


Abstract
Cytochrome P450 3A4 (CYP3A4), the most abundant human cytochrome P450 in liver, participates in the metabolism of ∼50% of clinically used drugs. The pregnane X receptor (PXR), a member of the nuclear receptor superfamily, is the major activator of CYP3A4 transcription. However, because of species differences in response to PXR ligands, it is problematic to use rodents to assess CYP3A4 regulation and function. The generation of double transgenic mice expressing human PXR and CYP3A4 (TgCYP3A4/hPXR) would provide a solution to this problem. In the current study, a TgCYP3A4/hPXR mouse model was generated by bacterial artificial chromosome transgenesis in Pxr-null mice. In TgCYP3A4/hPXR mice, CYP3A4 was strongly induced by rifampicin, a human-specific PXR ligand, but not by pregnenolone 16α-carbonitrile, a rodent-specific PXR ligand. Consistent with CYP3A expression, hepatic CYP3A activity increased ∼5-fold in TgCYP3A4/hPXR mice pretreated with rifampicin. Most antihuman immunodeficiency virus protease inhibitors are CYP3A substrates and their interactions with rifamycins are a source of major concern in patients coinfected with human immunodeficiency virus and Mycobacterium tuberculosis. By using TgCYP3A4/hPXR mice, human PXR-CYP3A4-mediated rifampicin-protease inhibitor interactions were recapitulated, as the metabolic stability of amprenavir, nelfinavir, and saquinavir decreased 52, 53, and 99%, respectively, in the liver microsomes of TgCYP3A4/hPXR mice pretreated with rifampicin. In vivo, rifampicin pretreatment resulted in an ∼80% decrease in the area under the serum amprenavir concentration-time curve in TgCYP3A4/hPXR mice. These results suggest that the TgCYP3A4/hPXR mouse model could serve as a useful tool for studies on CYP3A4 transcription and function in vivo.
The human cytochrome P450 3A (CYP3A) family is located on chromosome 7q22.1, and consists of four members (3A4, 3A5, 3A7, and 3A43) (Gellner et al., 2001). Of these, CYP3A4 is the most abundant cytochrome P450 (P450) in liver and small intestine and participates in the metabolism of approximately 50% of all prescribed drugs and some endogenous substrates, such as steroids and bile acids (Guengerich, 1999). CYP3A5 expression in humans is highly variable and only ∼20% of livers express CYP3A5 (Xie et al., 2004). CYP3A7 is predominantly expressed in fetal liver with a specific role in hydroxylation of retinoic acid and 16α-hydroxylation of steroids (Kitada et al., 1987; Chen et al., 2000). CYP3A43, the most recently discovered CYP3A, is expressed in prostate and testis with a low level of expression in liver (Westlind et al., 2001). Among the CYP3A members, CYP3A4 is the most critical P450 for drug metabolism. Because of its broad substrate spectrum, CYP3A4 contributes to many adverse drug-drug interactions (Guengerich, 1999).
Pregnane X receptor (PXR) is the dominant activator controlling CYP3A4 transcription (Lehmann et al., 1998; Xie et al., 2000). After ligand binding, human PXR forms a heterodimer with the retinoid X receptor and subsequently binds to PXR response elements in the 5′-flanking region of the CYP3A4 gene, resulting in increased transcription (Goodwin et al., 2003). PXR is activated by a large number of prescription drugs, herbal supplements, vitamins, and some endobiotics (Carnahan and Redinbo, 2005). It is interesting to note that there are significant species differences in response to PXR ligands between humans and rodents (Jones et al., 2000). Drugs such as rifampicin (RIF), clotrimazole, and troglitazone activate human PXR but are weak activators of rodent PXR. In contrast, dexamethasone and pregnenolone 16α-carbonitrile (PCN) activate rodent PXR but are weak activators of human PXR. Therefore, the CYP3A4-transgenic mouse models that have been generated are of limited utility for transcriptional and drug interaction studies because of their mouse PXR background (Granvil et al., 2003; Robertson et al., 2003; Zhang et al., 2003, 2004; Cheung et al., 2006). The generation of double transgenic mice expressing human PXR and CYP3A4 would provide a solution to this problem.
In the current study, a double transgenic mouse model was generated by bacterial artificial chromosome (BAC) transgenesis with human PXR and CYP3A4 in Pxr-null mice and designated TgCYP3A4/hPXR mice. Treatment of TgCYP3A4/hPXR mice with the PXR ligands mimicked the human response, because CYP3A4 was strongly induced by RIF, a human-specific PXR ligand, but not by PCN, a rodent-specific PXR ligand. Most antihuman immunodeficiency virus (HIV) protease inhibitors (PI) are CYP3A substrates, and their interactions with rifamycins is a major issue in patients coinfected with Mycobacterium tuberculosis (TB) and HIV (Breen et al., 2006; Swaminathan et al., 2006; Ribera et al., 2007). By using TgCYP3A4/hPXR mice, human PXR-CYP3A4 mediated RIF-PIs interactions were illustrated, thus demonstrating the utility of this mouse model for studies on CYP3A4 transcription and function.
Materials and Methods
Chemicals. RIF, PCN, midazolam (MDZ), ketoconazole, and NADPH were obtained from Sigma-Aldrich (St. Louis, MO). 1′-Hydroxymidazolam (1′-OH-MDZ) was purchased from BD Gentest (Woburn, MA). Amprenavir (APV), nelfinavir (NFV), and saquinavir (SQV) were supplied by the National Institutes of Health AIDS Research and Reference Reagent Program. All other chemicals were of the highest grade commercially available.
