


Abstract
The glucuronidation of 17β-estradiol (β-estradiol) and 17α-estradiol (epiestradiol) was studied to elucidate how the orientation of the 17-OH group affects conjugation at the 3-OH or the 17-OH of either diastereomer. Recombinant human UDP-glucuronosyltransferases (UGTs) UGT1A1, UGT1A3, UGT1A7, UGT1A8, and UGT1A10 conjugated one or both diastereomers, mainly at the 3-OH. The activity of UGT1A4 was low and unique because it was directed merely toward the 17-OH of both aglycones. UGT1A10 exhibited particularly high estradiol glucuronidation activity, the rate and affinity of which were significantly higher in the case of β-estradiol than with epiestradiol. UGT1A9 did not catalyze estradiol glucuronidation, but UGT1A9-catalyzed scopoletin glucuronidation was competitively inhibited by β-estradiol. UGT2B4, UGT2B7, and UGT2B17 exclusively conjugated the estradiols at the 17-OH position in a highly stereoselective fashion. UGT2B4 was specific for epiestradiol; UGT2B7 glucuronidated both diastereomers, with high affinity for epiestradiol, whereas UGT2B17 only glucuronidated β-estradiol. UGT2B15 glucuronidated both estradiols at the 3-OH, with a strong preference for epiestradiol. Human UGT2A1 and UGT2A2 glucuronidated both diastereoisomers at both hydroxyl groups. Microsomal studies revealed that human liver mainly yielded epiestradiol 17-O-glucuronide, and human intestine primarily yielded β-estradiol 3-O-glucuronide, whereas rat liver preferentially formed β-estradiol 17-O-glucuronide. Of the three recombinant rat UGTs that were examined in this study, rUGT2B1 was specific for the 17-OH of β-estradiol, rUGT2B2 did not catalyze estradiol glucuronidation, whereas rUGT2B3 exhibited high activity toward the 17-OH in both diastereoisomers. The results show that although many UGTs can catalyze estradiol glucuronidation, there are marked differences in their kinetics, regioselectivity, and stereoselectivity.
UDP-glucuronosyltransferases (EC 2.4.1.17; UGTs) are important conjugation enzymes that catalyze the glucuronidation of a large number of xenobiotics and physiological compounds. In the glucuronidation reaction, the glucuronic acid moiety is transferred from UDP-glucuronic acid (UDPGA) to the aglycone substrate. The aglycone substrates are mainly small lipophilic molecules, and conjugation with glucuronic acid increases their water solubility and accelerates their excretion through bile or urine (King et al., 2000; Tukey and Strassburg, 2000; Wells et al., 2004). The UGTs are membrane proteins of the endoplasmic reticulum that are expressed in the liver and many other tissues, including intestine, kidney, breast, placenta, testis, and prostate (King et al., 2000; Tukey and Strassburg, 2000).
The UGTs are divided into three subfamilies: UGT1A, UGT2A, and UGT2B (Mackenzie et al., 2005). The UGT1 gene locus encodes the nine members of the UGT1A subfamily: UGT1A1 and UGT1A3 through UGT1A10 (Gong et al., 2001; Mackenzie et al., 2005). All these enzymes share exons 2 through 5; therefore, the amino acid sequence of their C-terminal half is identical. The first exons are specific for the individual UGT1As, and they encode the variable N-terminal domains of the proteins. Nonetheless, there are high homologies also among the N-terminal domains of many UGT1As, particularly among UGT1A3 through UGT1A5 and UGT1A7 through UGT1A10. Within the UGT2A subfamily, there is probably exon sharing between UGT2A1 and UGT2A2 that leads to identical C-terminal halves. In UGT2A3 or among the UGT2Bs there is no exon sharing, and each enzyme is encoded by a separate gene that contains all the exons (Mackenzie et al., 2005). The human UGT2B subfamily contains seven members: UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15, UGT2B17, and UGT2B28 (Lévesque et al., 2001; Turgeon et al., 2001). UGT2B15 and UGT2B17 are 90% identical, whereas the percent identity among the other UGT2B isoforms is 70 to 79% (Tukey and Strassburg, 2001).
UGT substrate specificity is complex as many UGTs can glucuronidate several different compounds that vary significantly in their chemical structure. There is often a partial overlap in the substrate specificity of many human UGTs, perhaps reflecting the high sequence homology among them. On the other hand, there are also sharp differences in the activity even between highly similar isoforms such as UGT1A3 and UGT1A4, UGT1A9 and UGT1A10, or UGT2B15 and UGT2B17 (Dubois et al., 1999; Sten et al., 2006; Kubota et al., 2007). One of the major challenges in current UGT research is to better understand the factors that determine the substrate specificity of the different UGTs to be able to predict the glucuronidation of new compounds. Few pharmacophore-based models for the prediction of substrate specificity of individual UGTs have already been developed (Smith et al., 2003a,b; summarized by Miners et al., 2004), but there is a clear need for additional detailed information on the preference of these enzymes with respect to regioselective and stereoselective conjugation.
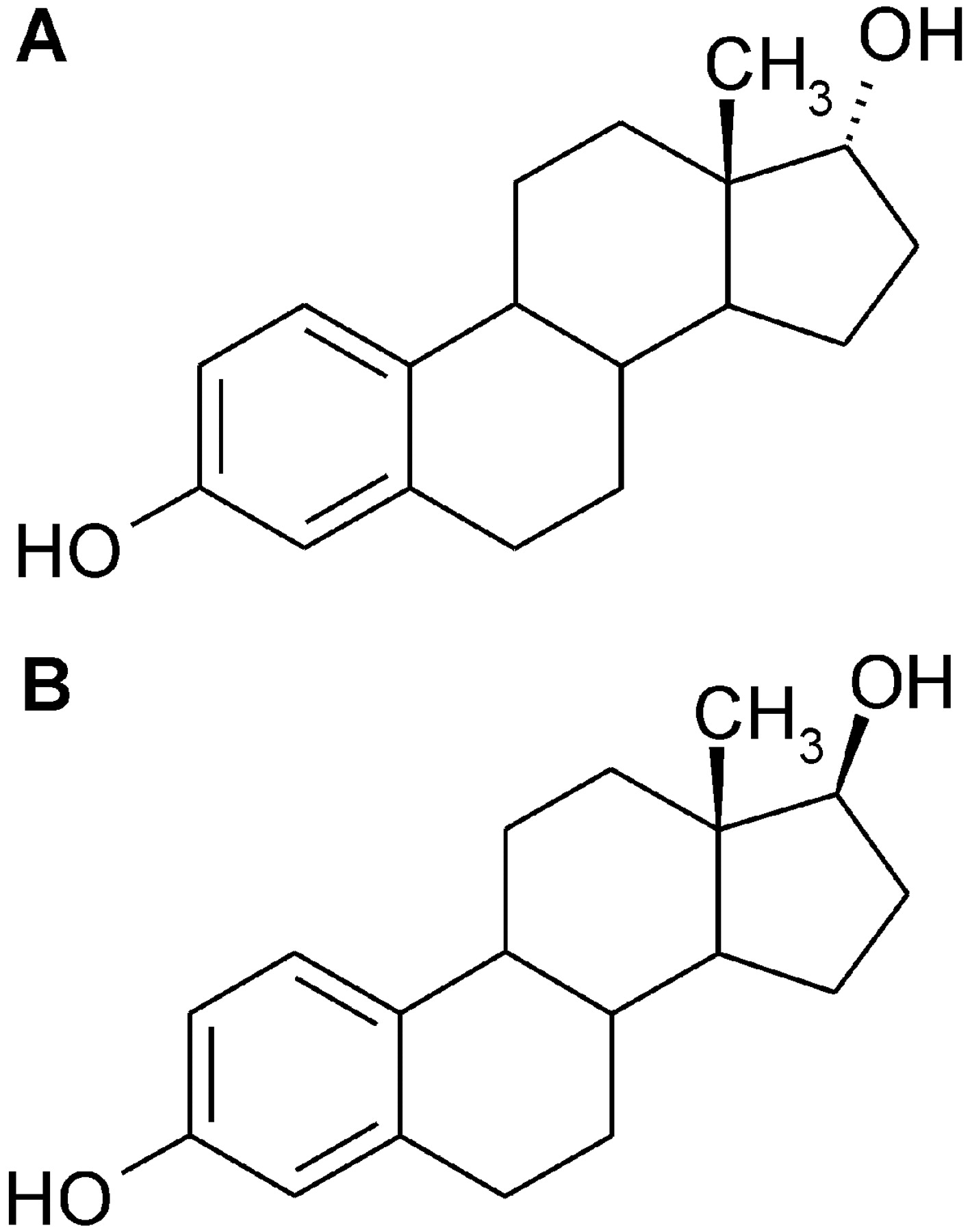
Chemical structures of epiestradiol (A) and β-estradiol (B).
