


Abstract
Recent regulatory guidance suggests that metabolites identified in human plasma should be present at equal or greater levels in one of the animal species used in safety assessments. In this report, a high-performance liquid chromatography-tandem mass spectrometry method is described whereby quantitative comparisons of exposures to metabolites between species can be obtained in the absence of authentic standards of the metabolites, calibration curves, and other attributes of standard bioanalytical methods. This novel method was tested using six drug-metabolite combinations. Plasma samples from animals are mixed with control plasma from humans and vice versa to remove possible differential effects of matrices. Through multiple ion monitoring-triggered enhanced product ion (EPI) scans, all metabolites were qualitatively confirmed, and daughter ions were selected for the most sensitive mass transitions to trigger EPI scans. Direct comparisons of metabolites in animal versus human plasma were achieved by calculating the peak area ratios of the metabolites versus an internal standard. Linearity of instrument responses was established by serial dilution. A statistical analysis demonstrated that experimentally measured ratios of the parent and metabolites in rat versus human correlated well with the nominal ratios of concentrations using linear regression with an average slope of 0.99 ± 0.08 (r = 0.994 ± 0.005). This analysis showed that if the experimentally determined ratio of mass spectrometer responses is ≥2.0, then the actual exposure ratio is unity or greater (p < 0.01). This method offers time- and resource-sparing advantages to ascertaining metabolite exposure comparisons between humans and laboratory animal species. A strategy for application of this approach within standard drug development processes is described.
Introduction
The use of laboratory animal species to predict the safety of new drug candidates is based on the condition that the animals have been safely exposed to the new compound at multiples of exposure to which humans will be subjected under ordinary therapeutic application. This strategy has recently been extended to include exposure to circulating metabolites of the new compound, because it is possible that toxicity could be effected by metabolites. Perspectives on the topic of drug metabolites and their relevance in safety testing, commonly known as “MIST” (Metabolites in Safety Testing) have been offered (Baillie et al., 2002; Hastings et al., 2003; Smith and Obach, 2005, 2006, 2009; Davis-Bruno and Atrakchi, 2006; Humphreys and Unger, 2006; Atrakchi, 2009; Robison and Jacobs, 2010).
The U.S. Food and Drug Administration and the International Conference on Harmonization have issued guidances defining expectations regarding the demonstration of the exposure of laboratory animals to human drug metabolites and how studies should proceed to evaluate human safety in the event that adequate exposure to human metabolites in the toxicological species is not attained through administration of the parent drug (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079266.pdf, http://www.ich.org/LOB/media/MEDIA5544.pdf). In general, for those metabolites that exceed a defined threshold in humans (i.e., 10% of total drug-related material or 10% of the exposure to parent drug in plasma, in the International Conference on Harmonization and Food and Drug Administration guidances, respectively), it is required that at least one animal species demonstrates equal or greater steady-state exposure to the metabolite than humans. If this is not achieved, then additional toxicology studies or an alternate toxicological species must be chosen to achieve the recommended exposure to the metabolite(s). Although it is possible that adequate exposure to metabolites will not be achieved in safety studies, it is more frequently the case that animals are exposed to greater concentrations of human drug metabolites, even if these metabolic pathways are relatively minor in animals (Smith and Obach, 2006). This result is probably due to the fact that the doses of parent compound administered to animals in toxicology studies far exceed the pharmacologically active doses given to humans, when corrected for relative body weight.