Generation of TgCYP3A4/hPXR Mice. The TgCYP3A4/hPXR mouse line was generated by BAC transgenesis. The BAC clone RP11-757A13 (123,778 bp) contains the complete CYP3A4 and CYP3A7 genes including 5′- and 3′-flanking sequences (Fig. 1A), and the BAC clone RP11-169N13 (165,093 bp) contains the complete human PXR gene sequence including 5′- and 3′-flanking sequences (Fig. 1B). Both BAC clones were obtained from Resgen/Invitrogen Corporation (Huntsville, AL) and were purified using a maxi prep kit (QIAGEN, Valencia, CA). The BAC clone for CYP3A4 was verified by Southern blot analysis with 32P-end-labeled CYP3A4 cDNA and DNA oligonucleotide probes recognizing specific regions (exons 1 and 13, -10 kilobases upstream) of the human CYP3A4 gene (Cheung et al., 2006). The BAC clone for human PXR was verified by PCR using primers designed to amplify specific regions within exons 2 and 9 and the 5′-untranslated region (Ma et al., 2007). Two major steps were performed to generate the TgCYP3A4/hPXR mice. In step I, the CYP3A4-transgenic mice (TgCYP3A4) were bred with Pxr-null mice to generate the TgCYP3A4/Pxr-null mouse line; in step II, the PXR-humanized mice (hPXR) were bred with the TgCYP3A4/Pxr-null mice to generate the TgCYP3A4 mouse containing human PXR. Mice positive for the human PXR and CYP3A4 transgenes and containing the mouse Pxr-null allele, as determined by PCR genotyping, were designated TgCYP3A4/hPXR mice. Pxr-null, TgCYP3A4, and hPXR mice were described previously (Staudinger et al., 2001; Cheung et al., 2006; Ma et al., 2007).
PCR Genotyping. The presence of the CYP3A4 transgene was determined using the following primers: forward 5′-TGG AAT GAG GAC AGC CAT AGA GAC-3′ and reverse 5′AGA AGA GGA GCC TGG ACA GTT ACT C-3′, amplifying a PCR product of 521 bp in the samples only positive for human CYP3A4 transgene (Cheung et al., 2006). Mouse epoxide hydrolase 1 gene primers served as an internal positive control for amplification, yielding a fragment of 341 bp in all samples (Miyata et al., 1999). The presence of the human PXR transgene was determined using the following primers: forward 5′-GCA CCT GCT GCT AGG GAA TA-3′ and reverse 5′-CTC CAT TGC CCC TCC TAA GT-3′, amplifying a PCR product of 576 bp in the samples only positive for human PXR transgene (Ma et al., 2007). The following primers were used to identify the mouse Pxr wild-type (WT) and null alleles: forward 5′-CTG GTC ATC ACT GTT GCT GTA CCA-3′ reverse1 5′-GCA GCA TAG GAC AAG TTA TTC TAG AG-3′, and reverse2 5′-CTA AAG CGC ATG CTC CAG ACT GC-3′, amplifying a PCR product of 348 bp for the wild-type allele and 265 bp for the Pxr-null allele (Guo et al., 2003).
Animals and Treatments. TgCYP3A4/hPXR, TgCYP3A4, and WT mice were maintained under a standard 12-h light/dark cycle with water and chow provided ad libitum. Handling was in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. Because it was previously shown in TgCYP3A4 mice that age and gender were determinants of hepatic CYP3A4 expression (Cheung et al., 2006), both male and female TgCYP3A4/hPXR mice were used, and liver samples were collected from 2-, 4-, 6-, 8-, 16-, and 24-week-old mice. For analysis of CYP3A4 tissue distribution, liver, lung, small intestine, kidney, heart, ovary, or testis were collected at 4 weeks of age. For studies on hepatic CYP3A4 regulation by xenobiotics, TgCYP3A4/hPXR and TgCYP3A4 mice (8–10 weeks old) were treated with 10 mg/kg PCN (i.p.) or with 10 mg/kg RIF (p.o.), once daily for 3 days. All mice were killed by CO2 asphyxiation, and samples were collected and frozen at -80°C for further analysis.
Microsome Preparation and Western Blot Analysis. All tissues were homogenized in ice-cold buffer (50 mM Tris-HCl, 150 mM KCl, 1 mM EDTA, and 20% glycerol). Microsomes were prepared by centrifugation at 10,000g for 20 min at 4°C, and the resulting supernatant was spun at 100,000g for 1 h at 4°C. Microsomal pellets were resuspended in the same ice-cold buffer used for homogenization. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce Chemical, Rockford, IL). For Western blot analysis, microsomal protein (10 μg) from each sample was loaded onto 10% SDS-polyacrylamide gels and electrophoretically transferred to nitrocellulose membranes (Whatman Schleicher & Schuell, Keene, NH). Immunoblot analysis was carried out using antibodies to human CYP3A4 [monoclonal antibody (mAb) 275-1-2], rat CYP3A1 (mAb 2-13-1) (Gelboin et al., 1995), and GAPDH (Millipore Corporation, St. Charles, MO). Corresponding secondary antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA). Immunoreactive proteins were detected with an ECL kit (Pierce Chemical) following the manufacturer's instructions.