17β-Estradiol (β-estradiol) is an important endogenous estrogen in humans that is also used clinically for the treatment of hormonal disorders in women, particularly hormone replacement treatment. Its xenobiotic diastereoisomer, 17α-estradiol or epiestradiol (Fig. 1), is of minor significance as its affinity for estrogen receptors is low compared with β-estradiol (reviewed by Dykens et al., 2005). Nevertheless, epiestradiol was found to have protective effects against toxic stressors in cells that are independent of estrogen receptors (Morin et al., 2002; Dykens et al., 2005). Dykens et al. (2005) have even suggested that epiestradiol could be a potential drug candidate for Alzheimer's and Parkinson's diseases. After p.o. administration most of the epiestradiol is rapidly conjugated (Hobe et al., 2002), and conjugated forms of β-estradiol are readily detectable in blood (Raftogianis et al., 2000). In estrogen target tissues, such as breast, β-estradiol is extensively converted by cytochrome P450, catechol-O-methyltransferase, sulfotransferases, and UGT enzymes before being released into the circulation (Guillemette et al., 2004). Hydroxylated and methoxylated derivatives are further conjugated by UGTs to glucuronides that are usually considered inactive. However, there were also reports that β-estradiol 17-O-glucuronide may be a toxic metabolite that causes cholestasis (Meyers et al., 1980; Mackenzie et al., 1996).
Estrogens can be glucuronidated by many human UGTs, and the glucuronidation of physiological estrogens, including β-estradiol, by individual UGTs was recently examined (Gall et al., 1999; Lépine et al., 2004; Starlard-Davenport et al., 2007). None of the studies, however, included all the human UGTs. Moreover, the glucuronidation of epiestradiol, the xenobiotic diastereoisomer of β-estradiol, has not yet been explored. Like previous studies with C19- and C21-hydroxysteroids (Jin et al., 1997; Bowalgaha et al., 2007), characterization of the glucuronidation of diastereoisomeric estrogens potentially provides important insights into structure-activity relationships of this class of compounds and the regioselectivity and stereoselectivity of human UGTs.
Materials and Methods
Materials. 17α-Estradiol, 17β-estradiol, 17β-estradiol-β-d-glucuronides (sodium salt), scopoletin, UDPGA trisodium salt, and d-saccharic acid 1,4-lactone were purchased from Sigma-Aldrich (St. Louis, MO). Sodium dihydrogen phosphate dihydrate and disodium hydrogen phosphate were from Fluka (Buchs, Switzerland and Steinheim, Germany). Radiolabeled [14C]UDPGA was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Pooled human liver and intestinal (from duodenum and jejunum) microsomes were purchased from BD Gentest (Woburn, MA), and male New Zealand rabbit liver microsomes were from In Vitro Technologies (Baltimore, MD). Rat liver microsomes were prepared in our laboratory from Aroclor 1254-induced male Sprague-Dawley rats and pig, bovine, and elk liver microsomes from untreated animals, as previously described (Luukkanen et al., 1997).
Recombinant human UGTs were expressed in baculovirus-infected insect cells as previously described (Kurkela et al., 2003, 2007). In recent studies we have noticed that our recombinant human UGT2B15 preparation contains a rather low level of active enzyme (M. Finel, unpublished observation); therefore, recombinant human UGT2B15 was also purchased from BD Gentest. The cDNAs for rat rUGT2B1 through rUGT2B3 were expressed in the same baculovirus system that was used for the human UGTs, including the addition of a short C-terminal fusion peptide that ends with six His residues (His-tag) (Kurkela et al., 2003). The cDNAs for the rat UGT2B1 and UGT2B2 were isolated from an expression library as previously described (Mackenzie et al., 1984, 1986a,b). The cDNA of rUGT2B3 was isolated by reverse transcription-polymerase chain reaction from total liver RNA of Wistar rat. The sense primer upstream to the first ATG included an Xba1 site, and the antisense primer downstream from the stop codon contained an Sph1 site. The amplified full-length DNA was subcloned as an Xba1-Sph1 fragment into the pUC118 vector and sequenced in both directions. The cDNAs for the three rat UGTs were transferred to the modified shuttle vector pFBXHA following insertion of a Sal1 restriction site just upstream to the original stop codon by polymerase chain reaction. Thereafter, virus preparation and protein production were performed as previously described (Kurkela et al., 2003). The relative expression levels of all the recombinant UGTs, with the exception of commercial UGT2B15, were determined by dot-blot analyses using anti-His-tag antibodies (Kurkela et al., 2004, 2007).
Screening of Microsomes and Recombinant UGTs. The activity of 19 human recombinant UGTs and three rat recombinant UGTs toward the two estradiol diastereomers, β-estradiol and epiestradiol, was initially measured using three protein concentrations (40, 100, and 200 μg/ml) and three aglycone concentrations (1, 10, and 100 μM). The samples were incubated for 30 min at 37°C as described below. Glucuronidation of the estradiol diastereomers by human intestinal microsomes and human, rat, rabbit, pig, bovine, and elk liver microsomes were studied at a substrate concentration of 100 μM. The protein concentration in the incubations was between 0.4 and 3.0 mg/ml, and the incubations were carried out in duplicate at 37°C for 60 min.
