grapefruit-, apple-, and orange juice-containing products for 7 days before study initiation and for the duration of the study. Subjects with a history of regular alcohol consumption exceeding one drink per day within 6 months of screening or intolerance/hypersensitivity to benzodiazepines or imidazoles were excluded.
Study Design and Procedures. The study was a sequential, open-label crossover trial using oral midazolam as a biomarker for CYP3A activity at baseline and after inhibition. The full protocol has been described previously (Chung et al., 2006) and is briefly described below.
Each subject completed the baseline phase and then proceeded to the inhibition phase, following a washout period of at least 1 week. Subjects arrived at the research unit between 6:00 AM and 10:00 AM following an overnight fast of at least 8 h. An intravenous cannula was placed in the forearm for serial blood draws. During the baseline phase, a single oral dose of midazolam (0.075 mg/kg) (Versed, 2 mg/ml syrup; Roxane Laboratories, Inc., Columbus, OH) was administered alone. Blood samples (7 ml) were collected at predose, 0.5, 1, 1.5, 2, 3, 4, and 6 h after midazolam administration.
In the inhibition phase, oral ketoconazole (400 mg, two ketoconazole 200-mg tablets; Taro Pharmaceuticals, Hawthorne, NY) was self-administered daily for 10 days. Ketoconazole tablets were dispensed in unit dose packages. Subjects were instructed to take the dose in the morning at the same time daily. Adherence to the ketoconazole regiment was checked by unit dose package count. A single oral dose of midazolam (0.075 mg/kg) was administered on either day 6 or day 9 of ketoconazole 2 h before ketoconazole administration. Randomization of midazolam to day 6 or day 9 was done because of an additional probe administration in the original study. However, because peak inhibition with ketoconazole occurs after 48 h of continuous dosing, subjects were assumed to be inhibited at either study day. Blood samples for midazolam concentrations were collected at predose, 0.5, 1, 2, 4, 6, 8, 12, 24, and 30 h after midazolam dosing.
Subjects were required to remain seated for a minimum of 2 h after midazolam administration. All blood samples were immediately centrifuged at approximately 2800 rpm at 4°C for 15 min. Harvested plasma samples were kept frozen at –80°C until analysis. Vital signs and oxygen saturation by pulse oximetry were monitored for 2 h after midazolam administration during each phase.
Assay and Pharmacokinetic Analysis. Midazolam plasma concentrations were analyzed at Prevalere Life Sciences, Inc., Whitesboro, NY. Liquid chromatography-tandem mass spectrometry using alprazolam as the internal standard was used. The assay has been described previously (Kashuba et al., 1998; Chung et al., 2006). Pharmacokinetic parameters were analyzed by noncompartmental analysis using WinNonlin version 3.2 (Pharsight Corporation, Mountain View, CA) (Chung et al., 2006). Apparent oral total body clearance (CL/F, where F represents bioavailability) data are presented as weight-adjusted clearance. A 10,000-subject Monte Carlo simulation using the mean and standard deviation of weight-adjusted CL/F and a log-normal distribution was performed to determine probability distribution of area under the concentration-time curves (AUCs) using Crystal Ball, version 7 (Decisioneering, Inc., Denver, CO). AUC was simulated using the equation AUC = F · dose/CL. A midazolam dose of 5 mg was chosen for the oral midazolam simulation study. The Monte Carlo simulation distributions were reviewed and confirmed to be an accurate reflection of the data used to generate them.
Statistical Analysis. A priori sample size calculations determined that 16 subjects were required to detect a minimum difference of 25% in area under the concentration-time curve AUC(0-∞) between phases with 80% power and an α of 0.05. SYSTAT version 11 (Systat Software Inc., Point Richmond, CA) was used for statistical analyses. Log-transformed CL/F, peak concentrations (Cmax), and AUC(0-∞) between phases was evaluated using a paired t test. An F test was used to compare variance in midazolam apparent oral clearance during baseline and inhibition phases. Percent coefficient of variation (CV%) was calculated to assess interindividual variability in CL/F in each phase. Data are presented as geometric mean with 95% confidence interval (CI). Data for the Monte Carlo simulation are presented as the median and the range. A p value of <0.05 was considered significant.
Results
Twenty subjects (all Caucasians) were enrolled in the study. Nineteen subjects (10 women, nine men) completed both baseline and inhibition phases. One male subject withdrew during the inhibition phase because of gastrointestinal adverse effects attributed to ketoconazole. Demographic data for the 19 subjects are shown in Table 1. Both phases were well tolerated. Sixteen of 19 subjects were homozygous for CYP3A5*3/*3, and the other three were heterozygous. No clinically significant adverse events were noted.