CYP3A Activity Analysis. Hepatic CYP3A activity was detected by using liver microsomes. MDZ 1′-hydroxylation was used as a probe for CYP3A activity, and the method for CYP3A activity analysis was described previously (Thummel et al., 1994; Ma et al., 2007). In brief, incubations were carried out in 100 mM sodium phosphate buffer (pH 7.4) containing microsomes with 100 μg of protein and 50 μM MDZ. Ketoconazole (5 μM) was used as CYP3A inhibitor and preincubated at 37°C for 5 min. The reaction was initiated by the addition of 20 μl of 20 mM NADPH, 37°C for 10 min and terminated by the addition of 1 ml of ethyl acetate and 1 ml of methyl t-butyl ether mixture. Samples were centrifuged at 3000 rpm for 5 min at 4°C. The organic layer was then transferred to a new tube, dried with N2, and reconstituted in 100 μlof 70% methanol and 30% H2O containing 0.1% formic acid. Six microliters of the reconstituted solution was analyzed by liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS) for 1′-OH-MDZ detection. All reactions were performed in duplicate.
Metabolic Stability of HIV PIs. Metabolic stability of the PIs was analyzed by using liver microsomes of TgCYP3A4/hPXR and WT mice pretreated with or without RIF. The incubations were performed in 100 mM sodium phosphate buffer (pH 7.4) containing microsomes with 100 μg of protein and 10 μM concentrations of PIs (APV, NFV, or SQV) in a final volume of 200 μl. The reactions were initiated by the addition of 20 μl of 20 mM NADPH at 37°C for 10 min and terminated by the addition of 200 μl of methanol. The mixtures were vortexed and centrifuged at 12,000 rpm for 10 min at 4°C. Six microliters of the deproteinized mixtures were analyzed by LC-MS/MS for detection of the parent drugs. By monitoring parent drug disappearance, the metabolic stability of PIs was determined according to their metabolic rates. Metabolic stability of MDZ was used as a positive control for these experiments.
Pharmacokinetic Study of Amprenavir in TgCYP3A4/hPXR Mice. TgCYP3A4/hPXR mice were fed with AIN-93G purified rodent diet (control) or modified AIN-93G rodent diet with 100 mg/kg RIF (Dyets Inc., Bethlehem, PA) for 6 days. On the 7th day, TgCYP3A4/hPXR mice were administered 50 mg/kg APV p.o. by gavage. Blood samples were collected from suborbital veins using heparinized tubes at 0, 0.25, 0.5, 1, 2, 4, 8, and 18 h after administration of APV. Serum was separated by centrifugation at 8000g for 10 min. For APV analysis, 15 μl of serum was mixed with 85 μlofH2O and 100 μl of methanol. The mixture was vortexed and centrifuged at 12,000 rpm for 10 min at 4°C. Six microliters of the deproteinized mixture was analyzed by LC-MS/MS for detection of the parent drugs. Pharmacokinetic parameters for APV were estimated from the plasma concentration-time data by a noncompartmental approach using WinNonlin (Pharsight, Mountain View, CA). The maximal concentration in serum (Cmax) was obtained from the original data. The area under the serum concentration-time curve (AUC0–18 h) was calculated by the trapezoidal rule.
LC-MS/MS Analysis. MDZ, MDZ metabolites, and PIs were analyzed using previously described LC-MS/MS methods with slight modification (Crommentuyn et al., 2003; Granvil et al., 2003). In brief, LC-MS/MS analysis was carried out using a high-performance liquid chromatography system consisting of a PerkinElmer Series 200 quaternary pump, vacuum degasser, Luna C18 50 mm × 4.6 mm i.d. column (Phenomenex, Torrance, CA), and autosampler with a 100-μl loop interfaced to an API2000 SCIEX triple-quadrupole tandem mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA). The flow rate through the column at ambient temperature was 0.25 ml/min with 70% methanol and 30% H2O containing 0.1% formic acid. Each analysis lasted for 5.0 min. The mass spectrometer was operated in the turbo ion spray mode with positive ion detection. The turbo ion spray temperature was maintained at 300°C, and a voltage of 4.8 kV was applied to the sprayer needle. N2 was used as the turbo ion spray and nebulizing gas. The detection and quantification of analysts were performed using the multiple reaction monitoring mode, with m/z 326/291 for MDZ, m/z 342/203 for 1′-OH-MDZ, m/z 506/245 for APV, m/z 568/330 for NFV, and m/z 671/570 for SQV. For these chemicals, the detection limits were <0.2 pmol. The curve range was 0.039 to 10 μM with linearities >98%.
Statistical Analysis. All values are expressed as the means ± S.D., and group differences were analyzed by Student's t test.
Results
Generation of TgCYP3A4/hPXR Mice. TgCYP3A4/hPXR mice were generated by BAC transgenesis with the complete CYP3A4, CYP3A7, and human PXR gene sequences in a mouse Pxr-null background (Fig. 1, A and B). Typical PCR genotyping results are shown in Fig. 1C. TgCYP3A4 mice were positive for the human CYP3A4 transgene and contained the mouse Pxr allele. The TgCYP3A4/hPXR mice were positive for both the human PXR and CYP3A4 transgenes and containing the mouse Pxr null allele.
CYP3A4 Expression in TgCYP3A4/hPXR Mice. CYP3A4 protein was analyzed by Western blot. Four-week-old TgCYP3A4/hPXR male and female mice demonstrated expression of CYP3A4 in liver and small intestine but not in the lung, kidney, heart, or testis/ovary (Fig. 2, A and B), which is consistent with the tissue distribution of Cyp3a in mice and CYP3A4 in humans (Guengerich, 1999). It is interesting to note that the developmental expression patterns for hepatic CYP3A4 protein showed significant gender difference between the ages of 2 to 24 weeks. In TgCYP3A4/hPXR male mice, hepatic CYP3A4 was detected in 2- and 4-week-old mice but not in mice older than 6 weeks (Fig. 2C), which suggests transcriptional down-regulation of basal CYP3A4 expression after sexual maturity in males. However, female mice expressed hepatic CYP3A4 at all time points assessed from 2 to 24 weeks of age (Fig. 2D). In contrast to the absence of expression of hepatic CYP3A4 in adult males, human PXR was constantly expressed in the liver of TgCYP3A4/hPXR mice (data not shown).