UGT Activity Assays. Recombinant UGTs that exhibited activity toward either estradiol diastereoisomer in the initial screening were subjected to kinetic analyses. All the enzyme assays, performed in triplicate, contained 1 mM UDPGA, 5 mM saccharolactone, 50 mM phosphate buffer, pH 7.4, 5 mM MgCl2, and 5% dimethyl sulfoxide in total volume of 250 μl. The aglycone concentrations in the kinetic analysis assays were selected using approximate Km values from the initial screening. Hence, for each UGT we have selected eight different β-estradiol and eight different epiestradiol concentrations, all of which were in the range of 0.5 and 300 μM. In most cases, the aglycone concentration range was at least from 0.3 to 5 times the Km value of the tested UGT, but in few cases the range was more narrow because of a solubility problem at substrate concentrations over 300 μM and detection limitation at very low substrate concentrations. The incubation time and protein concentration varied between enzymes and aglycones, from 10 to 45 min and from 8 to 200 μg/ml, respectively, and they were within the linear range for the tested UGT. The glucuronidation reactions were terminated by adding 25 μl of chilled 4 M perchloric acid and cooling the tubes in a cold block. The precipitated proteins were removed by centrifuging the samples for 5 min at 16,100g, and a 50-μl aliquot of the supernatant fraction was injected into the high-performance liquid chromatography (HPLC).
Inhibition of UGT1A9. Inhibition of UGT1A9 by epiestradiol and β-estradiol was studied using scopoletin as the aglycone. The concentration of scopoletin was 25 μM, which is close to its Km with UGT1A9 (see under Results). The estradiol concentrations were 5, 25, and 150 μM, and the protein concentration was 40 μg/ml. The samples were incubated at 37°C for 15 min; the reaction was terminated with 4 M perchloric acid; and an 80-μl aliquot of the supernatant fraction was injected into the HPLC.
To determine the Ki and the mode of inhibition by β-estradiol, the kinetics of scopoletin glucuronidation was studied in the absence and presence of three concentrations of β-estradiol (5, 10, and 20 μM). Seven scopoletin concentrations, between 5 and 500 μM, were used, and all the samples were prepared in duplicate. The data were fitted to the Michaelis-Menten equation and to the competitive inhibition equation (global fit) using GraphPad Prism version 4.03 for Windows (GraphPad Software Inc., San Diego, CA).
Quantification of Estradiol Glucuronides. β-Estradiol glucuronides were quantified using authentic standards. Seven concentrations between 10 and 1000 nM were used to construct calibration curves, and the linearity was excellent for both the 3-O- and 17-O-glucuronides (R2 > 0.99). For epiestradiol glucuronides, for which authentic standards were not available, radiochemical and fluorescence detection was combined to quantify these metabolites. Samples were incubated in the presence of 6 μM radiolabeled [14C]UDPGA and 50 μM unlabeled UDPGA for 60 min. The appropriate amount of human liver microsomes or recombinant UGT1A10 was used to catalyze the glucuronidation of epiestradiol. Following centrifugation, aliquots, 2 to 50 μl, of the supernatant fraction were injected into two different HPLC systems, which were separately equipped with radioactivity and fluorescence detectors. First, the amount of glucuronides formed during the incubation was determined by HPLC coupled with a radioactivity detector and calculated using the following equation: 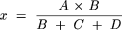 where x is the amount of one of the glucuronides formed (picomoles), A is the amount of UDPGA (labeled + unlabeled) added to the reaction mixture before incubation (picomoles), B is the peak area of the glucuronide under examination, C is the peak area of the other glucuronide formed, and D is the UDPGA peak area in the radioactivity detector (Kaivosaari et al., 2001). Same amounts of the same samples were subsequently injected to the HPLC with fluorescent detector, and the linearity of each glucuronide standard curve was determined. The linearity of the epiestradiol 17-O-glucuronide standards determined this way was good (R2 > 0.9), and that of epiestradiol 3-O-glucuronide was acceptable (R2 > 0.7). The reaction products were proven to be glucuronides because they gave signals on the radioactivity detector. The epiestradiol glucuronides were well separated by the HPLC, and they were assumed to elute from the column in the same order as the respective β-estradiol glucuronides. The latter assumption was supported by the type of glucuronides formed by most UGTs of subfamily 1A (see Discussion).
where x is the amount of one of the glucuronides formed (picomoles), A is the amount of UDPGA (labeled + unlabeled) added to the reaction mixture before incubation (picomoles), B is the peak area of the glucuronide under examination, C is the peak area of the other glucuronide formed, and D is the UDPGA peak area in the radioactivity detector (Kaivosaari et al., 2001). Same amounts of the same samples were subsequently injected to the HPLC with fluorescent detector, and the linearity of each glucuronide standard curve was determined. The linearity of the epiestradiol 17-O-glucuronide standards determined this way was good (R2 > 0.9), and that of epiestradiol 3-O-glucuronide was acceptable (R2 > 0.7). The reaction products were proven to be glucuronides because they gave signals on the radioactivity detector. The epiestradiol glucuronides were well separated by the HPLC, and they were assumed to elute from the column in the same order as the respective β-estradiol glucuronides. The latter assumption was supported by the type of glucuronides formed by most UGTs of subfamily 1A (see Discussion).
HPLC Analyses. The formation of estradiol glucuronides was analyzed by HPLC (Shimadzu, Kyoto, Japan) with fluorescence detection (excitation 216 nm, emission 316 nm) using a Chromolith SpeedRod RP18e column (50 × 4.6 mm) (Merck, Darmstadt, Germany). The mobile phase consisted of 50% 25 mM phosphate buffer, pH 3.0, and 50% methanol, and the flow rate was 1 ml/min. The column temperature was 30°C. The retention times were (mean ± S.D.): 2.4 ± 0.05 min for epiestradiol 3-O-glucuronide, 4.2 ± 0.17 min for epiestradiol 17-O-glucuronide, 2.2 ± 0.04 min for β-estradiol 3-O-glucuronide, and 3.0 ± 0.04 min for β-estradiol 17-O-glucuronide. The retention times of β-estradiol and epiestradiol were 7.6 and 8.3 min, respectively.
Radioactive samples, used for quantification of epiestradiol glucuronides, were analyzed using two separate HPLC systems. The Shimadzu equipment combined with fluorescence detector was used as described above, and an Agilent 1100 series HPLC (Agilent Technologies, Waldbronn, Germany) combined with a radioactivity detector (model 9701; Reeve Analytical, Glasgow, UK) was also used. In the latter case, the mobile phase consisted of 55% 25 mM phosphate buffer, pH 3.0, and 45% methanol. Two- to 50-μl aliquots of the supernatant fraction of centrifuged samples were injected into each HPLC systems.
The formation of scopoletin glucuronide by UGT1A9 was analyzed using the Agilent 1100 series instrument fitted with a Chromolith SpeedRod column and fluorescence detector (excitation 335 nm, emission 455 nm). The mobile phase was 90% 50 mM phosphate buffer, pH 3.0, and 10% methanol. The initial flow rate was 0.8 ml/min for 5 min, which was then increased to 2.5 ml/min for an additional 6 min. The retention times of scopoletin glucuronide and scopoletin were 4.6 and 8.9 min, respectively. The column temperature was 40°C.