Demographics, laboratory, and weight-based oral midazolam dose for 19 healthy volunteers
Data are presented as arithmetic mean ± S.D.
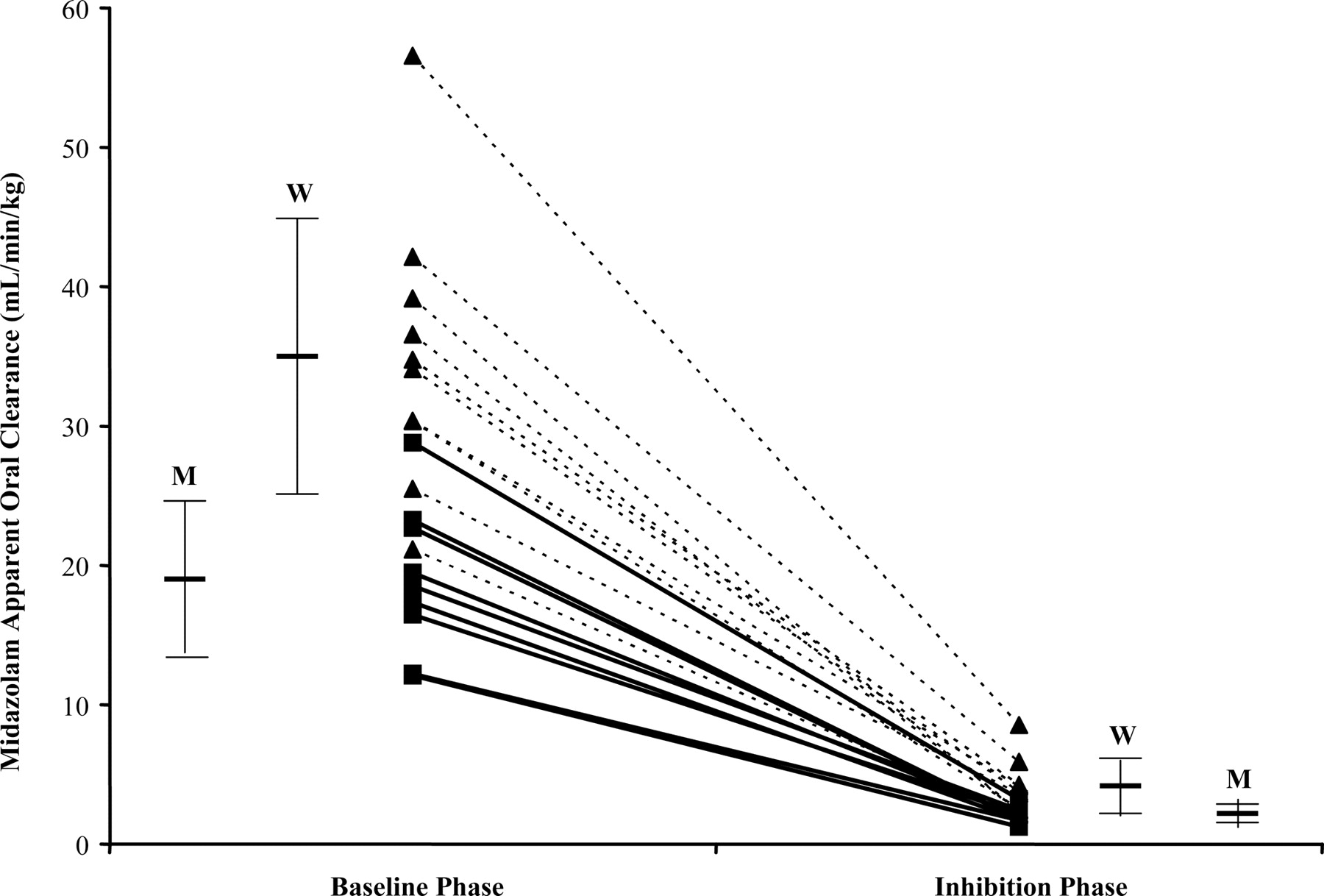
Mean midazolam plasma concentration versus time at baseline and inhibition are shown in Fig. 1. Coadministration of ketoconazole, a potent inhibitor of CYP3A, increased midazolam Cmax from a geometric mean 23 ng/ml (95% CI 19–29 ng/ml) to 55 ng/ml (95% CI 46–66 ng/ml), p < 0.0001. CV% increased from 41 to 58% from baseline to ketoconazole inhibition. Midazolam AUC(0-∞) increased 9.5-fold from 49 ng · h/ml (95% CI 40–60 ng · h/ml) to 467 ng · h/ml (95% CI 370–589 ng · h/ml), p < 0.0001, with CV% of 41 and 61%, respectively. Midazolam apparent oral clearance decreased by a mean of 89% (p < 0.0001) as shown in Table 2. There were no differences in midazolam CL/F on the basis of the order of drug administration during any phases. Figure 2 displays the drop in midazolam CL/F from baseline to inhibition phase. Variance in CL/F also decreased from 128.8 to 3.1 from baseline to inhibition phase, representing a decrease in variance of approximately 98% (p < 0.001).