Regulation of Hepatic CYP3A4 Expression in TgCYP3A4/hPXR Mice. The regulation of CYP3A4 expression in TgCYP3A4/hPXR mice was compared with TgCYP3A4 mice. The BAC clone used for establishing the TgCYP3A4/hPXR and TgCYP3A4 mice was the same in both lines. In TgCYP3A4 mice, endogenous mouse PXR is present, whereas TgCYP3A4/hPXR mice contain human PXR with a mouse Pxr-null allele. TgCYP3A4/hPXR and TgCYP3A4 mice were treated with the human PXR ligand RIF and rodent PXR ligand PCN. Hepatic CYP3A4 was detected by Western blot analysis. In adult male mice, CYP3A4 was not detected in the control TgCYP3A4/hPXR and TgCYP3A4 mice. After PCN treatment, CYP3A4 was induced strongly in TgCYP3A4 mice, but only slightly in TgCYP3A4/hPXR mice. RIF treatment did not increase CYP3A4 in TgCYP3A4 mice, whereas induction of CYP3A4 was robust in TgCYP3A4/hPXR mice (Fig. 3A). The regulation of CYP3A4 expression in female TgCYP3A4/hPXR mice was similar to that in male mice (Fig. 3B). The human PXR in TgCYP3A4/hPXR mice was also functional in regulation of mouse PXR target genes. After RIF treatment, mouse hepatic Cyp3a11, glutathione S-transferase A1, and organic anion-transporting polypeptide 2 were all induced significantly in TgCYP3A4/hPXR mice (data not shown).

Generation and genetic characterization of TgCYP3A4/hPXR mice. A, structure of the BAC clone containing the complete human CYP3A4 and CYP3A7 (exons 1–13) genes and the 5′- and 3′-flanking sequences. B, structure of the BAC clone containing the complete human PXR (exons 1–9) gene and the 5′- and 3′-flanking sequences. C, a representative genotyping result for TgCYP3A4/hPXR mice. Mouse epoxide hydrolase 1 gene (MEH) primers served as an internal positive control. TgCYP3A4 mice were positive for the human CYP3A4 transgene and contained the mouse Pxr allele. TgCYP3A4/hPXR mice were positive for both the human PXR and CYP3A4 transgenes and contained the mouse Pxr-null allele.
