


Abstract
Conventional methods to forecast CYP3A-mediated drug-drug interactions have not employed stochastic approaches that integrate pharmacokinetic (PK) variability and relevant covariates to predict inhibition in terms of probability and uncertainty. Empirical approaches to predict the extent of inhibition may not account for nonlinear or non-steady-state conditions, such as first-pass effects or accumulation of inhibitor concentration with multiple dosing. A physiologically based PK model was developed to predict the inhibition of CYP3A by ketoconazole (KTZ), using midazolam (MDZ) as the substrate. The model integrated PK models of MDZ and KTZ, in vitro inhibition kinetics of KTZ, and the variability and uncertainty associated with these parameters. This model predicted the time- and dose-dependent inhibitory effect of KTZ on MDZ oral clearance. The predictive performance of the model was validated using the results of five published KTZ-MDZ studies. The model improves the accuracy of predicting the inhibitory effect of increasing KTZ dosing on MDZ PK by incorporating a saturable KTZ efflux from the site of enzyme inhibition in the liver. The results of simulations using the model supported the KTZ dose of 400 mg once daily as the optimal regimen to achieve maximum inhibition by KTZ. Sensitivity analyses revealed that the most influential variable on the prediction of inhibition was the fractional clearance of MDZ mediated by CYP3A. The model may be used prospectively to improve the quantitative prediction of CYP3A inhibition and aid the optimization of study designs for CYP3A-mediated drug-drug interaction studies in drug development.
Metabolism-based pharmacokinetic (PK) interactions are well recognized as a source of clinically significant adverse drug reactions (Huang et al., 1999; Huang and Lesko, 2004). The early forecast of clinically significant metabolism-based pharmacokinetic drug-drug interactions is an increasingly important aspect of drug development. The assessment of drug-drug interaction potential before the conduct of a clinical trial often involves simple algebraic calculations. One such calculation is the ratio of the expected clinical exposure to the in vitro inhibition constant (Ki) of a new drug entity for a specific cytochrome P450 (Sahajwalla et al., 1999; Bjornsson et al., 2003). The primary sources of quantitative errors associated with this prediction approach are attributed to the experimental procedures used to determine the in vitro parameters, the variability in the intrinsic factors associated with PK properties of the inhibitor (e.g., absorption kinetics, plasma protein binding, and tissue partition coefficients), or the substrate (i.e., the fraction of total clearance attributed to the elimination pathway of interest), the extrinsic factors associated with the clinical study designs (e.g., dosing scheme, sampling design, and population demographics), and, most notably, the uncertainty in the effective concentration of the inhibitor at the enzyme site. In addition, complex drug disposition properties, such as saturable first pass kinetics and bidirectional transporter processes, are not adequately characterized by these simple linear models. Therefore, a definitive effort to forecast metabolism-mediated PK drug-drug interactions should take these factors into consideration.
Physiologically based PK modeling is a time-tested approach to investigate the interaction between drugs and biological systems (Ito, 1998; Tucker et al., 2001; Rowland and Peck, 2002; Chien et al., 2003; Rostami-Hodjegan and Tucker, 2004; Schmitt and Willmann, 2004; Ito and Houston, 2005). The precision of the prediction can be increased by incorporating into the model information about the PK of the interacting drugs and the associated variability terms. More importantly, the model, once adequately qualified, may be used to answer very specific questions through simulations, such as the selection of an optimal dosing regimen (i.e., the amount of the dose, dosing frequency, and duration of dosing) for the substrate and the inhibitor, and the most efficient PK sampling scheme to optimize parameter estimation and minimize volume of blood drawn. Finally, the pharmacokinetic and pharmacodynamic modeling and the associated clinical trial simulations may be used to support the optimal (safe, efficacious, and cost-effective) prescriptive recommendations in patients and in special populations (Lesko et al., 2000; Tucker et al., 2001; FDA, 2004).
The work presented in this article illustrates the development and qualification of a simulation model for predicting the inhibitory effect of ketoconazole (KTZ) on midazolam (MDZ) PK. MDZ is a widely used probe substrate for the activity of CYP3A. The ultimate application of this model will be to predict the magnitude of PK changes due to CYP3A inhibition for new drugs metabolized by CYP3A and to select the optimal dosing scheme for clinical studies (e.g., drug-drug interactions or high dose QTc prolongation studies) using KTZ as a potent inhibitor of CYP3A. Physiologically relevant parameters and significant covariates were included in the model to simulate the MDZ concentration-time profile under various KTZ dosing scenarios. The sensitivity of the predictions to key model parameters were assessed and ranked by their influence on the magnitude of prediction. The robustness of the model prediction was validated by comparing the results of the predictions to five published KTZ-MDZ drug-drug interaction studies. The iterative process of model development, refinement, and application for the purpose of predicting drug-drug interaction has been reviewed elsewhere (Chien et al., 2003).
Materials and Methods
A complete set of individual KTZ and MDZ PK data from a KTZ-MDZ interaction study would facilitate simultaneous modeling of the substrate-inhibitor pair. However, these data were not available and have not been published. Furthermore, no single study has been conducted to delineate the changes in MDZ PK using different doses of KTZ or varying the duration of KTZ dosing. Therefore, to simulate the effect of a KTZ dosing regimen on MDZ PK, an interaction model needed to be developed in stages. PK models for the substrate, MDZ, and the inhibitor, KTZ, were developed separately, using data from several different clinical studies. Furthermore, because these models were initially developed as descriptive models (viz., selected based on fit of the models to the observed data), it was necessary to refine specific features of these models, so that they may be used for prediction. Finally, these PK models were linked via a competitive Michaelis-Menten (MM) inhibition model. This model, modified from the original KTZ and MDZ descriptive models, was used to simulate the baseline (control phase) MDZ and KTZ plasma concentration-time profiles to validate the predictive performance of these models for the observed data.
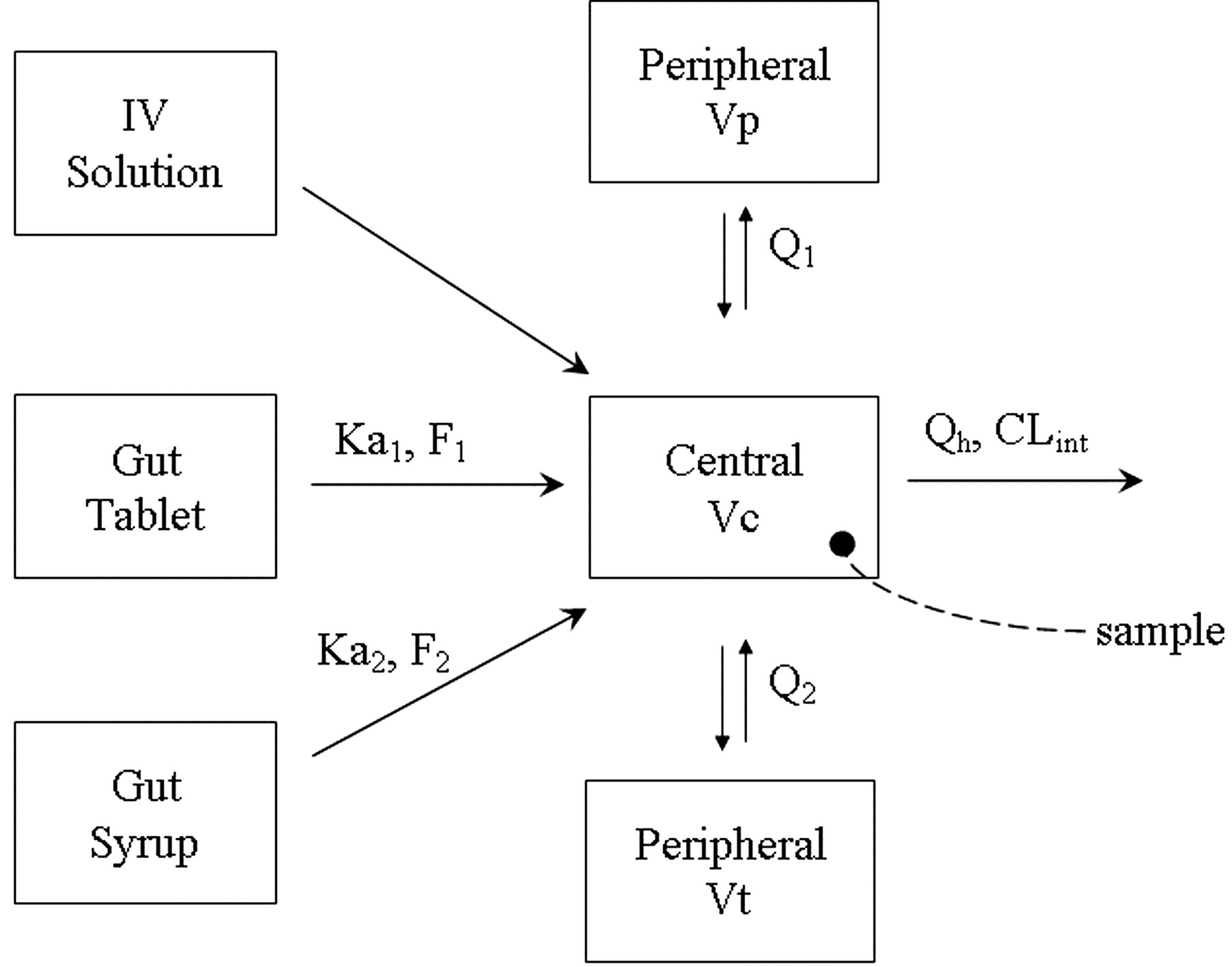
MDZ PK Model Development. A population PK model for MDZ was developed using data from healthy adult subjects (Ernest et al., 2004). The MDZ individual concentration-time data from 112 subjects were combined from five clinical studies of similar study design and population demographics (Gorski et al., 1998, 2003; Belle et al., 2002; Haehner-Daniels et al., 2004; Sauer et al., 2004; Ring et al., 2005). The data included intravenous and oral administration of MDZ in doses ranging from 1 to 15 mg. The population PK modeling was performed using the nonlinear mixed effects approach (Boeckmann et. al., 1994).