Kinetic Analyses. Kinetic constants were estimated by fitting the experimental data to either the Michaelis-Menten equation [v = Vmax × S/(Km+ S), where v is the initial velocity of the reaction, S is the substrate concentration, Vmax is the maximal velocity, and Km is the substrate concentration at 0.5 Vmax], substrate inhibition equation [v = Vmax × S/(Ks + S + S2/Ki), where Ki is the constant describing the substrate inhibition interaction], or the Hill equation [v = Vmax × Sn/(KAn + Sn), where KA is the substrate concentration at 0.5 Vmax] using nonlinear least-squares regression (GraphPad Prism version 4.03 for Windows, GraphPad Software Inc. or EnzFitter; Biosoft, Cambridge, UK). Goodness of fit was evaluated by considering the randomness of the residuals, R2, and standard error estimates, and the best equation to describe the data was selected after that. To be able to compare the results of different enzymes, the initial velocity data and thereby also the Vmax values were normalized for the relative expression of recombinant UGTs. The expression level of UGT1A1 was given the reference value 1, and those of the other enzymes were proportioned to that.
Results
The analyses of β-estradiol and epiestradiol glucuronidation at both the 3-OH and 17-OH sites by 19 human and three rat recombinant UGTs were carried out in two rounds. First, all the enzymes were assayed with both aglycones, as described under Materials and Methods. The UGTs that did not exhibit detectable activity were the human UGT1A5, UGT1A6, UGT1A9, UGT2A3, UGT2B10, UGT2B11, and UGT2B28, as well as the rat UGT2B2. The UGTs that catalyzed the glucuronidation of at least one of the estradiol diastereoisomers were subjected to kinetic analyses. The kinetic curves for the different UGTs are presented in Figs. 2 through 5, and the derived kinetic constants, where applicable, are listed in Table 1. The reactions generally followed either Michaelis-Menten or substrate inhibition kinetics. There were several exceptions, however, where the initial velocities data could not be fitted to any equation, e.g., glucuronidation of epiestradiol by UGT1A8, UGT2A2, and UGT2B7 and glucuronidation at the 3-OH of β-estradiol by UGT2B15 (Figs. 2, 3, 4, 5; Table 1). The glucuronidation of β-estradiol by UGT2A2 was the only reaction that was best-fitted to the Hill equation, but also in this case substrate inhibition was obvious at substrate concentrations above 150 μM.
The enzyme kinetic parameters of glucuronidation of epiestradiol and β-estradiol
The Km, Ks, KA, Ki, and n values are best-fit values ± S.E.
UGT1A1 exhibited prominent regioselectivity toward the 3-OH of both substrates, whereas glucuronidation at the 17-OH position was not detected. However, the rate of glucuronidation at the 3-OH was dependent on the configuration of the 17-OH so that UGT1A1 catalyzed β-estradiol glucuronidation about 6 times faster than epiestradiol glucuronidation (Fig. 2). Nevertheless, the kinetic analyses indicate that the Clint values of UGT1A1 for both diastereoisomers were similar because of higher affinity but lower Vmax value for epiestradiol compared with β-estradiol.
UGT1A3 revealed clear regioselectivity for the 3-OH position in both substrates but also conjugated the two estradiols at the 17-OH position, albeit at low rates. Similar to UGT1A1, the α-orientation of the 17-OH in the substrate leads to lower Ks value in UGT1A3 when glucuronidation at the 3-OH position is compared (Table 1). In contrast to UGT1A1, however, the Vmax values of UGT1A3 for epiestradiol and β-estradiol were similar, resulting in a higher Clint value for epiestradiol (Table 1).

Glucuronidation of estradiol diastereomers by recombinant human UGTs of subfamily UGT1A. The normalized initial rates were fitted to either Michaelis-Menten or substrate inhibition equation. See Table 1 for kinetic constants.