Hepatic CYP3A Activity in TgCYP3A4/hPXR Mice. In TgCYP3A4/hPXR mice pretreated with RIF, both hepatic human CYP3A4 and mouse Cyp3a were induced. Consistent with CYP3A expression, hepatic CYP3A activity increased significantly in TgCYP3A4/hPXR mice after PXR ligand treatment. In WT and TgCYP3A4 mice, RIF produced no significant effect on CYP3A activity (Fig. 4, A and B). However, in TgCYP3A4/hPXR mice pretreated with RIF, CYP3A activity increased ∼5-fold compared with the vehicle-treated control group (Fig. 4C), and it was markedly inhibited by the CYP3A inhibitor ketoconazole (Fig. 4D).
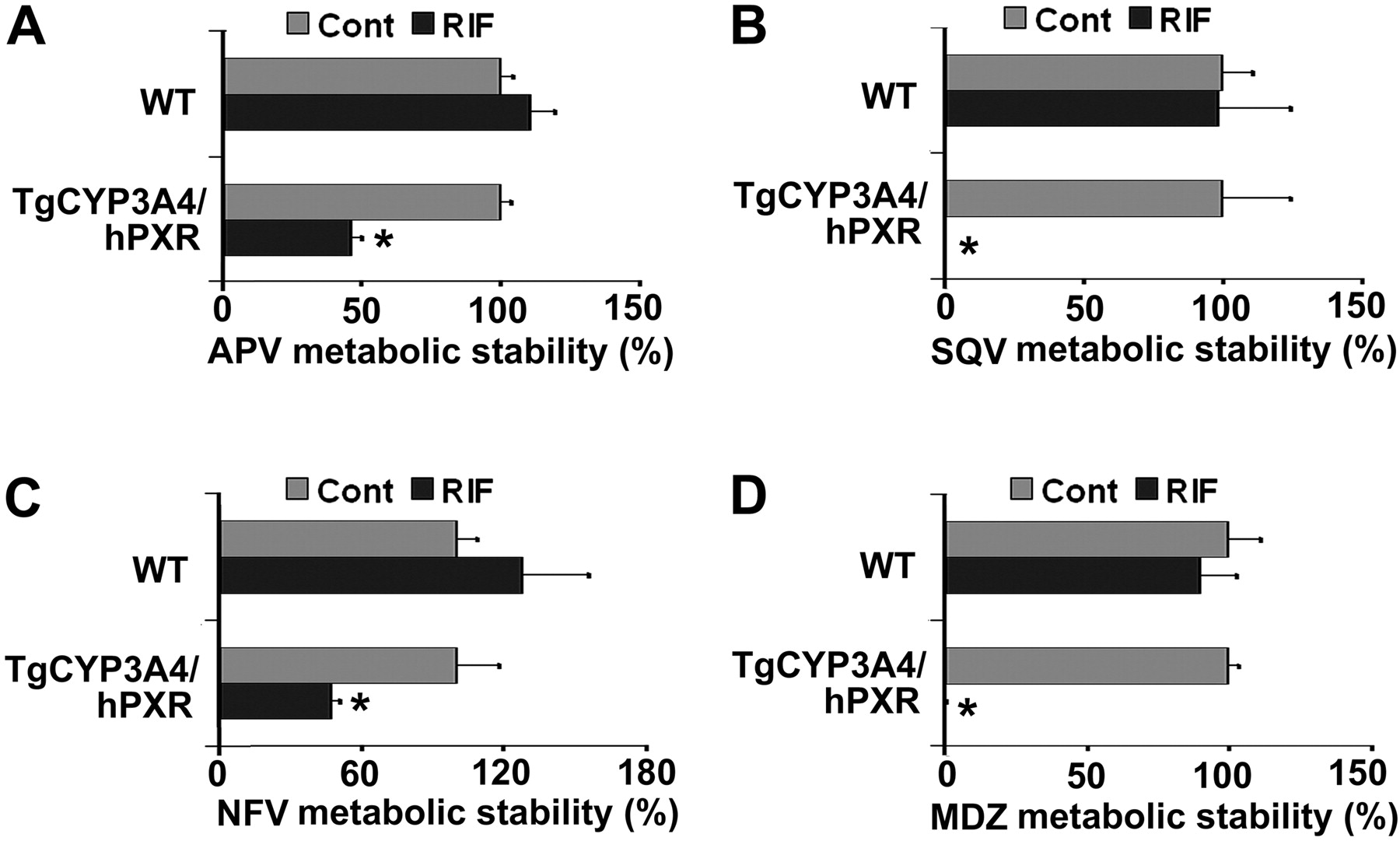
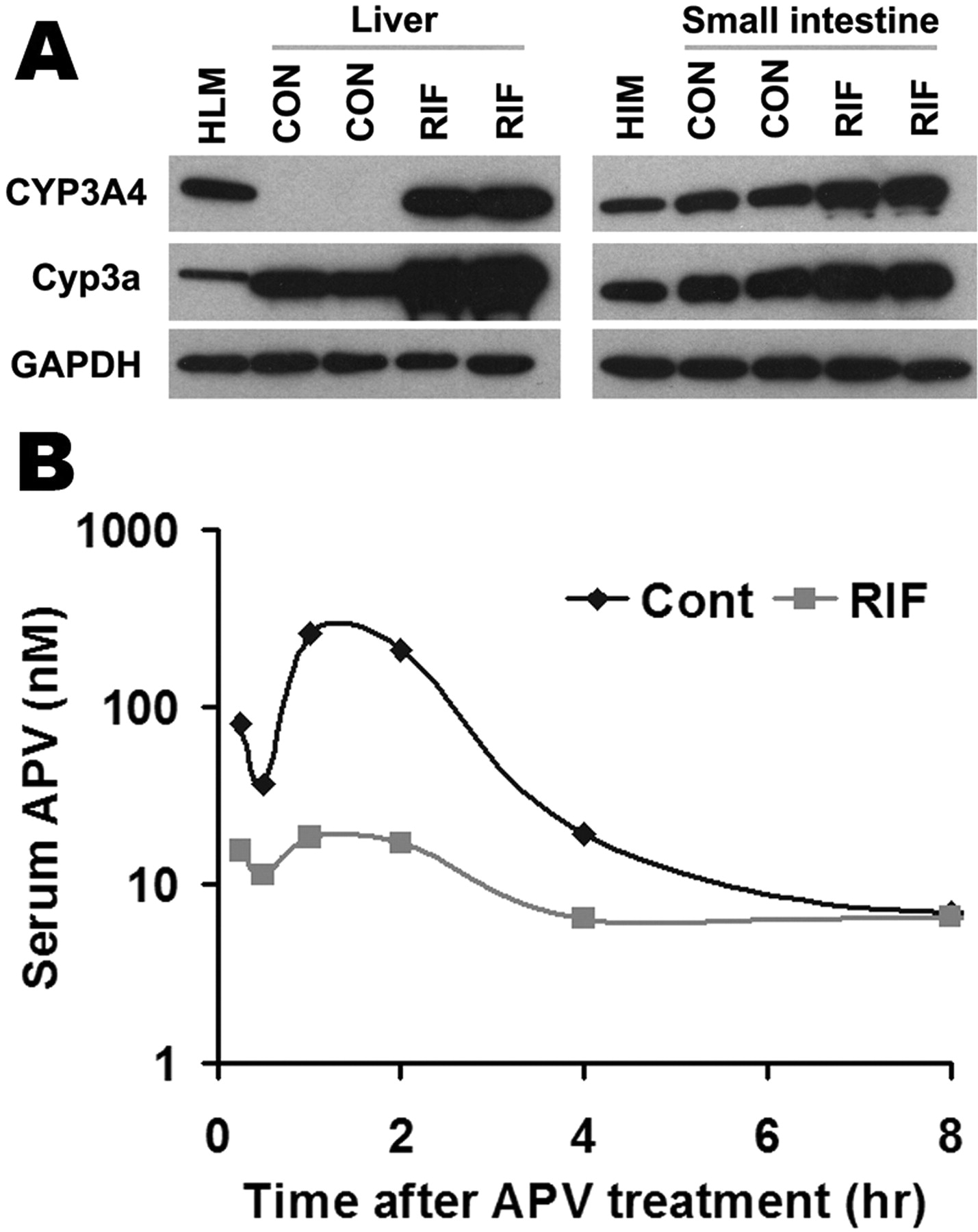
PXR-CYP3A-Mediated Drug-Drug Interactions in TgCYP3A4/hPXR Mice. Most clinically used HIV PIs, such as APV, SQV, and NFV, are CYP3A substrates, and their interactions with rifamycins are of concern in patients coinfected with TB and HIV (Breen et al., 2006; Swaminathan et al., 2006; Ribera et al., 2007). Human PXR-CYP3A4-mediated RIF-PIs interactions were assessed in TgCYP3A4/hPXR mice. PXR activation and CYP3A induction accelerated the metabolism of PIs. In liver microsomes from TgCYP3A4/hPXR mice pretreated with RIF, the metabolic stability of APV, NFV, and SQV decreased 52, 53, and 99%, respectively, but no significant change was observed in liver microsomes of WT mice (Fig. 5). In TgCYP3A4/hPXR mice pretreated with RIF, both human CYP3A4 and mouse Cyp3a in the liver and small intestine were induced; most significantly, hepatic CYP3A4, which was not expressed in the control group, was highly expressed after PXR activation (Fig. 6A). Consistent with the in vitro study, the pharmacokinetic analysis demonstrated that the Cmax of serum APV concentration decreased by ∼93% and the AUC0–18 h of APV decreased by ∼80% in TgCYP3A4/hPXR mice pretreated with RIF (Fig. 6B). These data revealed a significant effect of RIF on human PXR activation and CYP3A regulation and indicated that RIF PIs interactions were mediated by human PXR and CYP3A.