MDZ blood concentration was converted from plasma concentration by multiplying plasma concentration by a blood to plasma partitioning ratio of 0.86, which was determined ex vivo (Gorski et al., 1998). The clearance of MDZ was described by the well stirred liver model (eq. 1, where CL was the systemic clearance of MDZ, CLint,u was the MDZ unbound intrinsic clearance, fu was the fraction of MDZ unbound in the blood, and Qh was the hepatic blood flow. The hepatic blood flow was incorporated as an allometric expression of total body weight (eq. 2) (Brown et al., 1997) with the allometric exponent fixed to a typical value of 0.75. Gender, age, and body weight were covariates found to influence clearance, volume, and bioavailability. 
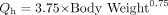
KTZ PK Model Development. A semiphysiologic KTZ PK model was developed (NONMEM, version V) using data from the literature. Mean KTZ plasma concentration-time data were combined from five clinical studies (Daneshmend et al., 1981, 1983, 1984; Gascoigne et al., 1981; Huang et al., 1986). In these studies, the subjects were administered KTZ orally in single and multiple doses ranging from 100 to 800 mg.
Since the model was developed using mean data from each study, for the purpose of simulation, the between- and within-subject variance terms assumed a typical 30% coefficient of variation (CV).
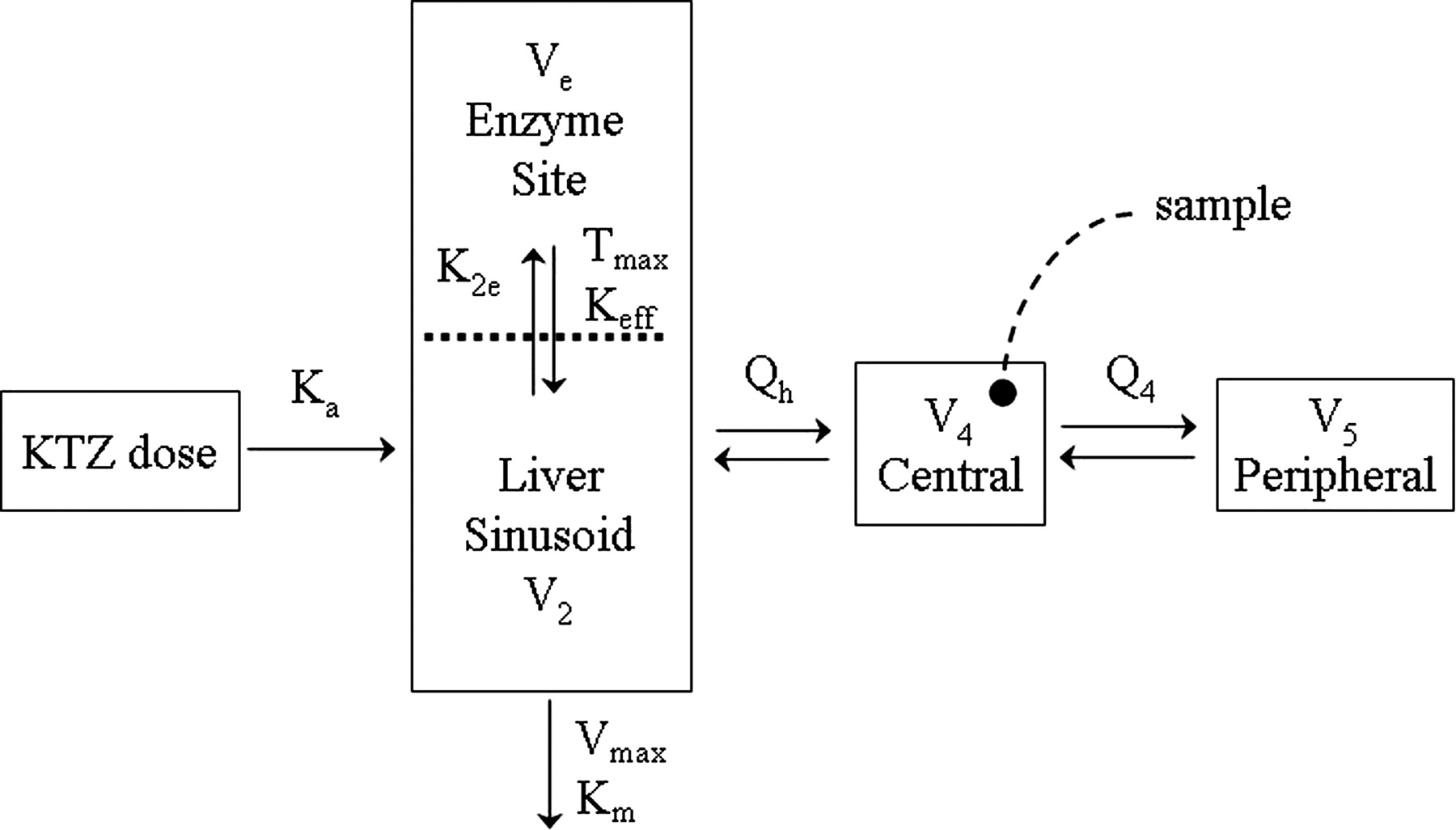
Previous work on KTZ disposition suggested that the model should incorporate elimination from the presystemic hepatic compartment occurring in parallel with a first-order uptake into an intracellular reservoir representing the enzyme site, followed by a saturable efflux from the enzyme site (Gibbs et al., 2000; Yamano et al., 2001; Kimura et al., 2003; Kimura et al., 2004). The parameters associated with the intrahepatic enzyme site compartment for KTZ were necessary for the simulation of the effective KTZ concentration-time data at the enzyme site following oral administration. However, the enzyme site compartment parameters cannot be estimated from plasma data alone; thus, they were allowed to vary over a wide range in the simulations to test the influence of these variables on the prediction of inhibition.
KTZ-MDZ Interaction Model Development.The Inhibitor Submodel. For simulation purposes, the effect of KTZ inhibition on MDZ intrinsic clearance was assumed to be driven by the total and unbound concentrations of KTZ in the gut and enzyme site within the hepatocyte, respectively, following oral administration of KTZ. Due to the absence of absolute bioavailability study data, the dose of KTZ was assumed to be completely absorbed. A competitive Michaelis-Menten inhibition model was used to simulate the effect of inhibition. Both competitive and noncompetitive enzyme inhibition models were tested. The type of inhibition model did not seem to have an impact on the extent of inhibition because substrate concentration was low relative to the Michaelis constant (results not shown). Consequently, the competitive and noncompetitive mechanisms of inhibition collapsed to the same relationship; thus, the competitive model was used. For the purpose of investigating influential variables that may have an impact on the prediction of inhibition of KTZ on MDZ PK, additional variables were incorporated into the model. The additional variables included KTZ protein binding (fu), the maximum KTZ efflux from the liver (Tmax), the saturable Michaelis constant for the efflux (Keff), the unbound in vitro KTZ equilibrium inhibition constant (Ki), and the fraction of MDZ cleared by CYP3A metabolism (fm).
Several investigators have hypothesized that KTZ accumulates in the enterocytes or hepatocytes via tight binding to cellular constituents, which would explain the persistent inhibition of CYP3A by KTZ (Matthew et al., 1993; Gibbs et al., 2000). Two alternative simulation models may be considered that could account for the accumulation. The most commonly used model assumes that a constant tissue partitioning coefficient (Kp) drives the accumulation of the unbound intracellular KTZ concentration in the hepatocyte. An experimental estimate for the human hepatic tissue-plasma partition coefficient has not been reported. A Kp value of 3 was indirectly calculated from data obtained in dog and rodent (Gibbs et al., 2002; Kimura et al., 2004). A constant Kp (eq. 3) was initially tested in the KTZ model. 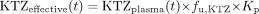
The effective concentration of KTZ (KTZeffective or KTZenzyme) at the enzyme site is a function of the total KTZ plasma concentration (KTZplasma), with a constant unbound fraction (fu,KTZ) of 0.01 and a constant Kp of 3.
The partitioning of the KTZ concentration to the site of inhibition represents the result of hepatic influx and efflux processes. This partitioning to the site of inhibition may be distinct from the conventional whole tissue to plasma ratio. Therefore, a second model (eq. 4) was considered in which unbound KTZ enters the hepatocyte following a first-order process, but the efflux of KTZ from the enzyme site (a separate compartment) back to the systemic circulation via the hepatic sinusoid was limited by a saturable process. 
The effective concentration of KTZ (KTZeffective) changes as a function of the unbound KTZ in the plasma (fu,KTZ× KTZplasma) entering the enzyme site; this process is governed by the first-order rate constant (K2e) and the saturable efflux process, which is governed by the Michaelis-Menten constant, Keff, and the maximum rate of efflux, Tmax.
Since the values of Tmax and Keff cannot be estimated from the available data and are highly correlated, Tmax assumed an arbitrary value of one. The value of Keff was allowed to vary over a wide range in the simulations to test the influence of this parameter on the prediction of inhibition. The initial value of Keff was assumed to be equivalent to the first-order uptake rate constant (K2e).
The Substrate Submodel. The MDZ population model was modified to allow simulation of first-pass (gut and liver) and systemic inhibition of CYP3A-mediated metabolism by KTZ. Specifically, the concentration of KTZ was introduced as the inhibitor in the form of a competitive MM inhibition model, and the gut and hepatic availability and systemic clearance terms were expressed as functions of free intrinsic clearance of MDZ (vide infra).