The measurement of steady-state plasma concentrations of drug metabolites in animals and humans can be complex and resource-intensive. The preparation of adequate quantities of characterized authentic standards of metabolites for use in assay development, validation, and analysis can be challenging and sometimes is impossible. Furthermore, development of a rigorous quantitative bioanalytical assay(s) for multiple analytes (i.e., drug + metabolites) can require considerable effort (Humphreys and Unger, 2006). Nevertheless, many drug candidates generate multiple metabolites in humans that exceed the recommended relative thresholds defined by regulatory agencies (e.g., 10% of the total drug-related material) and therefore require this investment. There have been approaches described that offer an ability to measure drug metabolites without the synthesis of an authentic standard. These have included the generation of a radiolabeled calibration standard of metabolite from a biological source (Zhang et al., 2007; Leclercq et al., 2009) or establishing a stock solution concentration of metabolite that has been isolated with 1H NMR and using that solution for the construction of standard curves (Vishwanathan et al., 2009). It has been proposed that LC-MS/MS peak areas can be used as a relative concentration comparison between animals and humans to demonstrate the coverage of metabolite exposure in animals, and some bioanalytical issues have been discussed and remain to be resolved (Leclercq et al., 2009; Walker et al., 2009).
The hybrid quadrupole linear ion trap mass spectrometer (QTRAP) has been used to quantitate the parent drug and simultaneously screen metabolites in biological matrix (Xia et al., 2003; Li et al., 2005). Multiple ion monitoring (MIM) can be used to trigger an enhanced product ion (EPI) scan to identify a large number of metabolites in a single chromatographic run (Yao et al., 2008). Likewise, multiple reaction monitoring (MRM) can be used to trigger EPI scans to profile predicted drug metabolites in in vitro samples (Shou et al., 2005; Gao et al., 2007).
In this report, we describe the use of these mass spectrometric capabilities along with a previously described algorithm for MIST (Walker et al., 2009) to produce a resource-sparing strategy for the confident determination of the relative metabolite plasma exposures between the toxicological species and humans. This strategy is based on the notion that it is unnecessary to make actual determinations of plasma concentrations of metabolites in animals and humans, but that it is only necessary to demonstrate that animals have greater exposures relative to humans. The experimental approach using LC-MS/MS peak area ratios to determine whether one species has a greater exposure to a metabolite is described herein using six drugs and their metabolites, along with a statistical analysis that demonstrates its utility and limits.