Tissue distribution and developmental expression patterns of CYP3A4 in TgCYP3A4/hPXR mice. A and B, liver, lung, small intestine (S.I.), kidney, heat, and testis/ovary were collected from transgenic male (A) and female (B) mice at 4 weeks of age. C and D, liver tissues were collected from transgenic male (C) and female (D) mice of different ages (2–24 weeks). All microsomes were prepared by differential centrifugation. Pooled microsomal samples (three to four in each group) were used for Western blot analysis. The monoclonal antibody against CYP3A4 (mAb 275-1-2) recognizes human CYP3A4 but not mouse Cyp3a or other liver proteins. The monoclonal antibody against Cyp3a (mAb 2-13-1) reacts with both mouse Cyp3a and human CYP3A4. Human intestine microsomes (HIM) and human liver microsomes (HLM) served as positive controls for CYP3A4. GAPDH was used as the loading control.

Regulation of hepatic CYP3A4 in TgCYP3A4/hPXR mice. TgCYP3A4 and TgCYP3A4/hPXR mice (8-week-old) were treated for 3 days with corn oil, 10 mg/kg/day PCN (i.p.), or 10 mg/kg/day RIF (p.o.) as detailed under Materials and Methods. Liver was collected from transgenic male (A) and female (B) mice 24 h after the last dose. Pooled liver microsomes (three samples) were prepared and analyzed by Western blot. The monoclonal antibody against CYP3A4 (mAb 275-1-2) recognizes human CYP3A4 but not mouse Cyp3a or other liver proteins. Human liver microsomes (HLM) served as a positive control for CYP3A4. GAPDH was used as the loading control.

CYP3A activity in liver microsomes of TgCYP3A4/hPXR mice. All mice (male, 8-week-old) were treated with corn oil or 10 mg/kg/day RIF (p.o.) for 3 days. Livers were collected 24 h after the last dose, and microsomes were prepared. MDZ 1′-hydroxylation was used as the probe for CYP3A activity and detected by LC-MS/MS. CYP3A activity in each control group (Cont) was set as 100%. Data are expressed as means ± S.D., n = 3. A, CYP3A activity in WT mice. B, CYP3A activity in TgCYP3A4 mice. CYP3A activity in TgCYP3A4/hPXR mice (C) and inhibition by ketoconazole (KCZ) (D). *, p < 0.05 compared with no KCZ group.
Discussion
In the current study, a double transgenic mouse model expressing human PXR and CYP3A4 was generated by BAC transgenesis in Pxr-null mice. Compared with the previously generated CYP3A4-transgenic mouse models (Granvil et al., 2003; Robertson et al., 2003; Zhang et al., 2003, 2004; Cheung et al., 2006), the major improvement in the current model was the inclusion of human PXR, the dominant activator of CYP3A4 transcription. All previous CYP3A4 mouse models were generated on a mouse PXR background, which does not mimic human CYP3A4 regulation because of the species differences in PXR in response to various ligands. The current mouse model improves upon these earlier mouse lines because the human CYP3A4 transgenic mouse was generated on a human PXR background. Treatment with PXR ligands in TgCYP3A4/hPXR mice mimicked the human response, as demonstrated by the robust induction of CYP3A4 by the human-specific PXR ligand RIF and weak CYP3A4 induction by the rodent-specific PXR ligand PCN. Consistent with CYP3A expression, hepatic CYP3A activity significantly increased in TgCYP3A4/hPXR mice treated with RIF. These results revealed that the TgCYP3A4/hPXR mouse model would be a useful tool for the study of CYP3A4 transcription and function.

Metabolic stability of PIs in liver microsomes of TgCYP3A4/hPXR mice. All mice (male, 8-week-old) were treated with corn oil or 10 mg/kg/day RIF (p.o.) for 3 days. Livers were collected 24 h after the last dose, and microsomes were prepared. PIs metabolic stability was determined according to their metabolic rates by monitoring parent drug disappearance. APV, NFV, SQV, and MDZ were detected by LC-MS/MS. Metabolic stability of APV (A), SQV (B), and NFV (C) was expressed as a percentage of control (n = 3). The metabolic stability in the control group was set at 100% for each chemical and each mouse line. Metabolic stability of MDZ (D) was used as a positive control for these experiments. *, p < 0.05 compared with the corresponding control.