The fraction of the MDZ dose absorbed through the intestinal lumen (Fa) was assumed to be complete. However, the MDZ gut and hepatic availability were incorporated as nonlinear functions of MDZ intrinsic clearance. The fraction of the MDZ dose that passes through the gut increases in the presence of KTZ, as described in eq. 5. 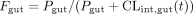 where the parameter Pgut represents a constant governing the absorption of MDZ from the gut, eliminating the need for a complex function of permeability, solubility, transport, and blood flow from the lumen. Pgut is assumed independent of gut intrinsic clearance (CLint,gut). The unbound fraction of MDZ in the gut is assumed to be 1. Therefore, if Pgut assumes a value equivalent to baseline intrinsic clearance of MDZ in the absence of the inhibitor, the baseline Fgut would be 0.5, which is consistent with the published values for MDZ (Thummel et al., 1996; Gorski et al., 1998). The intrinsic clearance of MDZ in the gut is expressed as shown in eq. 6.
where the parameter Pgut represents a constant governing the absorption of MDZ from the gut, eliminating the need for a complex function of permeability, solubility, transport, and blood flow from the lumen. Pgut is assumed independent of gut intrinsic clearance (CLint,gut). The unbound fraction of MDZ in the gut is assumed to be 1. Therefore, if Pgut assumes a value equivalent to baseline intrinsic clearance of MDZ in the absence of the inhibitor, the baseline Fgut would be 0.5, which is consistent with the published values for MDZ (Thummel et al., 1996; Gorski et al., 1998). The intrinsic clearance of MDZ in the gut is expressed as shown in eq. 6. 
The concentration of KTZ remaining in the gut at any time “t” is the amount of KTZ predicted in the KTZ gut compartment distributed into the volume of a glass of water, which is assumed to be 250 ml. Sensitivity analysis demonstrated that model prediction was insensitive to this volume term within a 10-fold range (results not shown); thus, its impact on the prediction was not evaluated further.
The availability of the MDZ dose through the liver (FLiver) is expressed as shown in eq. 7.  The arterial (Qarterial) and portal vein (Qportal) blood flow, expressed as fractions of total blood flow to the liver, were 0.15 and 0.75, respectively (Brown et al., 1997). The unbound intrinsic clearance of MDZ in the liver is described by eq. 8.
The arterial (Qarterial) and portal vein (Qportal) blood flow, expressed as fractions of total blood flow to the liver, were 0.15 and 0.75, respectively (Brown et al., 1997). The unbound intrinsic clearance of MDZ in the liver is described by eq. 8. 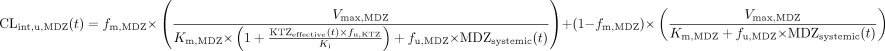 where fm,MDZ is the fraction of MDZ clearance attributed to CYP3A (assumed to be a population mean value of 0.9), fu,MDZ is the unbound fraction of MDZ in the blood, assumed to be a constant 0.04, and Vmax,MDZ and Km,MDZ are the MM constants describing the intrinsic clearance of MDZ. The intrinsic clearance of MDZ was estimated from the population PK model (63.8 l/h). Assuming the in vivo Km,MDZ is in the range of values determined from in vitro systems (1–5 μM), the value of Vmax,MDZ was derived to generate a baseline intrinsic clearance of approximately 60 l/h.
where fm,MDZ is the fraction of MDZ clearance attributed to CYP3A (assumed to be a population mean value of 0.9), fu,MDZ is the unbound fraction of MDZ in the blood, assumed to be a constant 0.04, and Vmax,MDZ and Km,MDZ are the MM constants describing the intrinsic clearance of MDZ. The intrinsic clearance of MDZ was estimated from the population PK model (63.8 l/h). Assuming the in vivo Km,MDZ is in the range of values determined from in vitro systems (1–5 μM), the value of Vmax,MDZ was derived to generate a baseline intrinsic clearance of approximately 60 l/h.
Model Prediction and Validation. The KTZ-MDZ interaction model was used to simulate five published KTZ-MDZ studies. These studies were designed to evaluate the effect of KTZ on MDZ AUC following a single 200-mg dose of KTZ, administered simultaneously or 2 h before MDZ (McCrea et al., 1999), 200 mg of KTZ every 12 h for 2 days, administered simultaneously (Tsunoda et al., 1999) or 12 h before MDZ (Eap et al., 2004), 200 mg of KTZ given once a day for up to 12 days (Lam et al., 2003), and 400 mg of KTZ administered once a day for 4 days (Olkkola et al., 1994). The ratio of MDZ AUC in the presence of KTZ to MDZ AUC in the absence of KTZ was used as a measure of inhibitory response. The simulations were conducted using the Pharsight Trial Simulator 2.2. (Pharsight Inc., Mountain View, CA). The distribution of MDZ AUC ratios under each study scenario was described using a probability density function (S-PLUS 6.2, Professional Pharsight edition; Insightful Corp., Seattle, WA), such that the integrated area under the probability density curve equals 1.
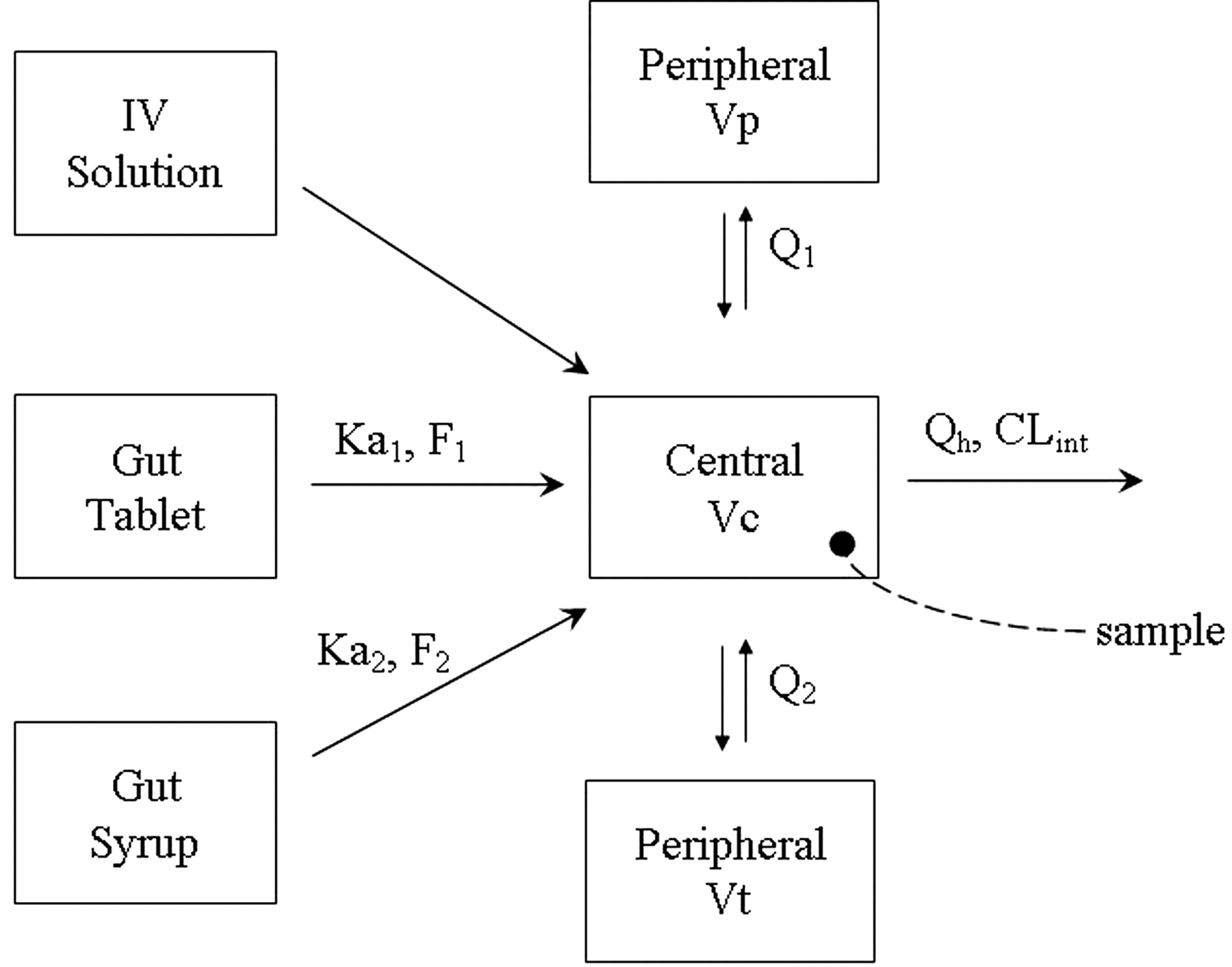
MDZ PK model. Ka1 and F1 are the first-order absorption rate constant and bioavailability from the tablet formulation; Ka2 and F2 are the first-order absorption rate constant and bioavailability from the oral solution formulation; Q1 and Q2 are the distributional clearance (product of intercompartmental rate constant and volume) from the central to the two peripheral compartments, Vp and Vt; Vc is the volume of the central compartment; Qh is the hepatic blood flow; and CLint is the intrinsic clearance of MDZ.