Materials and Methods
Chemicals and Reagents.
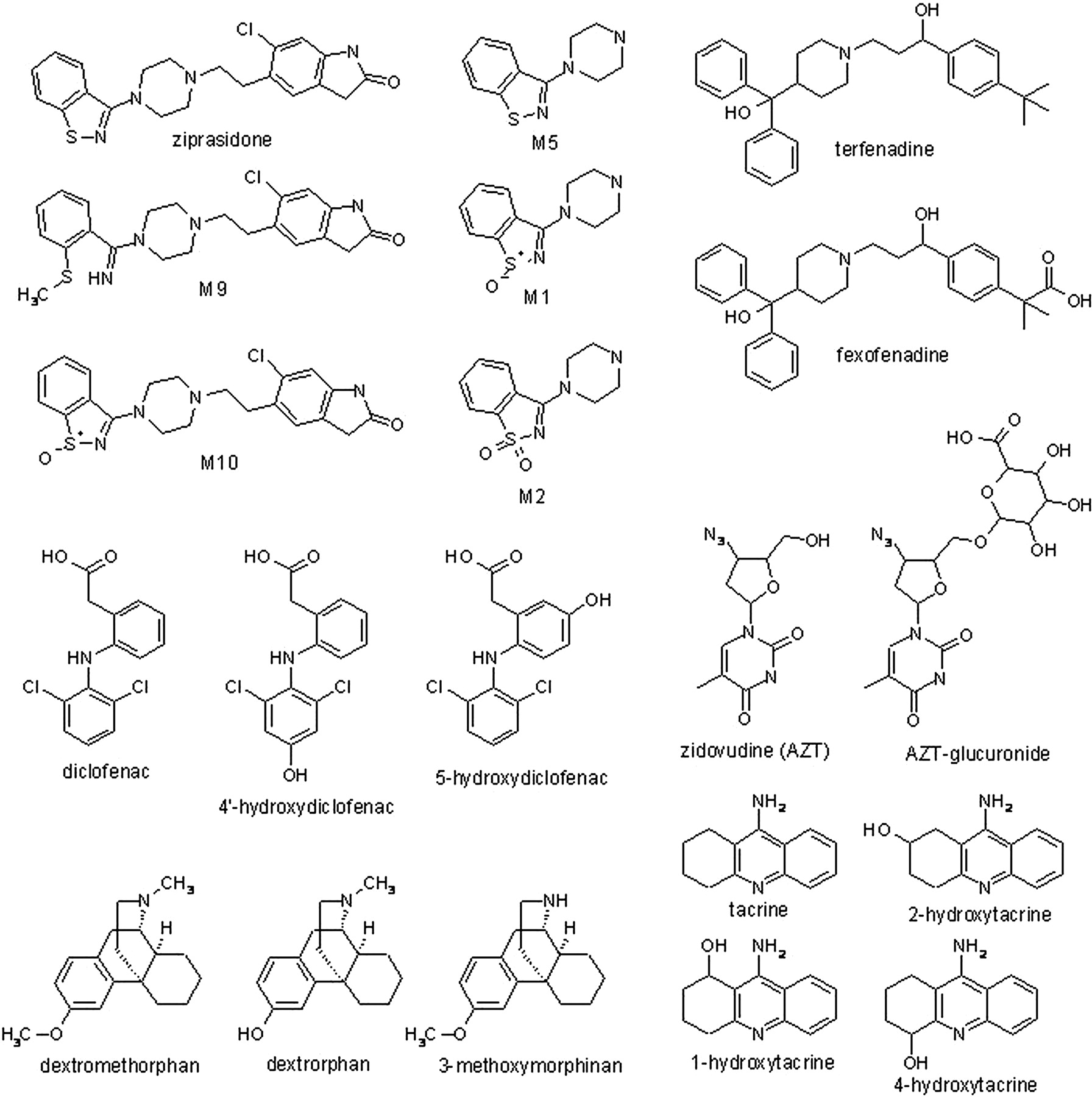
HPLC-grade water was purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). ACS reagent-grade acetonitrile was purchased from Honeywell Burdick and Jackson (Muskegon, MI). Formic acid, diclofenac, zidovudine (AZT), dextrorphan, dextromethorphan, terfenadine, and tacrine were purchased from Sigma-Aldrich (St. Louis, MO). Fexofenadine was purchased from Sequoia (Oxford, UK). Hydroxytacrine isomers, ziprasidone, and its metabolites were obtained from Pfizer Inc. (Groton, CT). 3-Methoxymorphinan was purchased from Sigma/RBI (Natick, MA). 5-Hydroxydiclofenac was purchased from Toronto Research Chemicals (Toronto, ON, Canada). AZT glucuronide and 4′-hydroxydiclofenac were prepared by Cerilliant Corporation (Round Rock, TX). Wistar Hanover rat and human K3EDTA plasma was purchased from Bioreclamation, Inc. (Westbury, NY).
Sample Preparation.
To evaluate the strategy and to mimic the types of samples one would analyze with this approach, a set of a drug and its metabolites was spiked into Wistar Hanover rat and human plasma. This was done for six drug/metabolite combinations, and each combination was spiked into rat or human plasma to generate four different sample sets to cover the range of ratios that may be produced in actual samples. (The concentrations of individual analytes are listed in the supplemental data.) Six replicates of the ziprasidone combination were prepared to access the interday assay variability of the strategy. The samples and the molecular weights of the drugs and metabolites were provided to the bioanalytical chemist, but she was blinded to the spiked metabolite concentrations.
To achieve a consistent matrix composition across all of the samples, plasma samples were diluted 1:1 with plasma of the opposite species to yield a 2-fold dilution of the samples. One sample set from each drug/metabolite combination was then serially diluted with 1:1 rat/human blank plasma to maintain the composition of the matrix and to yield 5, 10, 20, 50, and 100-fold diluted samples. One hundred microliters of all samples were then prepared for LC-MS/MS analysis by the addition of 4 volumes of 100 ng/ml sulfadimethoxine in acetonitrile as an internal standard (IS) and protein precipitation reagent except for the AZT/AZT glucuronide plasma samples for which 0.1% formic acid was added to the IS solution to stabilize the glucuronide conjugate. Samples were centrifuged, and the supernatants were dried under nitrogen and reconstituted with 20 μl of acetonitrile with 0.1% formic acid followed by 180 μl of water with 0.1% formic acid for sample analysis.