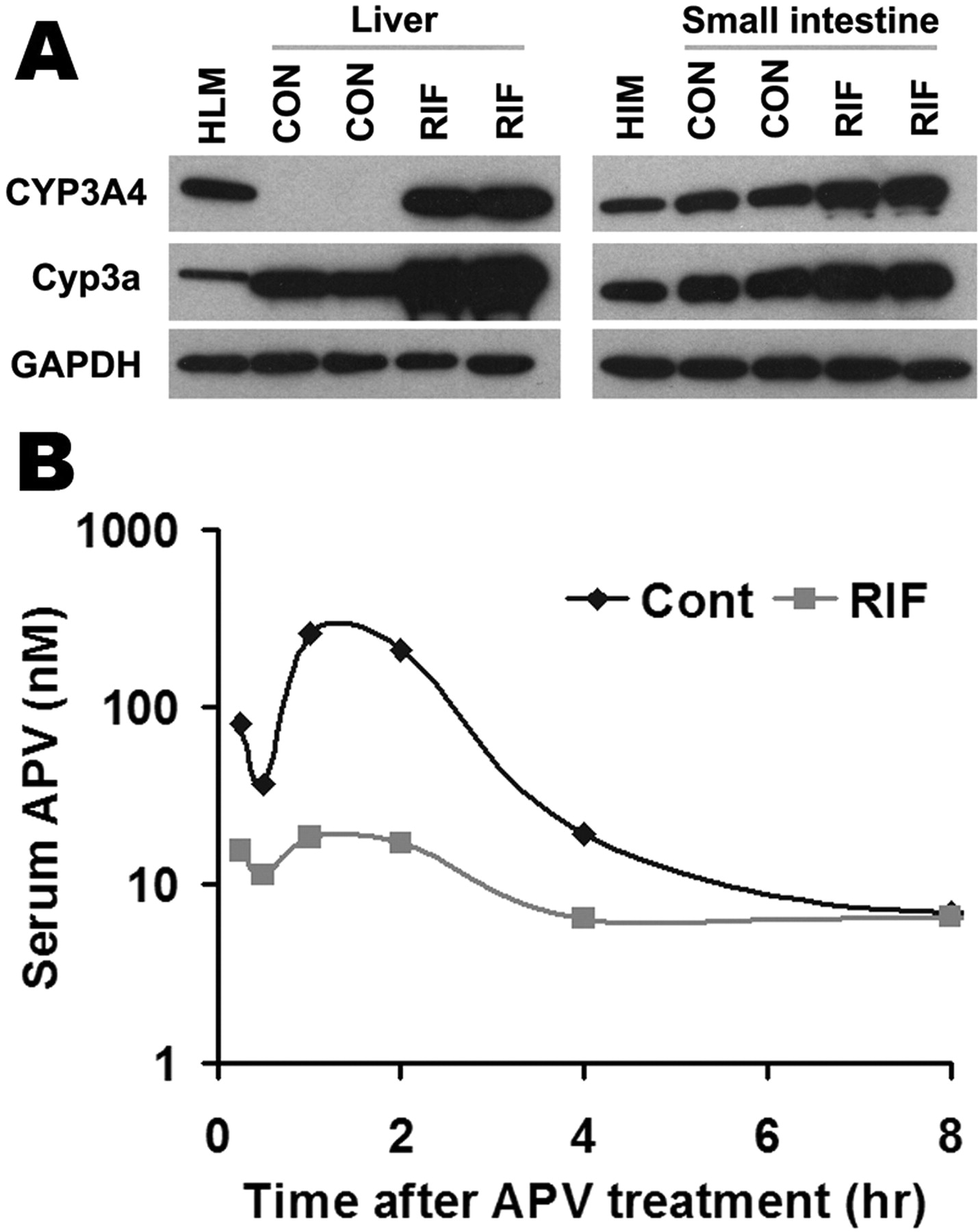
Effect of RIF on APV metabolism in vivo. TgCYP3A4/hPXR mice were treated with a purified diet (control) or a diet modified with 100 mg/kg RIF for 6 days. On the 7th day, TgCYP3A4/hPXR mice were administered 50 mg/kg APV p.o. by gavage, and blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, and 18 h after the treatment. A, expression of hepatic and intestinal CYP3A in TgCYP3A4/hPXR mice after 6 days of treatment with RIF diet. Liver and intestine microsomes were prepared and analyzed by Western blot. The monoclonal antibody against CYP3A4 (mAb 275-1-2) recognizes human CYP3A4 but not mouse Cyp3a or other liver proteins. The monoclonal antibody against Cyp3a (mAb 2-13-1) reacts with both mouse Cyp3a and human CYP3A4. Human liver microsomes (HLM) and human intestine microsomes (HIM) served as positive controls for CYP3A4. GAPDH was used as a loading control. B, time course of serum APV concentration (nanomolar) was expressed as means (pooled sample, n = 3). APV was detected by LC-MS/MS.
Because of the importance of CYP3A4 in drug metabolism, multiple in vitro and in vivo models had been generated for studies on CYP3A4. CYP3A4-expressing cell lines were produced using Chinese hamster CHL cells, yeast, and Escherichia coli (Brian et al., 1990; Zhou et al., 2001; Yamazaki et al., 2002). CYP3A4-expressing cell lines were demonstrated to be of value in the functional analysis of CYP3A4 but are limited for the study of CYP3A4 transcription and drug-drug interactions using PXR activators. By using primary cultures of human hepatocytes, transcriptional regulation and expression of CYP3A4 was recapitulated (Luo et al., 2004; Martinez-Jimenez et al., 2007). However, other CYP3A4 models should be considered because of the limited availability of human hepatocytes and the limitation of extrapolating in vitro findings to the in vivo situation. The development of CYP3A4-transgenic mouse models provides a means to overcome the limitations of in vitro models. CYP3A4-transgenic mouse models are valuable tools for studying the function and regulation of CYP3A4 in a whole animal system, and the functional significance of CYP3A4 expression can be evaluated under controlled conditions (Gonzalez and Yu, 2006). In previous studies, several CYP3A4-transgenic mouse models were generated (Granvil et al., 2003; Robertson et al., 2003; Zhang et al., 2003, 2004; Cheung et al., 2006). However, all of these mouse models are limited in transcriptional studies on CYP3A4 because of their mouse PXR background. The advantage of the TgCYP3A4/hPXR mouse line described in the current study is its human PXR background, which mimicked the human response to xenobiotics PXR activators.