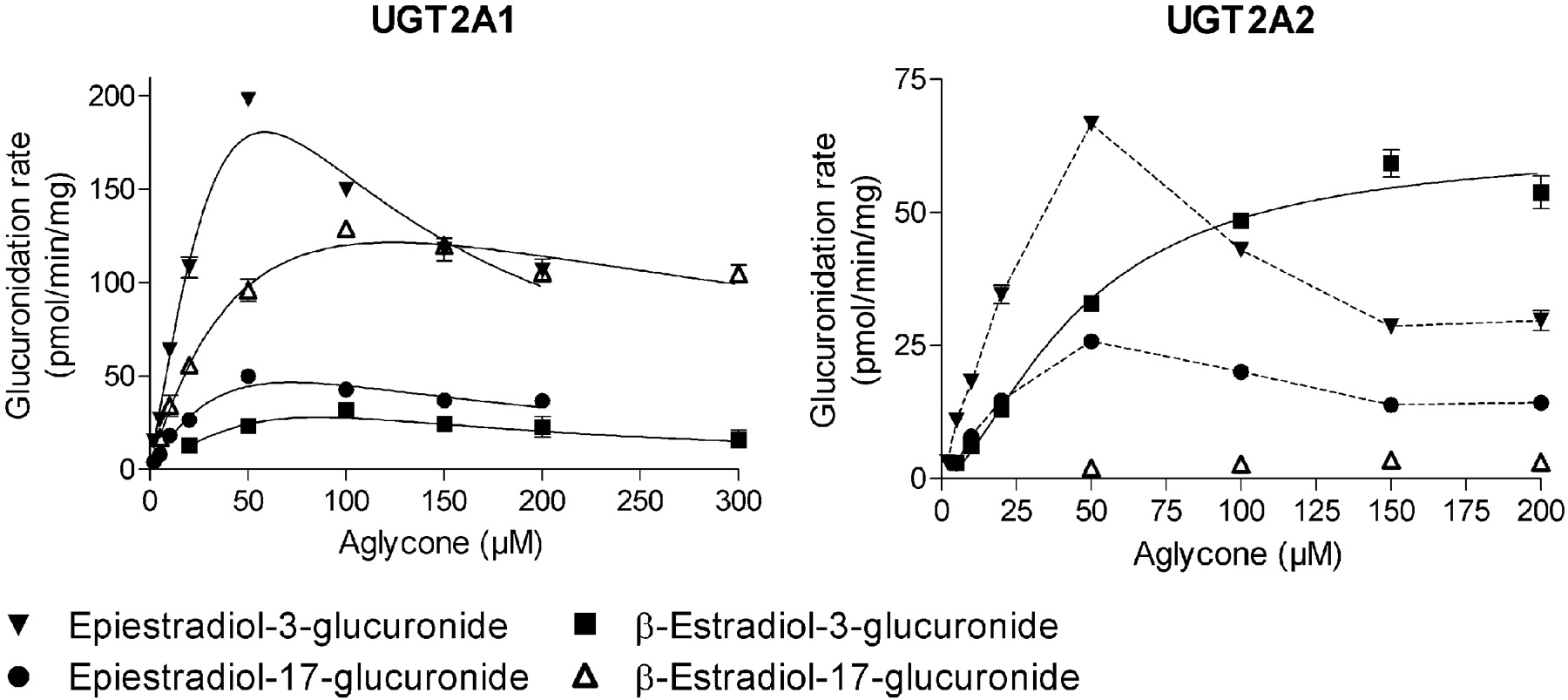
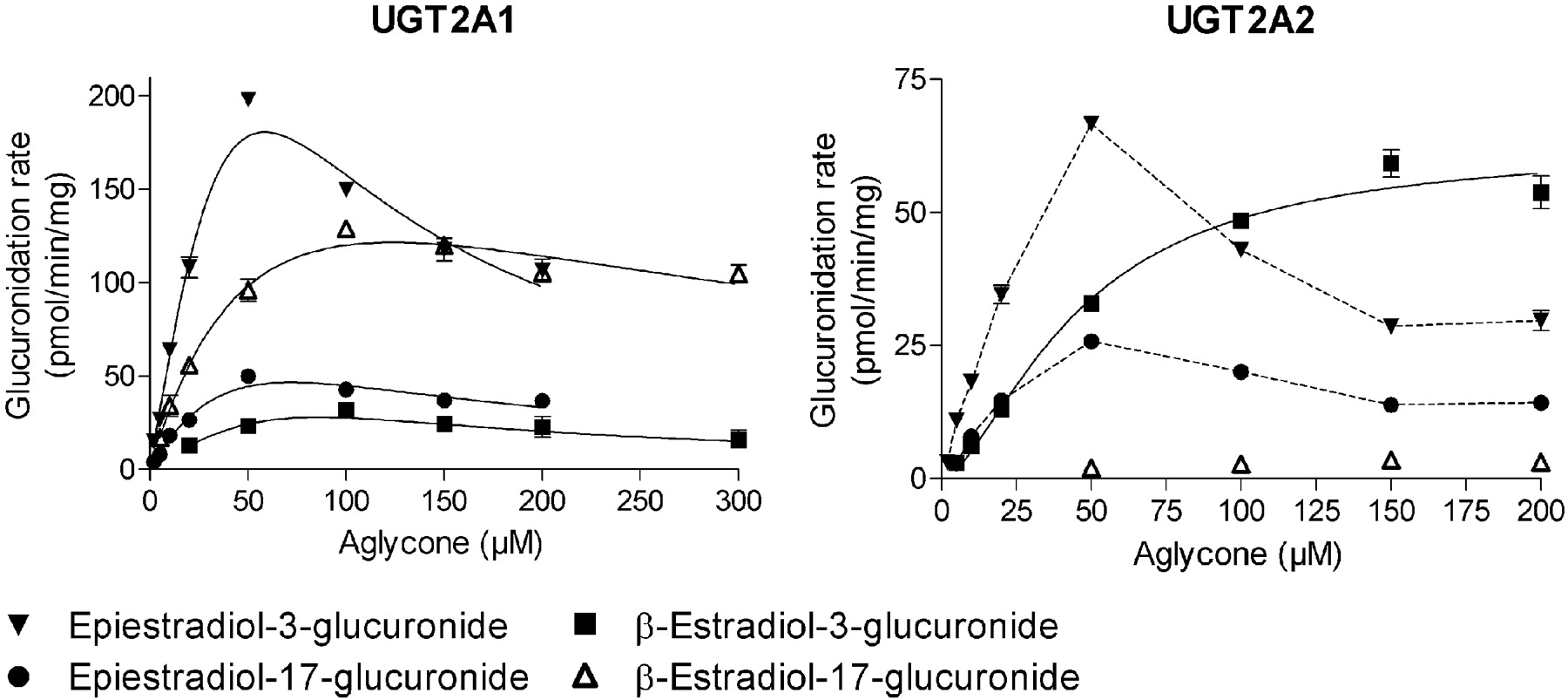
Glucuronidation of estradiol diastereomers by recombinant human UGTs of subfamily UGT2A. The normalized initial rates were fitted to either substrate inhibition or Hill equation. See Table 1 for kinetic constants.
Unlike most UGTs of the UGT1A family, UGT1A4 glucuronidated the two diastereoisomers solely at the 17-OH position but only at low rates. Interestingly, the glucuronidation of the two substrates at the 17-OH position by UGT1A4 was similar to the corresponding activities in UGT1A3, except that UGT1A3 glucuronidated β-estradiol at the 17-OH position slightly faster than it glucuronidated epiestradiol at this position, whereas in UGT1A4 this ratio was reversed (Table 1). However, the major difference between these two homologous enzymes was that UGT1A3 was significantly more active toward the 3-OH of the two substrates, whereas UGT1A4 did not conjugate the 3-OH position in either compound.

Glucuronidation of estradiol diastereomers by recombinant human UGTs of subfamily UGT2B. The normalized initial rates were fitted to the substrate inhibition equation. See Table 1 for kinetic constants.

Glucuronidation of estradiol diastereomers by rat recombinant UGT2B1 and UGT2B3. The normalized initial rates were fitted to either Michaelis-Menten or substrate inhibition equation. See Table 1 for kinetic constants.
UGT1A7 exhibited a low rate of activity, yielding only the 3-O-glucuronide of epiestradiol. The Ks value for this reaction was similar to the respective values for UGT1A3 and UGT1A10, but there were large differences in Vmax (Table 1). UGT1A8 exhibited considerable activity, but data of epiestradiol glucuronidation by this isoform could not be fitted in any enzyme kinetic equation. Thus, the Km or Ks could not be determined, and the maximum reaction rate was estimated by visual inspection (Fig. 2).
The most active human UGT in estradiol glucuronidation was UGT1A10, particularly when considering glucuronidation at the 3-OH position. The activity of UGT1A10 toward epiestradiol was very high, as was its activity toward the 3-OH group of β-estradiol (Fig. 2; Table 1). In the latter case, a combination of low Ks and, particularly, high Vmax value led to an exceptionally high Clint value. The very high estradiol glucuronidation activity of UGT1A10, much greater than that of UGT1A8 (Fig. 2; Table 1), was in disagreement with a previous significant report on this topic (Lépine et al., 2004). Therefore, we took further steps to ensure that our findings were correct and examined the estradiol glucuronidation activity of both UGT1A8 and UGT1A10 that were expressed in human embryonic kidney 293 cells, without any C-terminal fusion peptide (Uchaipichat et al., 2004). The results were broadly consistent with the findings presented in Fig. 2 and Table 1. In particular, the normalized (relative to UGT1A1 expression) Vmax value for UGT1A10 was 2 orders of magnitude higher than that of UGT1A8 (data not shown).
The human UGT2A subfamily enzymes are rarely included in glucuronidation studies. We have expressed these three enzymes as His-tagged proteins in baculovirus-infected insect cells in the same manner as the other UGTs that are included in the present study (N. Sneitz, X. Zhang, X. Ding, M. H. Court, and M. Finel, manuscript in preparation). The results revealed that UGT2A1 and UGT2A2 can catalyze glucuronidation of both β-estradiol and epiestradiol at both positions. UGT2A1 revealed stereoselectivity as it conjugated epiestradiol at the 3-OH position at much higher rate than β-estradiol. When considering the 17-OH position, the difference was smaller and with an opposite stereoselectivity (Fig. 3; Table 1).