Liquid Chromatographic and Mass Spectrometric Methods.
All sample analysis was performed on an API-4000 QTRAP triple quadrupole linear ion trap mass spectrometer equipped with a Turbo V IonSpray ionization source; all instrumentation control and quantitation were through the mass spectrometer Analyst software package (Applied Biosystems, Foster City, CA). The HPLC system consisted of two Prominence LC-AD20 components (Shimadzu, Columbia, MD). The autosampler was a CTC Analytics PAL autosampler (CTC Analytics AG, Zwingen, Switzerland). Chromatographic separation was performed on a Kinetex C18 column (2.6 μm, 150 × 4.6 mm; Phenomenex, Torrance, CA). The mobile phase consisted of two solvents: solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The following linear gradient was applied as a default gradient for all drug/metabolite combinations: 0 to 3 min, 2% B (modified to 20% B for diclofenac samples); 3 to 33 min, 2 to 80% B (modified to 20 to 70% B for diclofenac samples); 33 to 36 min, 80% B (modified to 70% B for diclofenac) to 98% B; 38 to 41 min, 98 to 2% B; and 41 to 45 min, 2% B. The flow rate was 0.5 ml/min. The total run time for each injection was 45 min. A VICI valve (Valco Instruments Co., Houston, TX) was used to divert the first 0.5 min and the last 7 min of HPLC effluent to waste. The injection volume was 10 μl.
For each analyte-metabolite combination, the analyte ionization parameters were optimized by infusing solutions of the parent drugs and internal standard in 50:50 0.1% formic acid in water-acetonitrile. Ionization and mass-dependent parameters for the parent drugs were used for their metabolites except for the AZT glucuronide conjugate for which the declustering potential was lowered to 50 V, and the source temperature was lowered to 350°C. The internal standard was monitored at the MRM transition of m/z 311> m/z 156 in positive ion mode and m/z 309 > m/z 122 in negative ion mode.
An information-dependent acquisition method was used to trigger the collection of enhanced product ion scans for method development and qualitative identification of the metabolites. The first experiment of the mass spectrometer was set in the MIM or MRM mode for the parent-metabolite combinations. Detection of an ion signal greater than 5000 cps then triggered collisionally activated EPI scans to generate structurally specific dissociation of the ion. EPI spectra were collected for three consecutive scans followed by an EPI scan mode exclusion time of 1 s. The parent ion for the IS was listed in the information-dependent acquisition exclusion list across the full LC run. The EPI scan was operated at a scan rate of 4000 atomic mass units/s using the dynamic fill option in the linear ion trap. The collision energy was set at 40 eV (−20 eV for diclofenac) with a spread of 30 eV. The dwell time for each MIM/MRM transition was 50 ms, and the total cycle time was approximately 0.7 s.
MIM_EPI: Qualitative Identification of Metabolites.
In the execution of this strategy, a list of the metabolite molecular ions, some fragmentation information, and HPLC behavior will be known from preliminary metabolite scouting studies and can be provided as input information to the bioanalytical scientist. The list of metabolites can be selected as ≥10% of total parent-related material based on the radiolabeled ADME study or the estimation from LC-UV-MS responses from a multiple-dose human study. In the latter case, the accuracy of the percentage of the total parent-related materials is not critical; rather, the list of metabolites should be comprehensive to cover all metabolites suspected to be important because the mass spectrometric method has the capability of simultaneously monitoring 50 mass transitions (Gao et al., 2007). The list of metabolites should be generated during routine metabolite identification studies and transferred to bioanalytical scientist. It will then be necessary for the bioanalyst to qualitatively confirm that the ions being quantitated are indeed the targeted metabolites. Qualitative confirmation that the targeted metabolites have been identified in both animal and human plasma is achieved by injecting the 2-fold diluted samples into the LC-MS/MS system using the MIM_EPI scan mode of the instrument. During the MIM mode of the experiment, the metabolite parent ions were selected in the first and third quadrupoles with the collision cell (Q2) set at a minimal collision energy of 5 eV. Once sufficient ion counts were detected, the linear trap (Q3) of the instrument switched to the EPI mode, triggering the fragmentation and detection of the resulting daughter ions. The resulting characteristic “fingerprint” fragmentation pattern of the metabolite provides qualitative confirmation of the metabolite.