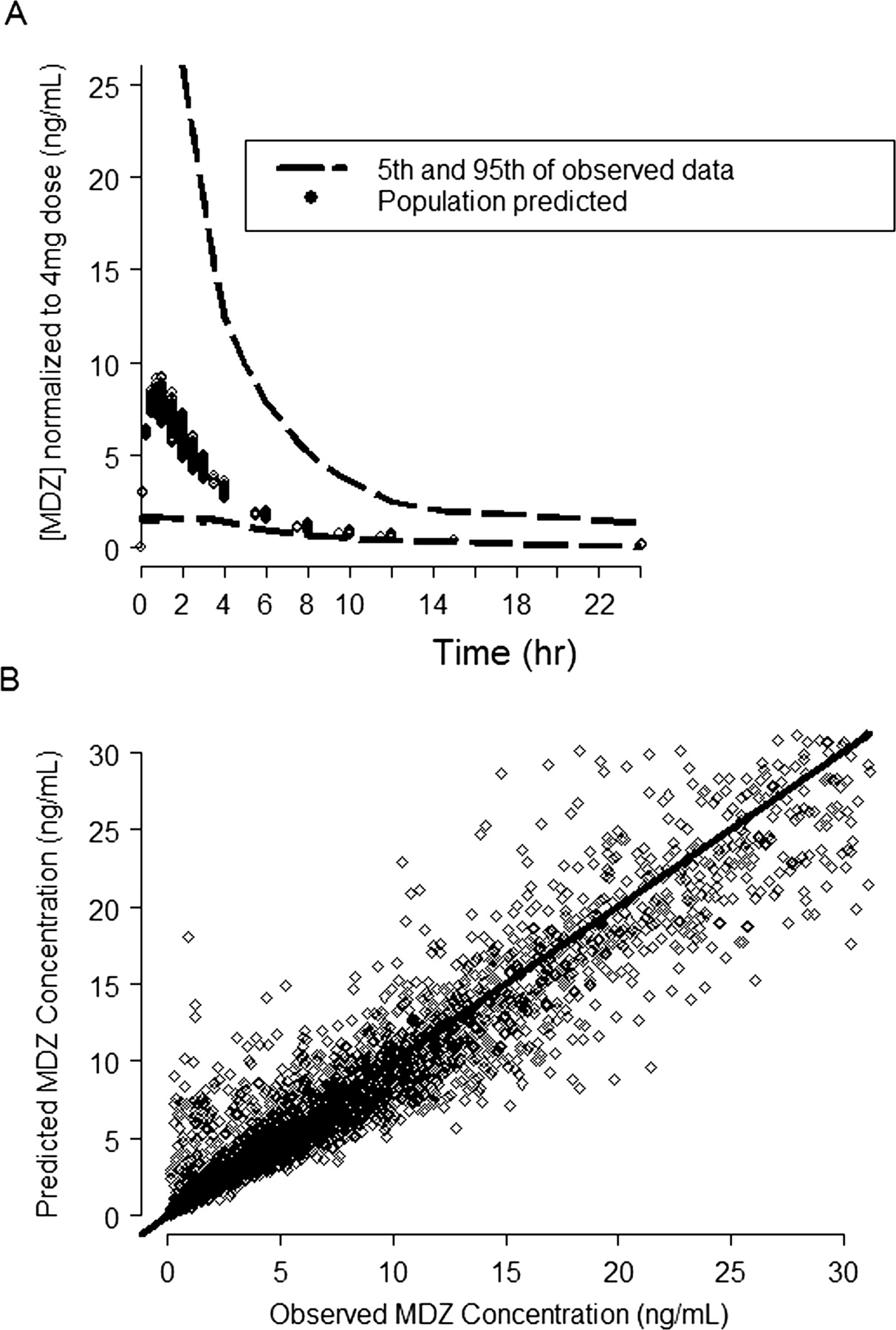
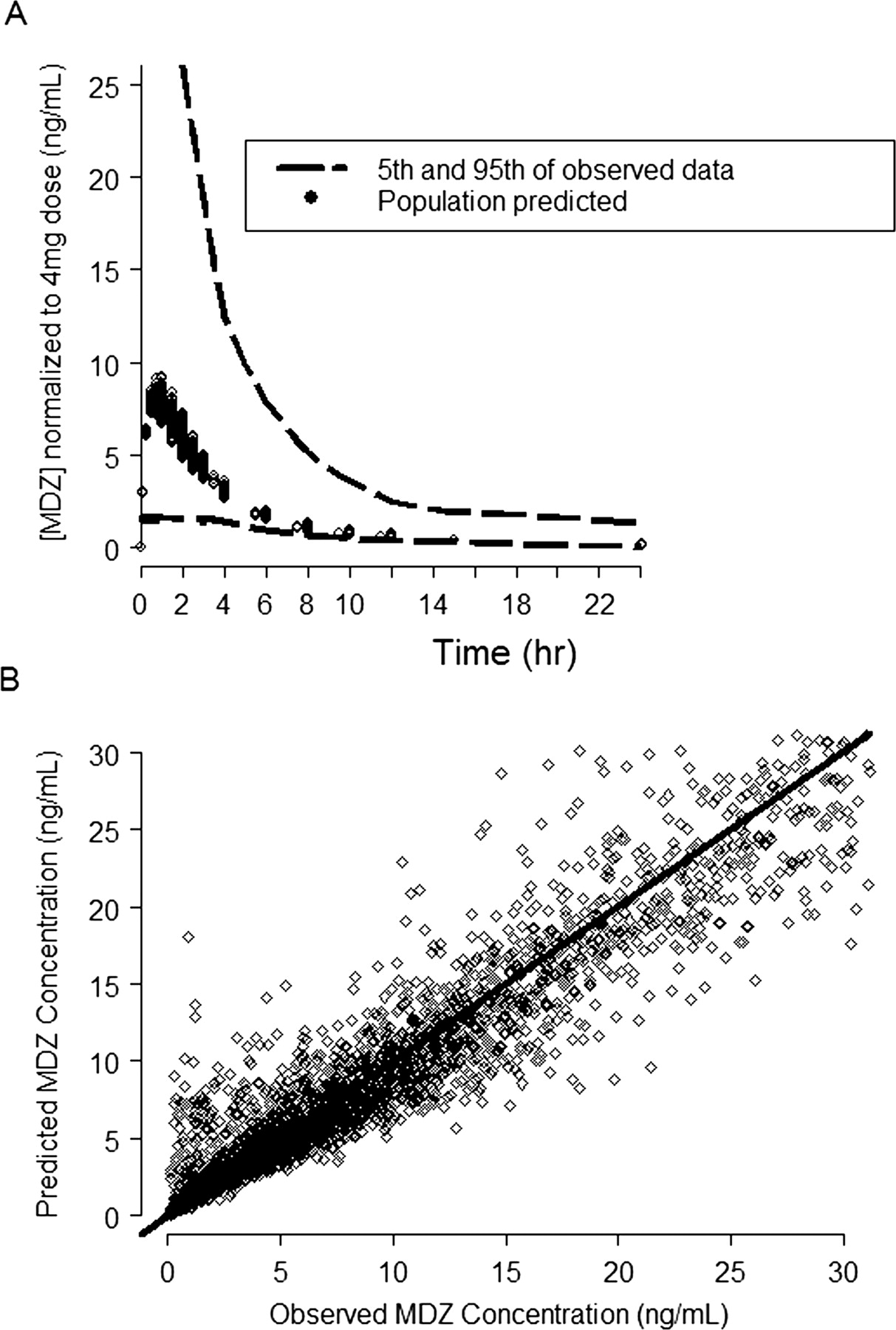
MDZ PK model predicted and observed concentration plots. A, the dashed lines represent the 5th and 95th percentile of the observed MDZ blood concentration (ng/ml) normalized to the 4-mg dose; the symbols represent the predicted mean population concentration from all the individuals in the data set. B, the symbols are the observed versus the model-predicted MDZ concentrations and the solid line represents the line of unity.
Based on the predicted results of the effect of KTZ on MDZ PK, measured in ratios of MDZ AUC, the model parameter with the highest uncertainty, Keff, was updated.
The values of five key model parameters (Ki and Keff for KTZ; fm, Fgut, and baseline intrinsic clearance of MDZ) were varied 10-fold within the simulation environment to rank order the importance of their influences on predicting KTZ inhibition of MDZ clearance.
Results
PK Models. A three-compartment model with first-order absorption using the NONMEM subroutines ADVAN12 and TRANS4 (Kramer et al., 1974; Boeckmann et al., 1994) best described the blood concentrations of MDZ following intravenous and oral administration of MDZ (Fig. 1). Figure 2 demonstrates that the model described the data well. Table 1 lists the blood PK parameter values and the associated intersubject and within-subject variability terms that could be estimated using the model. The effects of sex, age, and body weight were statistically significant in explaining the intersubject variability of MDZ intrinsic clearance and volumes of distribution.
Midazolam population pharmacokinetic parameters
A two-compartment model with first-order absorption using the NONMEM subroutines ADVAN6 and TRANS1 (Kramer et al., 1974; Sheiner and Beal, 1989) and saturable Michaelis-Menten elimination from a presystemic compartment (Fig. 3) was found to adequately describe the plasma PK of KTZ (Fig. 4). In addition, Fig. 4 shows that the model described the data well up to the 600-mg dose but under-predicted the concentrations following the 800-mg dose. Table 2 lists the PK parameter values and the associated “between-study” variability terms estimated based on these mean data. The values of the parameters associated with the intrahepatic enzyme site compartment, shown in Fig. 3, are included in Table 2. These parameters could not be estimated from observed data and, therefore, they were intentionally fixed to very small values, relative to Q2 (plasma flow constant between hepatic and central compartments) and intrinsic clearance, which would result in negligible contribution to overall plasma PK of KTZ. The optimal value of Keff was selected based on the agreement of the simulations with the results of clinical KTZ-MDZ interaction studies conducted using various durations of KTZ dosing (McCrea et al., 1999; Tsunoda et al., 1999; Lam et al., 2003; Eap et al., 2004).
Ketoconazole population pharmacokinetic parameters

KTZ PK model. Ka is the first-order absorption rate constant from the gut to the liver compartment; Vmax and Km are the Michaelis-Menten rate constants for the elimination of KTZ from the liver; K2e represents the first-order rate constant for the uptake of KTZ into the enzyme site compartment; Tmax and Keff are the Michaelis-Menten rate constants for the efflux of KTZ from the enzyme site; Qh is the hepatic plasma flow and Q4 is the central to peripheral intercompartmental flow constant. V4 and V5 are the volume of the central and peripheral compartments, respectively.