Apparent oral midazolam clearance by sex as a measure of CYP3A activity at baseline and after ketoconazole inhibition
Data presented as geometric mean.
Monte Carlo simulation distributions of area under the concentration-time profiles of midazolam at baseline and ketoconazole inhibition are displayed in Fig. 3, A and B, respectively. Estimated AUCs [median (range)] were 0.20 mg · min/ml (0.05–0.81 mg · min/ml) and 1.94 mg · min/ml (0.25–25.4 mg · min/ml) at baseline and inhibition phase, respectively. CV% was 61% at inhibition versus 41% at baseline.
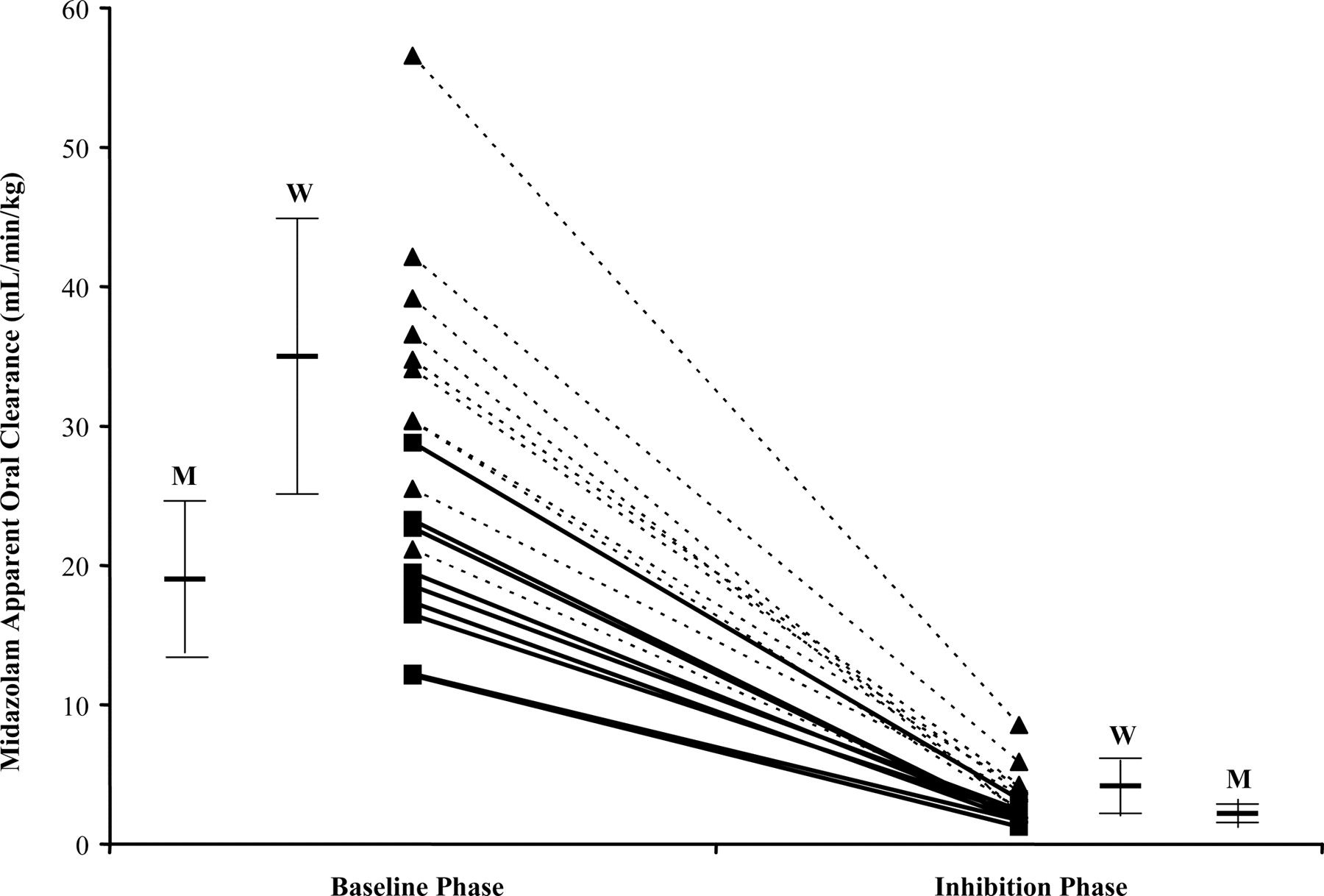
Midazolam apparent oral clearance at baseline and after ketoconazole inhibition in women (▴; n = 10) and men (▪; n = 9). Mean clearance for women and men at each phase is shown as a solid bar ± S.D. W denotes women, and M denotes men.