MRM_EPI and Quantitation of the Targeted Metabolites.
Assay sensitivity, selectivity from chemical noise, and quantitation were achieved by chromatographic peak quantitation in the MRM mode of the instrument using a second chromatographic injection. For each metabolite quantitated, Q1 was set at the parent ion of the metabolite, and Q3 was set to monitor one or more selected daughter ions that were observed for the metabolite in the MIM_EPI scan. The MRM transitions for the parent drug and the collision energy for the metabolites were determined from the authentic drug standard except when knowledge of a putative metabolite warranted a lower collision energy. For example, the collision energy would be scaled to lower values (e.g., 30 eV) for small molecules that are dealkylated (e.g., M1, M2, and M5 of ziprasidone). Similar to the prior experiment, the detection of sufficient ion counts triggers EPI scans so that at all times during the experiment there is qualitative confirmation that the targeted metabolite is being quantitated.
Chromatographic peaks were integrated and peak area ratios of metabolites versus internal standard at the MRM transitions were calculated. Direct comparisons of metabolites in rat versus human plasma were achieved by calculating the rat/human ratio of these peak area ratios.
Statistical Methods.
The statistical validity and limits of the strategy were determined from each dataset. The rat/human concentration ratio was calculated in a pairwise fashion for each drug and metabolite for both the experimentally determined mass spectrometric values and the known nominal concentration values. The ratios were log-transformed before analysis to stabilize the variability across the range of the ratios. The differences between log-transformed rat/human experimentally determined mass spectrometric ratios and the actual nominal ratios were calculated for all drugs and metabolites as well as for their 95% confidence intervals (CIs). These differences and CIs were anti-log-transformed back to obtain the experimentally measured mass spectrometric ratio: nominal ratios of actual concentration values and their 95% CIs. The 95% CIs were used to assess the equivalence of the two methods in determining the rat/human ratio. The absolute differences between log-transformed MS and nominal ratios were calculated for all drugs and metabolites to obtain the geometric average of the fold change between the two methods, as well as their upper 95% and 99% prediction confidence limits. The analysis was performed for both the replicated ziprasidone experiments and the multiple drug experiments. All data were analyzed in R version 2.9.1 (www.r-project.org).
Results
Daughter Ion Selection.