The Simulation Model. The MDZ simulation model refined from the descriptive model depicted in Fig. 1 is shown in the top part of Fig. 5. The additional parameters incorporated into the simulation model, with the associated mean values and variability terms, are shown in Table 3. The prediction of KTZ gut, enzyme site, and plasma concentration-time profiles following various KTZ dosing regimens are shown in the bottom part of Fig. 5. The arrows indicate the components of the MDZ model that require the input of KTZ concentrations (shown in equations), thus allowing the link between the two PK models. The concentration of KTZ in the gut drives the change in MDZ availability in the gut. Up to 4 h after KTZ administration, the concentration of KTZ in the gut is sufficiently high to inhibit all MDZ metabolism in the gut, resulting in a transient gut MDZ availability of nearly 100%. However, the amount of KTZ in the gut rapidly declines because of absorption. There was no apparent accumulation in the plasma concentration of KTZ, consistent with the published data from multiple dosing of KTZ (Daneshmend et al., 1983).
Values used in simulation for model parameters not estimated
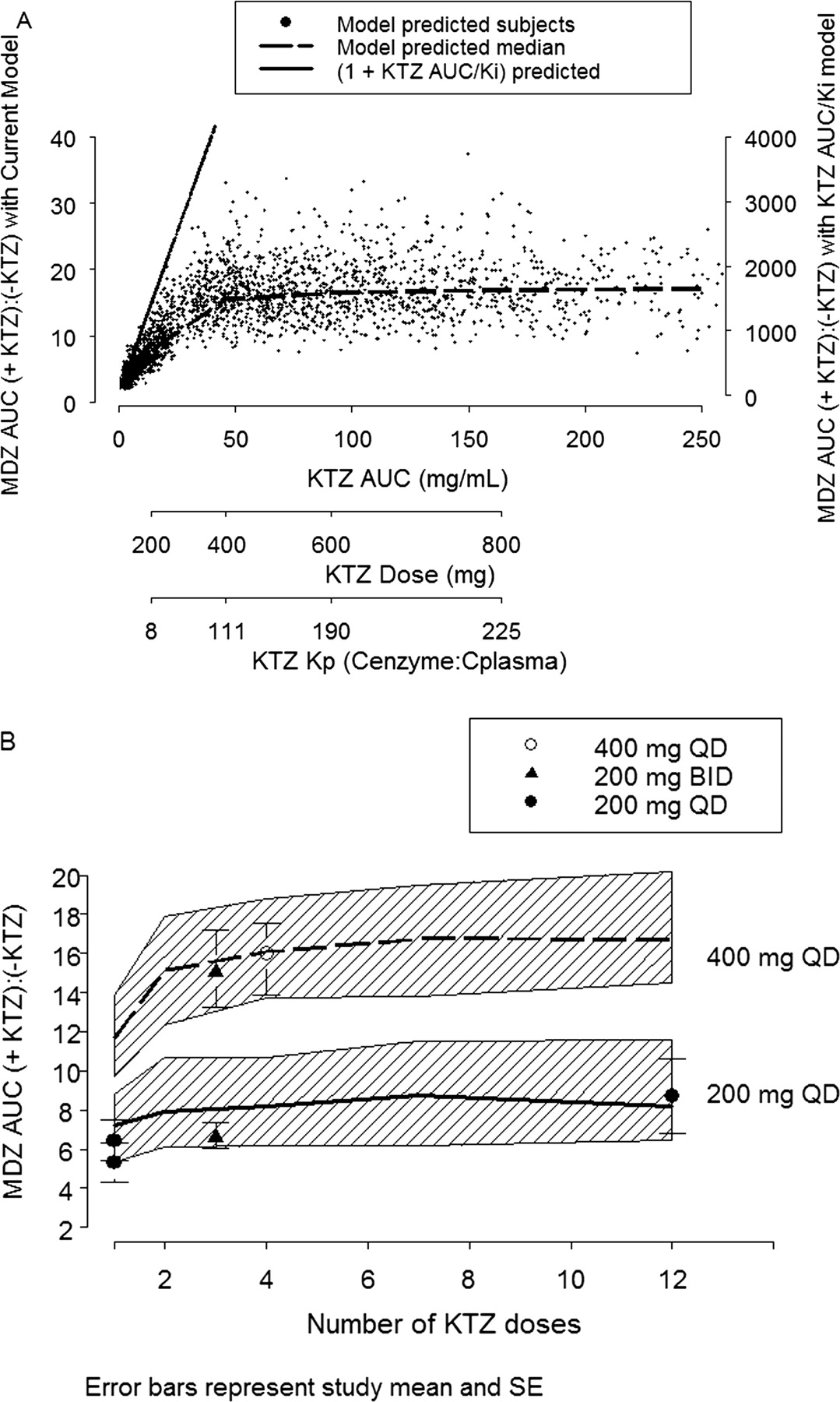
It was observed that the potency of KTZ inhibition varies with the dose of KTZ. A constant Kp value was found to be insufficient to predict the inhibition at the highest dose of KTZ (i.e., 400 mg), using the linear Kp model (eq. 3, relative to the lower dose of KTZ) (i.e., 200 mg). However, eq. 4, describing a first-order influx and saturable efflux process between the hepatic sinusoid and the enzyme site compartments, captures this dose-dependent feature and thus represents a more versatile and biologically plausible model. Consequently, the concentration of KTZ slowly accumulates at the site of the enzyme in the liver following each dose of KTZ; the concentration at the enzyme site drives the inhibition of first-pass hepatic and systemic MDZ clearance. Therefore, the inhibition by KTZ of both first-pass availability and systemic clearance of MDZ was predicted. The accumulation of KTZ at the enzyme site following the 400-mg KTZ q.d. for 12 days is shown in Fig. 5. A partitioning ratio, or Kp, calculated as AUC24 h of KTZ at the enzyme site to AUC24 h of KTZ in the plasma after the 12th KTZ dose, is dose-dependent, as is shown by the decreasing Kp with dose in the x-axis of Fig. 6A.

KTZ PK model predicted and observed concentration plots. A, the symbols are the observed mean concentration versus time from each study and the lines are model-predicted mean KTZ concentration versus time. B, the symbols are the observed versus the model-predicted KTZ concentrations and the solid line represents the line of unity.
The inhibitory response, defined as the ratio of MDZ AUC in the presence of KTZ to MDZ AUC in the absence of KTZ, as a function of increasing KTZ AUC (or dose), is shown in Fig. 6A. The model predicts that, with increasing inhibitor concentration, the MDZ AUC or AUC ratio increases to a maximum limit. In other words, there is a concentration of KTZ beyond which no more inhibition can occur. This is in contrast to the routinely used linear [1 + I/Ki] model (shown as a solid line in Fig. 6A), which predicts that the concentration of the substrate will increase indefinitely with increasing concentration of the inhibitor (Tucker et al., 2001).
The model also predicted that, for KTZ doses greater than 400 mg, the time-averaged (0–24 h) partitioning ratio (bottom axis, Fig. 6A) exceeds 100 and the inhibition reaches maximum response. Therefore, it is of interest to predict how rapidly after the start of KTZ dosing the effect will reach maximum. Figure 6B shows the simulation of MDZ AUC ratios following 200 mg and 400 mg of KTZ administered once a day for 1 to 12 days. The inhibitory response increases and reaches it maximum after the second dose of KTZ for both dosing regimens.
Model Evaluation.Predictive Performance. The results of five published KTZ-MDZ drug-drug interaction studies were used to validate the predictive performance of the model following six different KTZ dosing regimens. The simulations incorporated the variability terms for both MDZ and KTZ PK parameters, the body weight and age range typically observed in these studies, and the same sample size and proportion of male and female subjects. In addition, a 10% coefficient of variation in KTZ Keff was included in the simulations to account for uncertainty in the saturable efflux from the enzyme site. Using these variability and uncertainty terms, the probability that a new KTZ inhibition study would meet a certain acceptable target response (MDZ AUC ratio) for a given dosing regimen of KTZ was calculated. In these studies, KTZ was administered as a 200-mg single dose simultaneously with or 2 h before MDZ (McCrea et al., 1999), 200 mg twice a day for three doses with the last dose given 12 h before MDZ (Eap et al., 2004), 200 mg once a day for 12 days (Lam et al., 2003), 200 mg twice a day for three doses with the last dose given simultaneously with MDZ (Tsunoda et al., 1999), and 400 mg once a day for four doses (Olkkola et al., 1994). The model-predicted distribution of individual MDZ AUC ratios following oral administration of MDZ for each KTZ dosing regimen is shown in Fig. 7. The mean of the observed individual AUC ratios from each study fell within the 5th and 95th percentile of the model-predicted distribution of individual AUC ratios for all studies. The model overpredicted for one study in that individual AUC ratios within 1 standard deviation of the mean (not reported; assumed a typical 30% CV) fell outside the 5th percentile of the model-predicted individual AUC ratios in one study when KTZ and MDZ dosing was staggered by 2 h (Fig. 7B) (McCrea et al., 1999). The model predicted well the 200-mg b.i.d. regimen when dosing is staggered by 12 h (Fig. 7C) (Eap et al., 2004). The model underpredicted the mean and ∼25% of the subjects for the 200-mg b.i.d. regimen (Fig. 7E) (Tsunoda et al., 1999). Figure 8 shows the predictive performance of the model for the mean of each study corresponding to Fig. 7. The means of AUC ratios fell within the 5th and 95th percentiles of the predicted AUC ratios and approximated the central tendency of the distribution of four of six dosing regimens. The model did not predict the mean of the 200-mg single-dose study with a 2-h dosing stagger (Fig. 8B) and the 200-mg b.i.d. regimen (Fig. 8E).