Discussion
Ketoconazole is a known potent inhibitor of CYP3A activity. We investigated whether the use of ketoconazole (at the recommended inhibitory dose of 400 mg daily) (Bjornsson et al., 2003; Chien et al., 2006) would result in significant reduction in intersubject variability of CYP3A isozyme activity. Consistent with other findings, Cmax and AUC(0-∞) increased 2.4- and 9.5-fold, respectively, in our study (Olkkola et al., 1996). Apparent oral clearance decreased approximately 9-fold for all subjects and did not differ by sex. Those subjects with the highest clearance at baseline exhibited the greatest absolute change in clearance. The relative (percent) decrease in clearance was similar for each subject. The range in the decrease of CL/F was 85 to 94%. Subjects with the highest CYP3A activity at baseline continued to have the highest CYP3A activity during inhibition (Fig. 2). This illustrates that inhibition by ketoconazole, although reducing CYP3A activity in all subjects, did not convert all subjects to an equivalent level of CYP3A activity. Although variance of CL/F decreased as expected in the inhibition phase, the measure of variability between individuals (CV%) actually increased. Interestingly, the increase in variability was seen only in women (Table 2). CV% for men remained constant at 28% from baseline to ketoconazole inhibition, whereas CV% increased from 28 to 51% for females. Whether this observation is unique to our subjects or a sex difference in CYP3A activity is unknown; however, our previous data have suggested that women show modestly greater CYP3A isozyme activity compared with men when using oral midazolam as a probe (Chen et al., 2006).
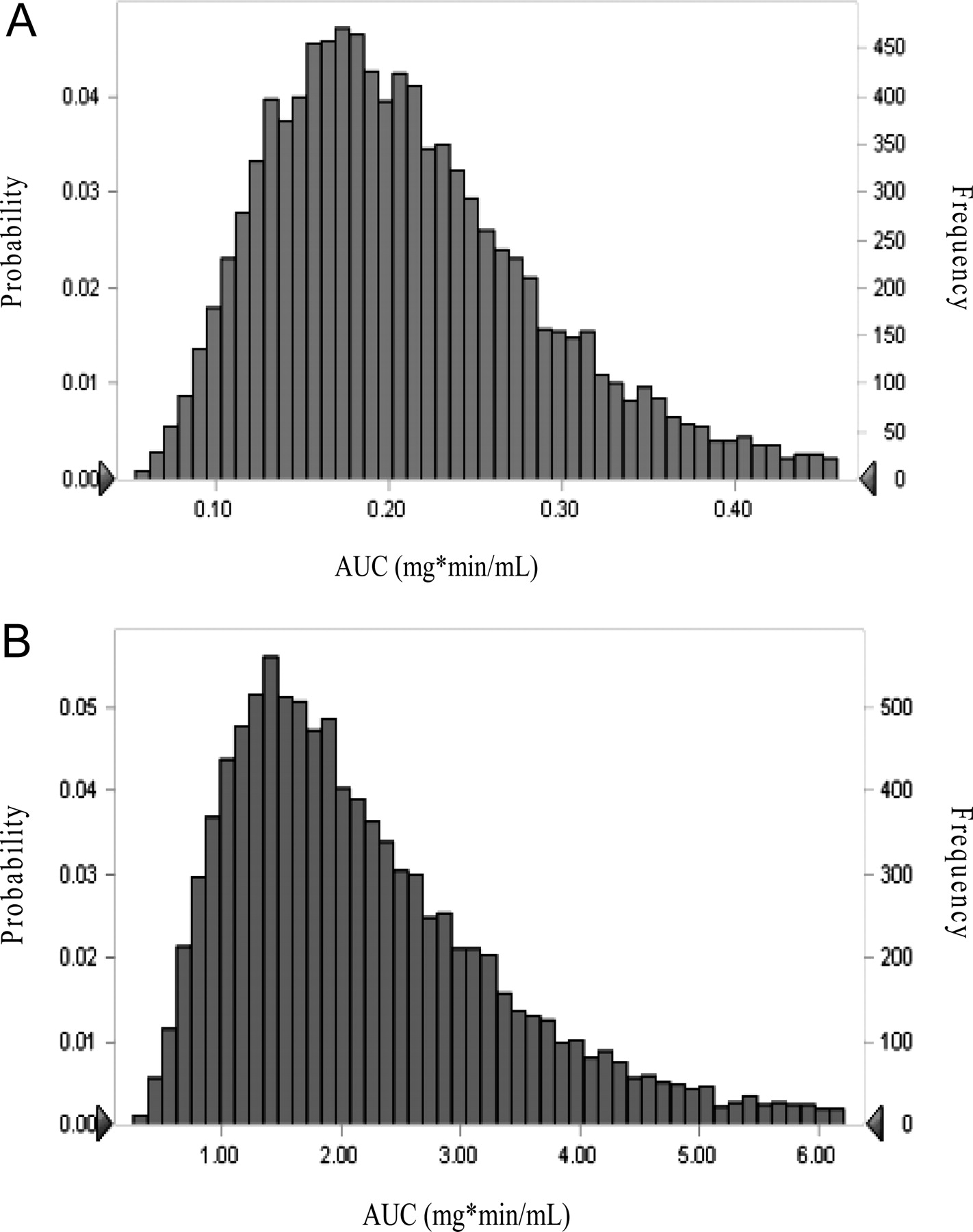
A, distribution of area under the concentration-time curves of oral midazolam administration at baseline via 10,000-subject Monte Carlo simulation. B, distribution of area under the concentration-time curves of oral midazolam after ketoconazole inhibition via 10,000-subject Monte Carlo simulation.
Monte Carlo simulations were performed to determine the distribution of exposure using a 10,000-subject simulation. As shown in Fig. 3, A and B, the range in exposure to midazolam increased from baseline to inhibition, consistent with findings form midazolam CL/F data showing an increase in interindividual variability with CYP3A inhibition.