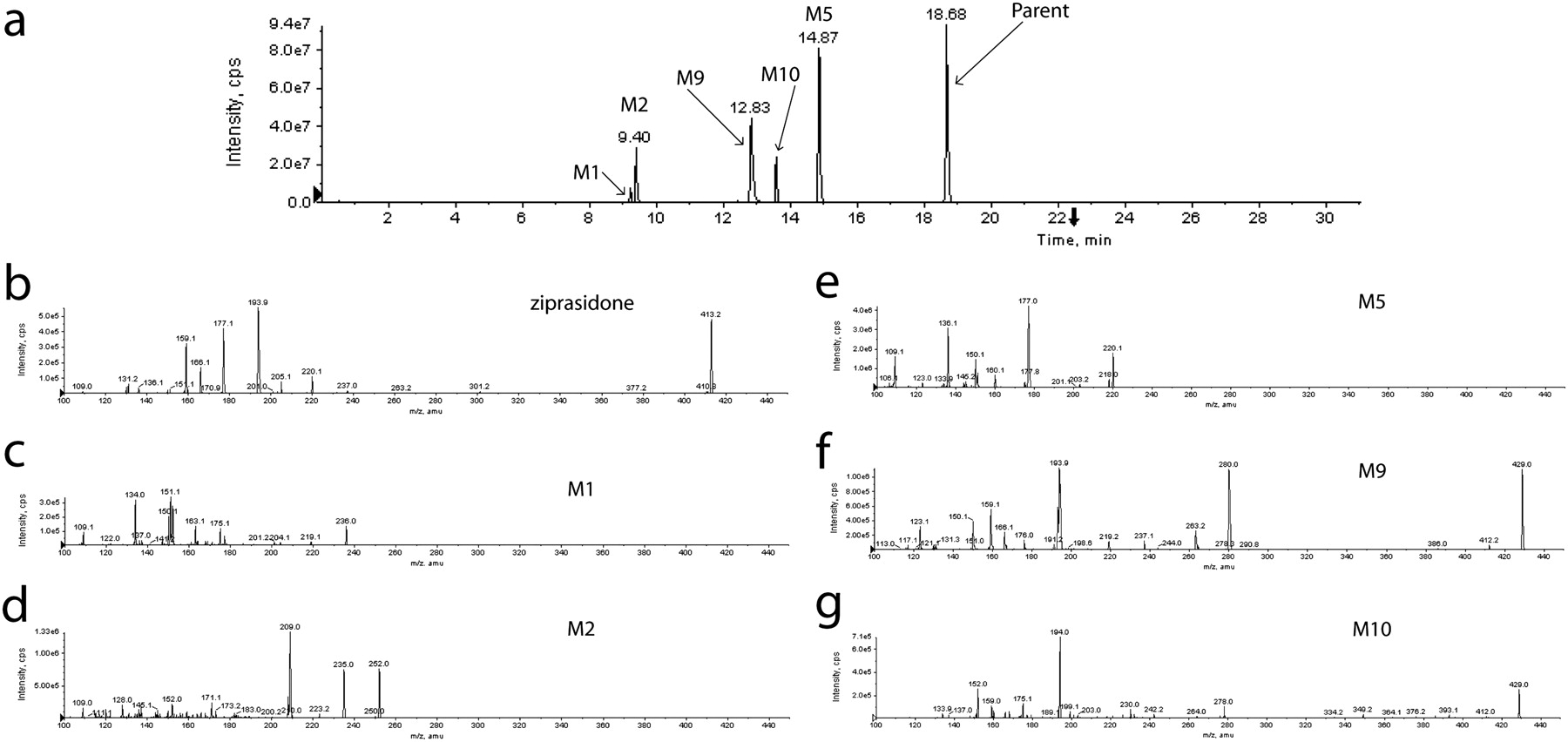
All target metabolites for the six combinations of parent drugs and their metabolites were found, and spectra were used to compare with the parent for structural confirmation. The compounds and their metabolites used for this analysis (Fig. 1) were selected to present various challenges to the strategy, such as multiple structural isomers (e.g., tacrine, diclofenac, and dextromethorphan metabolites), metabolites wherein major portions are removed (e.g., ziprasidone), phase 2 metabolites (AZT), and metabolites in which substantial changes in pKa are introduced (e.g., terfenadine and AZT). MRM transitions for the six combinations of parent drug/metabolites are listed in Table 1. The total ion chromatogram and the EPI spectra of ziprasidone and its five metabolites are shown as an example in Fig. 2, a to g.

Structures of drugs and metabolites. Glu, glucuronide.
Drug/metabolite combinations and their major daughter ions for MRM transitions

Total ion chromatogram and EPI spectra of ziprasidone and its five metabolites in rat plasma sample 1. a, total ion chromatogram of ziprasidone and five metabolites. b, EPI spectra of ziprasidone. EPI spectra of metabolites: c, M1; d, M2; e, M5; f, M9; g, M10.