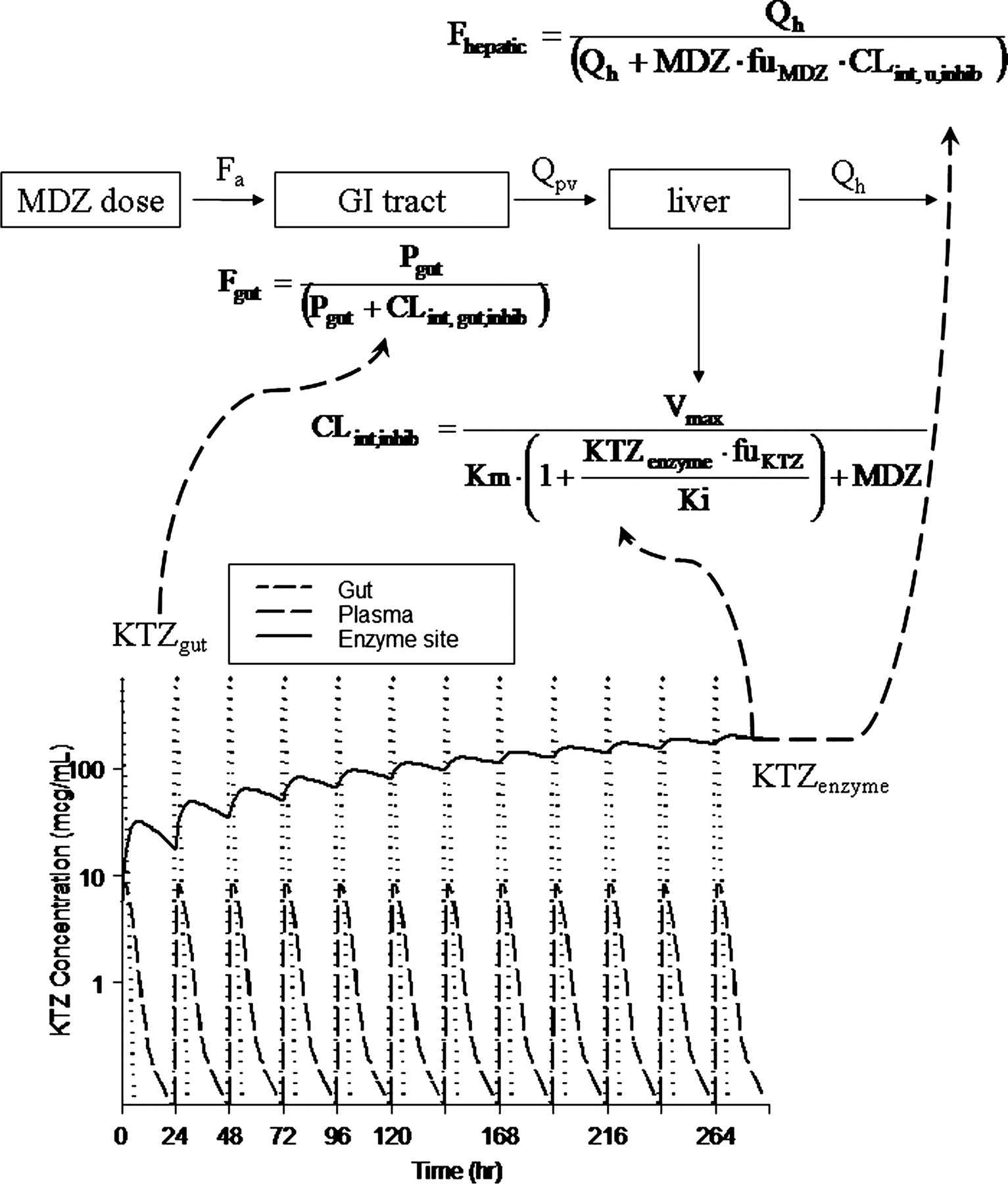
KTZ-MDZ interaction model and prediction of KTZ gut, enzyme site, and plasma concentration-time profiles. Total KTZ inhibits MDZ availability from the gut (Fgut) and unbound KTZ (KTZ · fu) inhibits liver (FLiver) and intrinsic clearance of MDZ in the liver (CLint,u,inhib). The terms are previously defined. Dashed arrows indicate the equations that KTZ concentration in each compartment act upon.

A, model-predicted inhibition of individual MDZ AUC ratios (indicated by +KTZ:–KTZ) versus KTZ AUC or dose. The symbols are 2500 simulated subjects given 100, 200, 400, 600, and 800 mg q.d. KTZ for 12 days with a dose of MDZ given on day 12. The dashed line is the median value of MDZ AUC ratio at each KTZ dose. The solid line is the MDZ AUC ratio predicted based on the [1 + I/Ki] equation (axis label to the right), where I is the plasma AUC of KTZ at each dose, fu is fixed at 0.01, and Ki is fixed at 0.01 μM. The KTZ dose on the second x-axis aligns with the corresponding median AUC of KTZ on the first x-axis. The KTZ partitioning ratio (Kp) on the third x-axis aligns with the corresponding KTZ dose on the second x-axis. B, model-predicted AUC ratios versus number of daily KTZ doses before MDZ dose. The number of KTZ dosing days ranges from 1 to 12 days. The solid and dashed lines are the median predicted AUC ratios for the 200-mg and 400-mg KTZ doses, respectively. The hatched bands are the 5th and 95th interquartile range of the prediction. The symbols and vertical bars are the observed mean and standard error of five clinical studies. The closed circles represent 200 mg q.d. (McCrea et al., 1999; Lam et al., 2003); the closed triangles represent 200 mg b.i.d. (Tsunoda et al., 1999; Eap et al., 2004); the open circles represent 400 mg q.d. (Olkkola et al., 1994).
In summary, the model predicted the reported interaction very well for the simultaneous 200-mg KTZ and MDZ dosing. The model predicted very well for the 200-mg q.d. and 400-mg q.d. KTZ following various dosing duration scenarios. Further refinement of the model may be needed to predict short-interval dosing stagger.
Based on distribution of MDZ AUC ratio as described by the probability density function, the model predicted a less than 3% chance that MDZ AUC ratio would exceed a mean value of 25 (highest -fold AUC reported with a CYP3A-metabolized drug) at 400 mg of KTZ for four doses (Fig. 8F). The model also predicted a small (14%) probability that a new clinical study conducted at the 400-mg q.d. regimen of KTZ would yield a mean MDZ AUC ratio equivalent to the 200-mg q.d. dosing regimen, suggesting that these KTZ regimens are not equivalent. Therefore, the simulations clearly indicated that, despite the fact that 200-mg b.i.d. and 400-mg q.d. dosing regimens result in the same daily dose of 400 mg, the 400-mg q.d. regimen is necessary to obtain maximum inhibition of CYP3A by KTZ after more than 2 days of dosing (Fig. 6B).
Sensitivity Analyses. Sensitivity analyses are particularly critical to the qualification of complex nonlinear models. The sensitivity of inhibitory response (MDZ AUC ratio) to the key model variables (variables determined to be unique to the linked inhibition model) under various study conditions were evaluated through simulations. Figure 9 illustrates the influence that the five key model variables would have on the prediction. There is no published information on the true population distribution of these parameters for drugs predominantly metabolized by CYP3A. However, plausible upper and lower limits (as indicated in parentheses for each variable) for each key variable were predefined, and the ratios of MDZ AUC at the 5th and 95th percentile of the range were calculated from 10,000 subjects in each set of simulations.
The influence of each variable on the predicted AUC ratio is shown following the two different dosing regimens of KTZ (Fig. 9). The solid vertical line depict the mean of AUC ratio, which was predicted using the final model. The horizontal bars depict the range of predicted AUC ratios at parameter values that span a 10-fold range and contain the mean value of each parameter used in the predictions presented earlier. The Ki and Keff values of the inhibitor vary from 0.01 to 0.1 μM and 1 to 10 mg/l, respectively. The fm and initial baseline intrinsic clearance values of the CYP3A substrate vary from 0.1 to 1 and 20 to 200 l/h, respectively. The most influential variable following the 200-mg q.d. and 400-mg q.d. KTZ dosing regimens was the fraction of metabolic clearance attributable to the inhibited pathway (fm). Interestingly, the variable with the greatest model uncertainty, the saturable parameter for the KTZ efflux from the hepatocyte (Keff), was not more influential than Ki at either KTZ dosing regimens. The second most influential parameter after fm is the fraction absorbed through the gut (Fgut); the remaining variables are equally influential within the 10-fold limits. Furthermore, inasmuch as this work is focused on within-subject changes in MDZ AUC, the population covariates (sex, age, and weight) and hepatic blood flow have negligible impact on the predicted inhibitory response (results not shown).
Discussion
The prediction of in vivo drug-drug interactions from in vitro enzyme inhibition data are fraught with false negatives and positives. The prediction of AUC ratio (AUC in the presence of the inhibitor to the absence of the inhibitor) using in vitro determined Ki values along with a single point estimate of systemic inhibitor concentration, originally derived from the well stirred liver model (Rowland and Matin, 1973; Wilkinson and Shand, 1975), has been extensively used in the pharmaceutical industry to assess the risk of drug-drug interactions for new chemical entities (Tucker et al., 2001; Bjornsson et al., 2003). This is a relatively simplistic model that fails to account for a number of important considerations for a priori prediction of drug-drug interactions, including the fraction of drug metabolized by the inhibited pathway, first-pass metabolism at the gut wall, unbound fraction of drug in microsomes and blood, time course of the inhibitor concentration in the portal vein during absorption, and the effective concentration of the inhibitor at the enzyme site. A number of modifications have been proposed to improve the performance of the simplistic approach by accounting for some of these phenomena, including the incorporation of arbitrary scaling factors, liver partition coefficient values, or the use of maximum plasma concentration to minimize underprediction. However, in spite of some notable successes (Yao and Levy, 2002; Ito et al., 2005; Riley et al., 2005), these modifications do not address time-dependent inhibitor concentrations for a system that is inherently nonlinear. This is particularly problematic when the temporal nature of inhibition is important (Yang et al., 2003) and when the distribution or elimination kinetics of the inhibitor are nonlinear. Thus, it was deemed necessary to develop novel and versatile nonlinear models to predict drug-drug interactions with sufficient accuracy and precision to be useful for designing definitive drug-drug interaction studies or to support dosing recommendations.