Human UGT2B4 exhibited both regioselectivity and stereoselectivity. It only catalyzed epiestradiol glucuronidation and solely at the 17-OH position. UGT2B7, in turn, revealed a clear preference for epiestradiol over the physiological steroid β-estradiol, but it also glucuronidated the latter substrate. Like UGT2B4, UGT2B7 was fully regioselective for the 17-OH site and did not catalyze the formation of 3-O-glucuronides. An interesting finding in the case of the human UGT2B7 was its exceptionally high affinity for epiestradiol (Fig. 4; Table 1). The precise Km value could not be determined for this reaction because when the incubation conditions were kept within the linear range with regard to both time and protein concentration, the amount of glucuronide formed was not detectable at aglycone concentrations lower than 0.5 μM.
UGT2B15 was the only member of subfamily UGT2B that conjugated the two estradiols at the 3-OH rather than the 17-OH position. Like UGT2B4 and UGT2B7, it exhibited a clear preference for epiestradiol over β-estradiol. In the initial studies with our recombinant human UGT2B15, only low glucuronidation activity of epiestradiol at the 3-OH position was detected (results not shown). Because this was in sharp contrast with the other human UGT2Bs that are largely restricted to the 17-OH position, and because other studies gave us reasons to suspect that the amount of active UGT2B15 in the insect cell microsomes prepared locally is significantly lower than in our other recombinant UGT preparations (M. Finel, unpublished observation), we purchased a commercial sample of the human UGT2B15. The results revealed that, qualitatively, the two enzymes are the same because the commercial sample exhibited strong preference for the 3-OH position of epiestradiol. The commercial UGT2B15 sample was clearly more active and also produced a trace amount of 3-OH glucuronide of β-estradiol, but 17-O-glucuronides were still not detected.
UGT2B17 was the only human UGT with a strict regioselectivity and stereoselectivity for the 17-OH of β-estradiol. Its activity was among the highest of all the human isoenzymes at this position, suggesting that it may be an important enzyme for steroid metabolism in tissues containing it. It may be noted that with respect to UGT2B17, our results also differ sharply from the observations of Lépine et al. (2004) as they reported that UGT2B17 lacks β-estradiol glucuronidation activity.
In addition to the 19 human UGTs, we have also tested three rat UGTs from the UGT2B subfamily, rUGT2B1 through rUGT2B3. rUGT2B2 was not active toward either estradiol diastereomer; rUGT2B1 only glucuronidated β-estradiol, whereas rUGT2B3 glucuronidated both diastereomers but only at the 17-OH position. The regioselectivity and stereoselectivity of rUGT2B1 were similar to those of the human UGT2B17, nearly restricted for the 17-OH of β-estradiol. Interestingly, rUGT2B3 was highly and similarly active toward the 17-OH of both diastereomers, a clear exception among all the tested UGTs (Fig. 5; Table 1). The only difference between the kinetics of these reactions was in the substrate inhibition that was slightly stronger in the case of epiestradiol (Fig. 5).
One of the intriguing findings of this work is the very large difference in estradiol glucuronidation activity between UGTs that are highly similar to each other at the level of amino acid sequence. The best example for this is the contrast between the high activity of UGT1A10 toward the 3-OH of β-estradiol (Fig. 2; Table 1) and the lack of estradiol glucuronidation activity by UGT1A9. To clarify whether UGT1A9 binds one or both estradiol diastereomers, we examined the inhibition of scopoletin glucuronidation activity of UGT1A9 by the two estradiols. The results reveal that β-estradiol can inhibit scopoletin glucuronidation activity substantially, whereas the inhibitory effect of epiestradiol on this activity was limited (Fig. 6A). To further clarify the mode of inhibition by β-estradiol, we have determined the apparent Km values for scopoletin glucuronidation in the absence and presence of three concentrations of the inhibitor. The outcome of this experiment (Table 2; Fig. 6, B and C) revealed that β-estradiol is a competitive inhibitor of UGT1A9 with a Ki value of 5.8 ± 1.1 μM, a very close value to the Ks of UGT1A10 for β-estradiol (Table 1).
The kinetic constants for scopoletin glucuronidation by UGT1A9 in the presence and absence of β-estradiol
The Km values are best-fit values ± S.E.
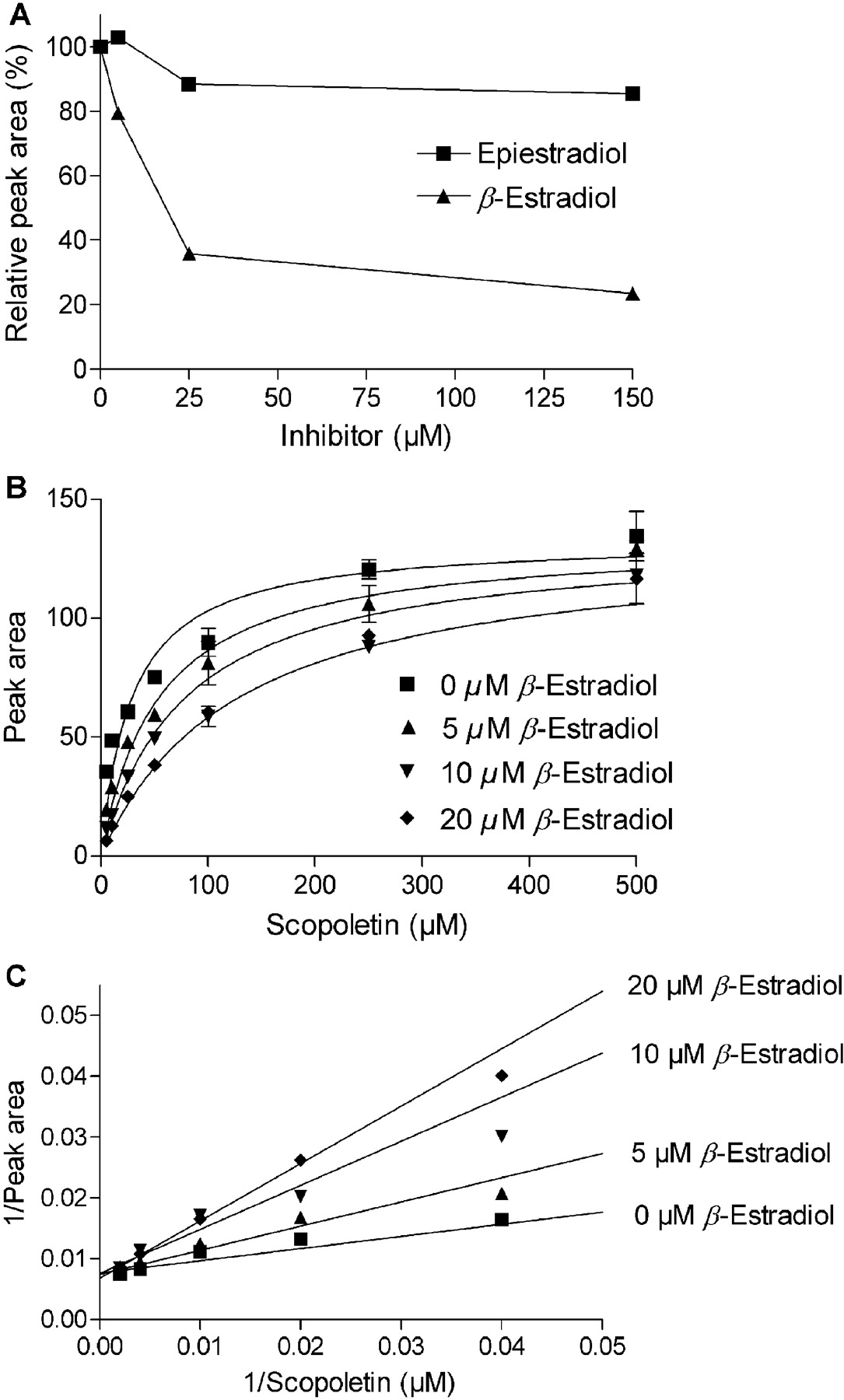
Inhibition of the scopoletin glucuronidation activity of UGT1A9 by estradiol diastereomers. A, inhibition at a single scopoletin concentration, 25 μM, by several concentrations of epiestradiol and β-estradiol. B, kinetic analysis and global fit of the data to competitive inhibition equation for the inhibition of scopoletin glucuronidation by β-estradiol. The derived Ki value was 5.8 ± 1.1 μM. C, determination of the mode of inhibition of UGT1A9 by β-estradiol. The apparent Km and Vmax values were first determined by fitting the data separately to untransformed Michaelis-Menten equation. The obtained values were substituted into the reciprocal transformation of the equation (1/v = Km/Vmax × 1/[S] + 1/Vmax), and the straight lines in the figure represent the equations that were obtained in this way [see Table 2 for the Km[infi](app) and Vmax(app) values]. Actual data points were eventually added onto the calculated straight lines.