CYP3A inhibitors such as ketoconazole have been used in an attempt to “standardize dose” of certain CYP3A substrates. Martin et al. (1999) evaluated the use of ketoconazole as an inhibitor of cyclosporine to reduce the dose and thus the cost of therapy. Tham et al. (2006) investigated the use of ketoconazole to reduce variability in clearance of midazolam and docetaxel. These authors reported a reduction in the variability in midazolam clearance but not of docetaxel in two separate groups of patients assessed during a constitutive state or during inhibited activity. A recent study using high-dose ketoconazole (400 mg three times a day) found intersubject variability of docetaxel (a drug that is not solely a CYP3A substrate) increased with inhibition (Engels et al., 2006). Another study by Desai et al. (2004) observed no significant decrease in interpatient variability of carboxyamidatriazole (CYP3A substrate) when a 200-mg dose of ketoconazole was coadministered.
The cause for the wide intersubject variability observed with CYP3A activity has not been fully elucidated. Factors such as age, sex, and genetic polymorphism of CYP3A5 have been investigated in an attempt to explain differences in CYP3A activity. Although some findings suggest these factors may play a role, knowledge in this area remains limited. Recent data from Bosch et al. (2006) showed that several single nucleotide polymorphisms are found in the pregnane X receptor (PXR) gene. The PXR receptor has been implicated in the up-regulation of CYP3A. These authors suggest that these single nucleotide polymorphisms may influence the PXR receptor and potentially CYP3A activity.
The concept of using an inhibitor to reduce the intersubject variability of CYP3A activity is an interesting one. Potentially, if inhibition resulted in individuals having a relatively “similar” clearance of a CYP3A substrate, standardized dosing could be used to achieve a narrow range of exposure. Our data suggest that inhibition of CYP3A activity with ketoconazole does not reduce intersubject variability. In fact, between subject variability actually increased with CYP3A inhibition. Use of a potent inhibitor in a clinical setting does not reduce the variability of CYP3A substrate exposure and thus, does not allow for use of a “one-dose-fits-all” approach.
Acknowledgments
We thank Dr. Paul G. Ambrose for assistance with Monte Carlo simulations.
Footnotes
-
This article was presented in part at the 107th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics in Baltimore, MD, March 11, 2006; and at the Food and Drug Administration Science Forum in Washington, DC, April 18, 2006.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011742.
-
ABBREVIATIONS: P450, cytochrome P450; CL/F, apparent oral clearance; AUC, area under the concentration-time curve; CI, confidence interval; PXR, pregnane X receptor.
- Received June 29, 2006.
- Accepted September 20, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}