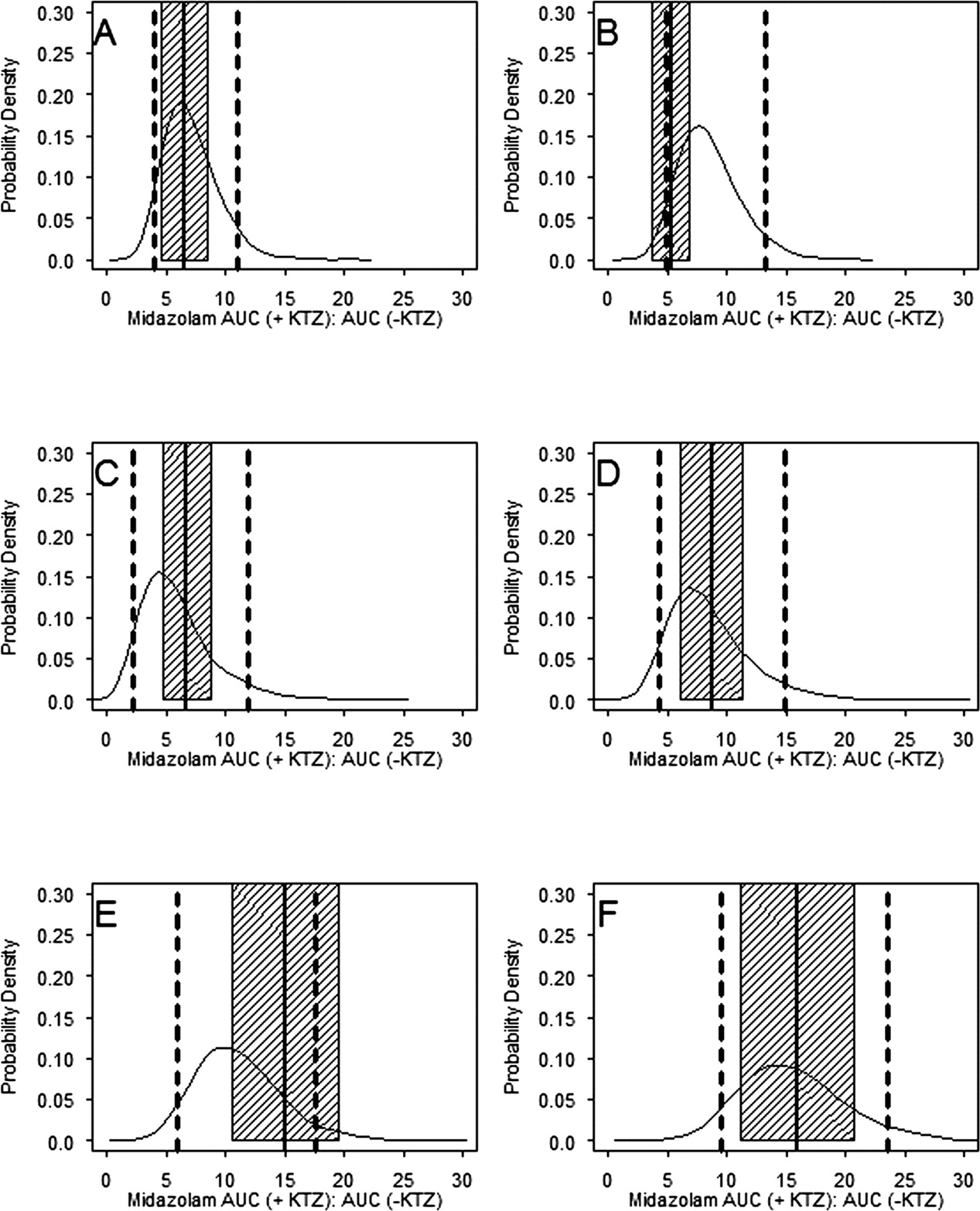
Probability density distribution of the individual subject-predicted ratio of MDZ AUC (indicated by +KTZ:–KTZ). The model-predicted distribution of the MDZ AUC ratio in 400 replicates for KTZ dosing regimens from each of five published studies are shown: A, 200-mg single dose, simultaneously with MDZ (McCrea et al., 1999); B, 200-mg single dose, 2 h before MDZ (McCrea et al., 1999); C, 200 mg every 12 h for three doses, last dose 12 h before MDZ (Eap et al., 2004); D, 200 mg q.d. for 12 days (Lam et al., 2003); E, 200 mg b.i.d. for three doses (Tsunoda et al., 1999); F, 400 mg q.d. for 4 days (Olkkola et al., 1994). The solid probability density curve represents distribution of the MDZ AUC ratio from 4000 subjects. The dashed vertical lines are the 5th and 95th percentile of the population distribution. The solid vertical lines are the literature reported mean of each study. The shaded regions represent 1 standard deviation from the reported means (a typical 30% CV was assumed if S.D. of the ratio was not reported).
Nonlinear models offer the advantage of allowing incorporation of biological processes that may be saturable in clinical dose range. The model presented in this work incorporated several important features that directly address the problems noted above. The model was developed in three parts. Initially, compartmental models, with relevant covariates, characterizing the PK of MDZ and KTZ, were developed using literature data. Literature values of the physiological parameters were incorporated into the model along with estimated or assumed population variability. Second, the models for the inhibitor and substrate were linked through established enzyme kinetic models. Finally, sensitivity analyses and validation of the model were performed to qualify the performance of the model against actual clinical trial data.

Probability density distribution of the study mean ratio of MDZ AUC (indicated by +KTZ:–KTZ). The histograms represent the distribution of the geometric means of 400 trial replicates using the six KTZ dosing regimens from the published studies (referenced in Fig. 7 legend). The dashed vertical lines are the 5th and 95th percentile of the predictions. The solid vertical lines are the literature reported mean of each study.
The prediction of drug-drug interactions following oral administration of agents primarily metabolized by CYP3A is rarely successful because it requires quantitative information or assumptions related to the influence of the highly complex and variable physiology of the first-pass processes on the observed plasma concentrations of the substrate. Complex physiological models are needed to predict the influence of permeability and solubility at the intestinal wall, concentration of the inhibitor and substrate at the intestinal lumen, regional distributions of the drug-metabolizing enzymes and transporters, and the balance between transit time and blood flow, which creates a sink condition against metabolism and efflux (Rostami-Hodjegan and Tucker, 2004; Schmitt and Willmann, 2004; Wu and Benet, 2005). Thus, a practical empirical approach (eq. 5) was used in this work as a preferred alternative to complex physiological models for the gut. The incorporation of the gut wall availability (Fgut) parameter into the model significantly improves the accuracy of predicting AUC following oral administration of a potent inhibitor. The baseline value of Fgut can be calculated indirectly based on data obtained from well designed experiments, as was done in this case for MDZ (Thummel et al., 1996; Gorski et al., 1998). Furthermore, a partition coefficient constant may be used to account for the underprediction of inhibitory effect observed with KTZ when unbound plasma concentration and in vitro Ki values are used. However, this approach assumes an underlying linear process; thus, it cannot be used if there is a dose and time dependence of the inhibitory effect. The incorporation of the saturable efflux process in the KTZ model is a novel approach to account for the persistent and supraproportional increase in inhibitory effect observed with potent inhibitors suspected of nonlinear tissue accumulation. A major advantage of the final linked model is that it accounts for the increase in inhibitory effect that is dependent on the duration and frequency of dosing the inhibitor. However, it is not possible to directly obtain reliable experimental data to assess the rate and extent of accumulation of the inhibitor at the enzyme site. Therefore, simulation of published clinical trials that used different duration and frequency of KTZ dosing can provide an indirect but reasonable range of values for the rate constants (i.e., K2e, Keff, and Tmax) that govern the observed accumulation of effect.