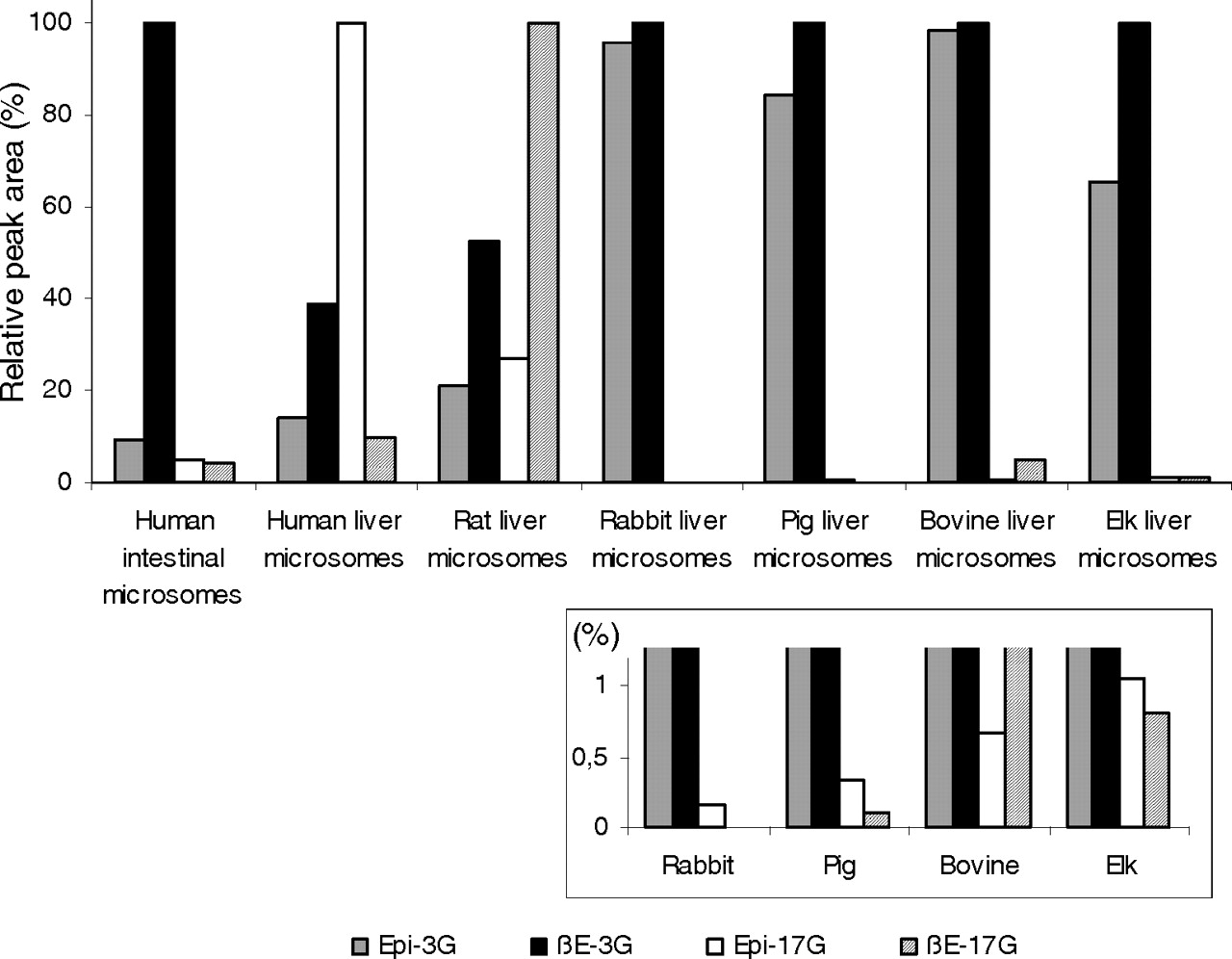
The results with individual human UGTs prompted us to compare human liver and intestinal microsomes to verify the applicability of the findings to the native UGTs. In addition, we have examined liver microsomes from several different animal species. The results (Fig. 7) show that both human and rat liver microsomes exhibit higher glucuronidation activity at the 17-OH position than at the 3-OH. However, the estradiol glucuronidation activity of microsomes from rabbit, pig, bovine, and elk was high, and glucuronidation occurred mainly at the 3-OH position. Looking more closely at the relative levels of 17-OH glucuronides formed by human and rat liver microsomes, it is interesting to note that whereas human liver microsomes catalyzed mainly epiestradiol glucuronidation, rat microsomes exhibited a clear preference for β-estradiol (Fig. 7). The results also show that in human intestinal microsomes there is a very high activity toward the 3-OH position of β-estradiol, in good agreement with the high activity of human UGT1A10 (Fig. 2; Table 1) and with the previous study by Czernik et al. (2000).
Discussion
The current study has revealed extensive new information on the activity of individual UGTs and, hence, their potential role in estradiol metabolism in different tissues. The focus of the present work was mainly on structure-activity relationships of the human UGTs. The sex hormone β-estradiol and its xenobiotic diastereoisomer, epiestradiol, were selected for this study because each of them can be glucuronidated at two different positions, providing an interesting system to examine the substrate specificity of individual UGTs and the overlap between them in this respect. Moreover, the use of these two diastereoisomers could reveal how the configuration of the 17-OH affects its own glucuronidation by different UGTs, as well as its longer-range effects on the glucuronidation at the 3-OH of these compounds.