In TgCYP3A4/hPXR mice, the basal expression of CYP3A4 in liver exhibited developmental expression characterized by sexual dimorphism in postpuberty. In female TgCYP3A4/hPXR mice, hepatic CYP3A4 was expressed constantly; however, in male mice, there was no hepatic CYP3A4 expression after sexual maturity. This phenomenon was also noted in the two previously generated TgCYP3A4 mouse lines (Yu et al., 2005; Cheung et al., 2006). In humans, CYP3A4 expression increases during development from childhood to adulthood, but it is not clear whether CYP3A4 expression is sex-dependent, and the conclusion on the gender difference of hepatic CYP3A activity remains uncertain although the trend exists for higher expression in females (Schmucker et al., 1990; Wolbold et al., 2003). Indeed, the sex specificity of CYP3A4 in humans is affected by multiple factors, such as dietary supplements, drinking, smoking, and medication history, which could affect expression levels through increased PXR-mediated induction. These factors can be controlled in the case of animals maintained under defined dietary and environmental conditions, suggesting that mouse models may be useful for investigation of the sexual dimorphism and developmental expression of CYP3A4. For example, it was demonstrated that growth hormone regulated sexual dimorphism in the developmental expression of the CYP3A4 transgene (Cheung et al., 2006). The major difference between the current mouse model and the previous TgCYP3A4 mouse models is the human PXR background; however, all TgCYP3A4 male mice showed similar hepatic CYP3A4 expression levels and regulation, thus suggesting that human PXR does not affect basal CYP3A4 expression.
The most common clinical implication for human PXR activation is the occurrence of drug-drug interactions mediated by the up-regulated CYP3A4. The adverse interactions of rifamycins and PIs for anti-HIV are typical examples of human PXR-CYP3A mediated drug-drug interactions. According to the World Health Organization, approximately 13 million people living with HIV infection worldwide are coinfected with TB, and TB accounts for up to one-third of deaths of patients with HIV infection. PIs are commonly used for anti-HIV treatment, and these drugs are generally metabolized by CYP3A4. Rifamycins are widely used drugs for TB treatment; however, they are also human PXR activators. Significant drug-drug interactions were noted in clinical treatment with rifamycins and PIs. Approximately 60 to 90% decreases were seen in plasma levels of PIs when combined with RIF (Niemi et al., 2003), which led to concern with the use of combined therapies for patients coinfected with HIV and TB (Breen et al., 2006; Swaminathan et al., 2006; Ribera et al., 2007). It is difficult to predict and manage drug-drug interactions mediated by human PXR and CYP3A4 in rodents because of the species differences in response to PXR ligands. By using TgCYP3A4/hPXR mice, human PXR and CYP3A4-mediated RIF-PIs interactions were revealed. A decrease in the AUC0–18 h of APV in TgCYP3A4/hPXR mice after RIF treatment was observed, similar to what was reported in humans (Polk et al., 2001). These data suggest that the TgCYP3A4/hPXR mouse is a useful tool for in vivo studies on the drug-drug interactions between PXR ligands and CYP3A substrates. However, it should be noted that data generated with this mouse model should be interpreted with caution because the hepatic blood flow rates and the distribution of CYP3A between hepatic and enteric sites may not precisely mimic those of humans, especially with the wide interindividual variation in expression in the human population.
In conclusion, a double transgenic mouse model expressing human PXR and CYP3A4 was generated successfully by BAC transgenesis. This mouse model provides a means for studying the regulation and function of human CYP3A4 gene in a whole animal model.
Acknowledgments
We thank the National Institutes of Health AIDS Research and Reference Reagent Program for providing the HIV protease inhibitors and John R. Buckley for technical assistance.
Footnotes
-
This study was funded by the National Cancer Institute Intramural Research Program. J.R.I. is grateful to the U.S. Smokeless Tobacco Company for a grant for collaborative research.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.022723.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; RIF, rifampicin; PCN, pregnenolone 16α-carbonitrile; BAC, bacterial artificial chromosome; TgCYP3A4, CYP3A4-transgenic mice; hPXR, PXR-humanized mice; TgCYP3A4/hPXR, double transgenic mice expressing human PXR and CYP3A4; HIV, human immunodeficiency virus; PI, protease inhibitor; TB, Mycobacterium tuberculosis; MDZ, midazolam; 1′-OH-MDZ, 1′-hydroxymidazolam; APV, amprenavir; NFV, nelfinavir; SQV, saquinavir; bp, base pair; PCR, polymerase chain reaction; WT, wild-type; mAb, monoclonal antibody; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LC-MS/MS, liquid chromatography-coupled tandem mass spectrometry; PCR, polymerase chain reaction; AUC, area under the concentration-time curve.
-
↵1 Current affiliation: Department of Pharmacology, Toxicology and Therapeutics, University of Kansas Medical Center, Kansas City, Kansas.
-
↵2 Current affiliation: Department of Chemical Biology, Laboratory for Cancer Research, College of Pharmacy, Rutgers University, Piscataway, New Jersey.
- Received June 5, 2008.
- Accepted September 12, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}