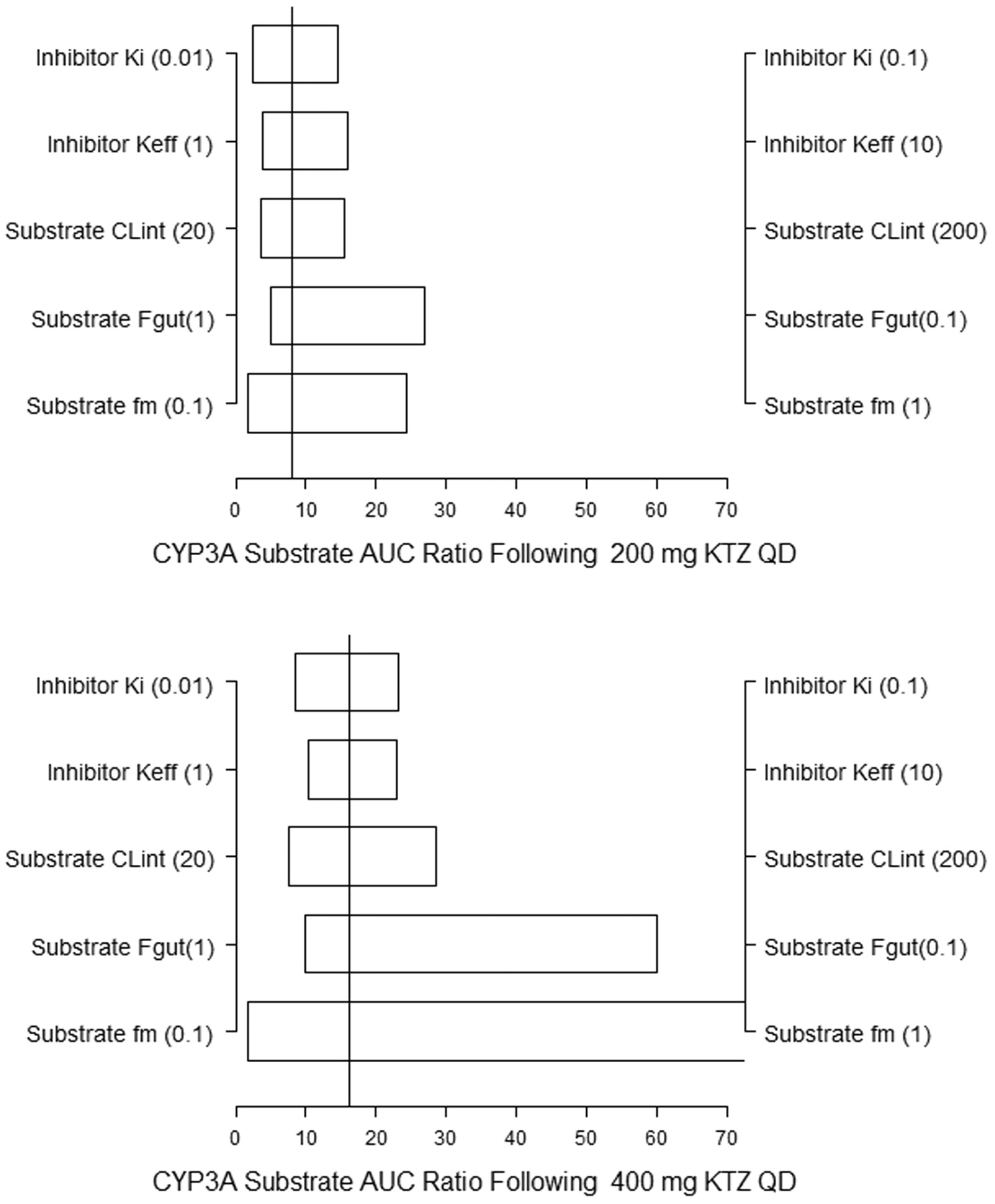
KTZ-MDZ model parameter sensitivity for 200 mg q.d. and 400 mg q.d. KTZ dosing regimens. The five variables, inhibitor Ki and Keff and substrate fm, Fgut, and baseline intrinsic clearance (CLint) are allowed to vary over a 10-fold range for calculation of the AUC ratios: Ki from 0.01 to 0.1 μM, Keff from 1 to 10 mg/l, CLint from 20 to 200 l/h, and fm from 0.1 to 1. The horizontal bars depict the AUC ratio at the 5th percentile of the lower value and the 95th percentile of the higher value (10×). The solid vertical lines are mean predicted AUC ratio using the mean values of the variables from Tables 1, 2, 3.
The success of a prediction is conditional on understanding the variability and capturing the uncertainty of the influential variables in the inhibitory model. Uncertainty in model parameters is common in all nonlinear systems. In general, a wide range of plausible clinical trial outcomes may be expected, depending on the choice of study design, study population, inclusion/exclusion criteria, and dosing regimen. Through stochastic simulation with the model, the impact of various model parameters with a high degree of uncertainty can be assessed (Nestorov et al., 2002; Kato et al., 2003). Thus, another novel and important feature of the current model is the incorporation of pharmacokinetic uncertainty in model estimation. In addition, the sensitivity analyses confirmed that the greatest inhibition for this substrate-inhibitor pair will be in individuals with highest fractional clearances for the substrate via the inhibited pathway (fm). Incorporating fm into the empirical I/Ki equation has been shown to have significant impact on the accuracy of predicting inhibition (Ito et al., 2004, 2005; Ito and Houston, 2005). However, fm is a parameter often associated with uncertainty because there is no convenient and robust approach to predict fm, particularly for a new drug candidate in early development. Thus, a model was developed in which data from in vitro and clinical 14C studies were used to provide reasonable estimates of likely range of fm values, which would be powerful for gaining a thorough understanding of the plausible outcomes of a drug-drug interaction study. Incorporating the entire range of this highly influential parameter in the current model is an important aspect of the prediction. In this example, the prediction of KTZ inhibition of MDZ clearance was presented as a distribution of plausible inhibitory outcomes (Fig. 8), instead of a single point estimate, allowing a projection of the probability of drug-drug interaction meeting a predefined target or threshold. The same rationale applies to all parameters with a high degree of uncertainty.
Another source of uncertainty comes from the reference data set used to develop the PK submodels. The MDZ model was developed using individual data from five studies; therefore, the model may not predict the baseline PK of a study that was not part of the reference data set and the PK parameters of that population fall outside the range estimated from the reference population. This may explain the underprediction observed for one of the studies (Tsunoda et al., 1999). The inhibition of MDZ clearance reported was greater than that reported by the other studies that used a similar 200-mg dosing regimen. It is interesting to note that the MDZ PK submodel under-predicted the clearance of MDZ for the control group of that study population (Greenblatt et al., 2004). Therefore, it may be postulated that the baseline intrinsic PK properties of MDZ may be different for that study population. By contrast, the model overpredicted the results of a study (McCrea et al., 1999) when the dose of KTZ was administered 2 h before MDZ. Further refinement of the model will be necessary to explore the impact of baseline intrinsic clearance of MDZ in different populations and KTZ dosing stagger.
The Ki values obtained using human liver microsomal systems are subject to a number of nonphysiological artifacts. Nonspecific binding of inhibitors and substrates to proteins, lipids, and the incubation vessels has been raised as potential sources of inaccuracy in Ki estimates, particularly for basic drugs. Nonspecific binding of the inhibitor can result in a higher estimate of the inhibitor Ki value and thus may contribute to underprediction of the inhibition. More often than not, in vitro determined Ki values cannot be meaningfully extrapolated to in vivo Ki (IC50). Several investigators have attempted to compare the in vivo calculated Ki values to the in vitro determined Ki values (Ito et al., 2002; Yao and Levy, 2002). The in vitro determined Ki value for KTZ appeared to be adequate for this model-based prediction, once the cumulative inhibitory effect was explained by the saturable efflux process. Understanding the local concentration of the inhibitor is important in establishing congruency between in vitro and in vivo Ki and to further minimize the prediction error.
Drug-drug interactions may alter the dose-response relationship of a pharmaceutical agent underlying its beneficial and adverse effects in patients. Unfortunately, most drug-drug interaction studies are designed in the context of quantitative measures of safety and efficacy, and they provide little practical prescriptive recommendations for optimal therapeutic dosing adjustment in patients. The empirical calculation, generating a point estimate based on the I/Ki ratio alone, has been proposed by the Pharmaceutical Research and Manufacturers of America (PhRMA) working group (Tucker et al., 2001; Bjornsson et al., 2003) as a convenient screening approach for qualitative assessment and rank-ordering of risks. Thus, caution is warranted in the extrapolation of these results in quantitative terms, in the absence of other relevant information to be obtained in the late stages of development. However, a major advantage of the semiphysiologically based PK drug-drug interaction model developed here is that it provides useful dosing recommendations by quantifying changes in exposure of a drug after inhibition of CYP3A by KTZ.
The prediction of drug-drug interaction in the real-world setting often involves the interplay of multiple pathways for clearance, metabolism, and transporters. They are, therefore, complex and difficult to evaluate using only simple empirical approaches. To more accurately predict the drug-drug interaction potential in patients taking multiple medications, the complexity and uncertainty can be initially addressed through simulations. Furthermore, the consequence of neglecting important variables, or incorporating erroneous model parameters, should be quantified through some form of sensitivity analysis as part of risk assessment, as described here.
A valid and meaningful prediction strategy for drug-drug interactions requires a database of in vitro enzyme kinetics and in vivo PK information for an integrated approach to modeling and forecasting. The current work has demonstrated the feasibility and utility of this approach. Thus, a priori planning and designing of experiments to maximize the amount of information learned from each in vitro study and clinical trial are required for thorough model development. Such models will also be used to minimize the risks of exposing healthy volunteers to unnecessarily high concentrations of the experimental agent. The ultimate goals of such modeling exercises are to improve study design, to maximize learning about the new pharmaceutical agent, and to maximize therapeutic benefit in patients through well informed dose or dosing regimen adjustments.
In summary, a drug-drug interaction model was developed and validated, which successfully predicted KTZ inhibition of MDZ for both 200-mg and 400-mg q.d. regimens. Simple models are not able to correctly predict AUC ratio for an “average” systemic KTZ concentration at different doses and following different durations of dosing. The final model developed here may be readily used to predict the magnitude and the range of inhibition by KTZ, at any KTZ dose and dosing frequency, for new drugs that rely on CYP3A for their elimination. The model may require further refinement to address short-interval dosing stagger. Finally, based on these results, 400 mg once a day for at least 3 days is the optimal dosing regimen to use in a definitive drug-drug interaction study to demonstrate maximum inhibition of CYP3A-mediated metabolism by KTZ.
Acknowledgments
We acknowledge all scientists and associates at the Indiana University and the Lilly Research Laboratories who contributed to the various aspects of study conduct, data analyses, and report writing to generate valuable data and information required for model development. Special thanks to Jeanne Geiser for technical assistance with clinical trial simulations, and Dr. Shiew-Mei Huang and Bob Temple of the Food and Drug Administration for providing valuable input in support of the modeling and simulation project.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008730.
-
ABBREVIATIONS: PK, pharmacokinetic(s); NONMEM, nonlinear mixed effects modeling; KTZ, ketoconazole; MDZ, midazolam; AUC, area under the concentration-time curve; MM, Michaelis-Menten; Tmax, MM constant of efflux; Keff, MM constant of saturable efflux; Ki, inhibitory constant; fm, fraction of clearance metabolized by a specific enzyme or enzyme system; fu, fraction of drug not bound to protein; Kp, partition coefficient constant.
- Received December 1, 2005.
- Accepted April 5, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}