The necessity for both the MIM_EPI and MRM_EPI runs for the qualitative confirmation and quantitation of the targeted metabolite is illustrated in these spectra. For example, the oxidative metabolite (M10) of ziprasidone produces fragmentation similar to that of the parent with some daughter ions common to both (e.g., m/z, 194 and 159). In this case, one can predict the MRM transitions for quantitation without the MIM_EPI experiment. However, some metabolites produce fragmentation patterns very different from those of the parent such as the N-dealkylated metabolite (M5) and the oxidized N-dealkylated metabolites (M1 and M2) of ziprasidone. In these cases the MRM transitions cannot be predicted from the parent fragmentation, and the MIM_EPI scan is required for assay development. In these experiments, we observed that the MIM_EPI scan mode produced sufficient product ions for the unambiguous confirmation of the targeted metabolites and therefore should be used as a method development tool to select the MRM transitions for semiquantitation of the metabolites. In the absence of authentic standards, we determined that multiple daughter ions for the same metabolite should be monitored with EPI scans to optimize the assay sensitivity and to ensure that the peak quantitated has been qualitatively confirmed as the targeted metabolite. This will be particularly important for the analysis of unknown samples in complex biological matrices.
Separation of Isomeric Metabolites.
Although EPI scans can produce structurally specific fragmentation patterns for the unambiguous identification of a metabolite, isomeric metabolites can and often do produce the same fragmentation patterns and are thus indistinguishable by mass spectrometry. 1-Hydroxytacrine, 2-hydroxytacrine, and 4-hydroxytacrine produced identical product ion scan spectra and major daughter ions of m/z 197 and 182 (Table 1). Likewise, 4′-hydroxydiclofenac and 5-hydroxydiclofenac produced identical daughter ions at m/z 266, 230, 194, and 166. For these sample sets, we found that the new shell particle technology provided the highest separation efficiency while maintaining a low chromatography system back pressure (Gritti et al., 2010). Adequate separation of five of the six tested drug-metabolites combinations was achieved, using a single, long default gradient on a Kinetex C18 column. The isomeric metabolites 1-hydroxytacrine, 2-hydroxytacrine, and 4-hydroxytacrine were baseline-separated using the default gradient as shown in Supplemental Fig. 1. Only 4′-hydroxydiclofenac and 5-hydroxydiclofenac could not be separated with the default gradient (Supplemental Fig. 2a), but this separation was achieved (Supplemental Fig. 2b) with minor modification of the gradient as described under Materials and Methods.
Matrix Effect.
Matrix-specific effects on ionization efficiency are a known and significant concern in the development of quantitative LC-MS/MS assays (Little et al., 2006; Ismaiel et al., 2008). Blank animal plasma was added to the human plasma samples and vice versa to normalize (but not eliminate) matrix ionization effects. The validity of matrix mixing to normalize matrices was confirmed in these experiments by monitoring the elution of phospholipids with MRM transitions (daughter ion 184) (Nouri and Wujcik, 2009) in blank human (Fig. 3a), rat (Fig. 3b), and mixed plasma (Fig. 3c). The long default chromatographic gradient with the efficient particles produced a wide chromatographic window between 3 and 26 min where no significant phospholipids elute. Most of the parent/metabolites eluted between 3 and 26 min; thus, matrix effects are minimal. Nevertheless, diclofenac and its metabolites coeluted with the phospholipids detected. Although the average of the animal/human ratio of peak area ratios was not significantly affected, it was observed that the measurement error was larger for this drug/metabolite combination.

Endogenous phospholipids in blank plasmas. a, human plasma; b, rat plasma; c, human + rat plasma (1:1).
Linearity of Instrument Response.
For this approach to provide sound cross-species comparisons, linearity in instrument response needs to be demonstrated. In this approach, the plasma sample itself represented the highest metabolite concentration that must fall within the linear range. Therefore, the linear response range of the instrument for the metabolite was defined by serial dilution of the plasma sample in the mixed blank plasma matrix and the determination of the dilution at which linear response is lost. It is the least diluted sample that falls in the linear range that is used for the calculation of the animal/human exposure ratio. Demonstration of response linearity using this approach is illustrated in Fig. 4a for the M1 metabolite of ziprasidone in rat plasma. In this example, the initial 2-fold dilution falls within the linear response of the instrument and can be used to calculate the exposure ratio. As another example, Fig. 4b illustrates the nonlinearity of response observed for the M9 metabolite of ziprasidone in rat plasma. Here, a 2-fold and even a 5-fold dilution of the sample resulted in a nonlinear response, and it was not until a 10-fold dilution of the sample was done that the linear range was defined and a peak area ratio was obtained for confident exposure comparison.