We have investigated the activity of all the known recombinant human UGTs and three recombinant rat UGTs. Seven human UGTs, namely, UGT1A5, UGT1A6, UGT1A9, UGT2A3, UGT2B10, UGT2B11, UGT2B28, and the rat UGT2B2, did not glucuronidate either estradiol at detectable rates. The other enzymes, human UGT1A1, UGT1A3, UGT1A4, UGT1A7, UGT1A8, UGT1A10, UGT2A1, UGT2A2, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 along with rat rUGT2B1 and rUGT2B3, glucuronidated at least one of the two substrates at one position. The regioselectivity and stereoselectivity among these enzymes varied considerably, revealing general patterns among the different UGTs but also interesting exceptions. The results are discussed here, keeping in mind the sequence homology among individual enzymes.
The general picture that emerges from this study is a distinct regioselectivity between the three subfamilies of the human UGTs. The UGT1As that glucuronidate estradiol do it preferentially on the 3-OH. The human UGT2Bs mainly glucuronidate estradiol at the 17-OH, depending on its configuration. The human UGTs of subfamily UGT2A that exhibit estradiol glucuronidation activity, UGT2A1 and UGT2A2, have more promiscuous binding sites because they can glucuronidate these steroids at both positions. These patterns of estradiol glucuronidation by members of the different UGT subfamilies are partial, however, and exceptions to these rules have been found. For example, UGT2B15 glucuronidated epiestradiol at the 3-OH rather than the 17-OH, and UGT1A4 glucuronidated both steroids at the 17-OH but not the 3-OH position (Table 1).
The rules of stereoselectivity among the different UGTs are even more complex than those governing regioselectivity and may be viewed as a dimension of the substrate specificity. This is particularly striking in the case of the three human UGT2Bs that glucuronidated either substrate at the 17-OH position. Hence, UGT2B4 is specific for the 17-OH of epiestradiol; UGT2B17 is strictly specific for β-estradiol, whereas UGT2B7 exhibited a clear preference, but not strict specificity, for epiestradiol (Table 1). In addition, UGT2B7 exhibited an exceptionally high affinity for epiestradiol (Fig. 4). It was much higher than what we have observed in other UGTs, and such low Km values are rarely encountered in glucuronidation studies. Another interesting example for complex stereoselectivity is provided by the three rat UGTs that were examined in this study. Whereas rUGT2B1 resembles the human UGT2B17 as far as stereoselectivity is concerned, rUGT2B3 stands out as an interesting exception because it was both highly active and practically insensitive to the configuration of the 17-OH, while still exhibiting a strict regioselectivity.

Formation of the four estradiol glucuronides by microsomes from different sources. The samples were incubated at 37°C for 60 min in the presence of 100 μM of the aglycone substrates. The relative activities were measured as peak areas, and the largest peak area of each microsomal sample was given the reference value of 100%. The subsection shows an enlargement of the smallest bars from the main chart.
It is also interesting to examine the estradiol glucuronidation activity results from the point of view of protein homology. The human UGTs of the UGT1A subfamily can be divided into four subgroups based on amino acid sequence identity. Two groups contain a single enzyme each, namely, UGT1A1 and UGT1A6. The third group is composed of UGT1A3 through UGT1A5, and the fourth group is composed of UGT1A7 through UGT1A10 (Gong et al., 2001; Mackenzie et al., 2005). It is noteworthy that the highly homologous UGT1A3 and UGT1A4 differ dramatically in their glucuronidation activity toward the 3-OH of the two steroids. On the other hand, both enzymes exhibited very similar activity, even if low, toward the 17-OH of both estradiols. These observations may provide a starting point for identifying the residue(s) responsible for the high activity of UGT1A3, but not UGT1A4, toward the 3-OH of the steroids, similarly to what was done previously for the glucuronidation of 1-naphthol (Kubota et al., 2007). Our findings on the activity of UGT1A3 are also relevant to previous observations on the role of UGT1A3 in the glucuronidation of estrone and therefore in regulating estradiol biosynthesis (Caillier et al., 2007). The present work shows that UGT1A3 may also play a direct role in the glucuronidation of estradiol, not only its precursor estrone. In addition, it may be pointed out that because UGT1A3 is expressed in the liver (Tukey and Strassburg, 2001), it may contribute to estradiol glucuronidation in this tissue, even if its affinity toward the 3-OH of β-estradiol is lower than that of UGT1A1.
There are large differences in estradiol glucuronidation activity between the human UGT1A7 through UGT1A10, despite the high degree of sequence similarity among them. UGT1A10 is by far the most active human enzyme in β-estradiol glucuronidation at the 3-OH position, and its activity toward the 3-OH of epiestradiol is also high (Fig. 2; Table 1). The estradiol glucuronidation activity of UGT1A10 was reportedly low in a previous study (Lépine et al., 2004), perhaps because of the use of a low activity batch of recombinant UGT1A10. With respect to the glucuronidation of β-estradiol by UGT1A7 through UGT1A9, however, our results are in agreement with the previous study (Lépine et al., 2004). The similarities include the lack of detectable activity of either UGT1A7 or UGT1A9 but significant activity of UGT1A8 toward the 3-OH of β-estradiol. In the present study, we have also detected high activity of UGT1A8 toward the 3-OH of epiestradiol, indicating that the α configuration of the 17-OH significantly increases its activity toward the 3-OH (Fig. 2). The configuration of the 17-OH also affects the activity of UGT1A10 toward the 3-OH, but in the case of UGT1A10 the direction of the effect was opposite, namely, higher activity toward β-estradiol than epiestradiol.
The configuration of 17-OH also affected the interactions between the estradiol diastereomers and UGT1A9 (Fig. 6), an enzyme that does not catalyze estradiol glucuronidation. This observation, along with the inhibition kinetics of β-estradiol (Table 2; Fig. 6, B and C), supports the suggestion that although UGT1A9 does not glucuronidate β-estradiol, it binds this steroid at nearly the same affinity as UGT1A10. Hence, no activity of a given UGT toward a test compound does not necessarily mean no binding.
One of the most striking findings of the present study is that whereas certain UGTs exhibit strict stereospecificity, others essentially lack specificity. Human UGT2B4 glucuronidates the 17-OH only in epiestradiol, whereas human UGT2B17 was highly specific for the β configuration of this hydroxyl group. In contrast, rUGT2B3 can catalyze the glucuronidation of the 17-OH in both steroids with efficiency. Future detailed structural information on the UGTs may help to understand the molecular basis for these features, as well as for the regioselectivity differences among the UGTs.
Taken together, our results show that many UGTs are capable of estradiol glucuronidation, but they catalyze these reactions with varying regioselectivities and stereoselectivities and differ in terms of catalytic efficiency. Hence, the assumption of broadly overlapping specificities of UGTs is not justified, particularly when dealing with compounds that may be glucuronidated at more than one position.
Acknowledgments
We thank David Elliot and Johanna Mosorin for skillful technical assistance.
Footnotes
-
This research was supported by the Academy of Finland (Project No. 210933), the Sigrid Juselius Foundation, and the Emil Aaltonen Foundation.
-
doi:10.1124/dmd.108.022731.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; UDPGA, UDP-glucuronic acid; HPLC, high-performance liquid chromatography.
- Received June 5, 2008.
- Accepted August 18, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}