Serial dilution curve of ziprasidone metabolites in rat plasma sample 1. a, metabolite M1; b, metabolite M9.
Ratio Measurements and Error.
Six interday replicate analyses (n = 6) of ziprasidone and its metabolites done over a 3-month period demonstrated an average interday assay variability (percent coefficient of variation) of rat/human concentration ratio measurements of 26.3%. Further statistical analysis of the six replicates using the geometric average fold change demonstrated that determination of the animal/human ratio from a singlicate measurement of the samples required that a ratio ≥2.23 or ≥2.71 be measured to reach the 95 or 99% confidence levels, respectively, that the actual nominal concentration of the metabolite in animal was equal to or greater than that in humans (Table 2). The ratio that established that the plasma concentration of animal/human is equal to or greater than unity decreased to ≥1.77 or ≥1.94 to meet the 95 or 99% confidence level, respectively, when three replicate analysis were performed. Of interest, there was diminishing return in the fold change of the confidence limits beyond three replicates because the ratio needed to establish unity or greater animal/human exposure only slightly decreased to ratios of ≥1.63 and ≥1.74 to produce confidence limits of 95 and 99%, respectively when six replicate analysis were used (Table 2).
Rat/human ratios determined from mass spectrometric peak area ratios with high confidence that the nominal concentration ratio of rat vs. human ≥1
The experimentally determined individual drug or metabolite rat/human concentration ratios using this method correlated well with the nominal rat/human concentration ratios with an average correlation coefficient of 0.994 and S.D. of 0.005 over a wide dynamic range of ratios (0.01–200) as shown in Fig. 5. The average slope of the regressions was 0.99 with an S.D. of 0.082. The quality of the correlation between the experimentally determined ratios versus the actual ratios and a regression slope that was near unity demonstrated that this strategy has yielded accurate individual measurements of the relative ratios of plasma concentrations in rat versus human without authentic metabolite standards.

Correlation of rat/human ratio of mass spectrometric peak area ratio with nominal ratio of actual spiked plasma concentrations in animals versus humans for individual analyte.
Figure 6 shows the statistical analysis for all 20 analytes used for this evaluation, covering a wide range of chemical space and isomeric metabolites. Here the experimentally determined ratio value/actual ratio value is plotted for a singlicate (n = 1) measurement per sample. Unity indicates that the experimentally determined ratio is equal to the true ratio value. Almost all data points fall over the range of 0.80 to 1.3. From this plot, statistical analysis of all 20 analytes using the geometric average fold change indicated that for a singlicate measurement (n = 1) there is 95% confidence that the exposure in rat is equal to or greater than that in human when the ratio determined is ≥1.83, and this confidence increases to 99% when the ratio determined is ≥2.07 (Fig. 6). One can use these ratios and confidence limits as a guide over the entire chemical space of the dataset. An assay run in singlicate can provide meaningful data regarding MIST coverage, especially if the animal/human ratio is very high. If the ratio is less than 2.0, then it is recommended that three interday assays covering the storage time duration should be performed to provide the measurement error and indication of stability of the samples. If the ratio is still below this value, it is possible that the actual ratio is below unity and that the metabolite requires further characterization of safety.

Error distribution of singlicate measurement of six drug-metabolite combinations (n = 1). The first measurement of ziprasidone and its five metabolites was used. Geometric average fold change was used for confidence level prediction.
Discussion
The regulatory guidances on human metabolite safety assessment emphasize providing assurance that human drug metabolites were also present in animal species used in toxicology studies of the parent drug. Only in rare instances is this not the case such that further toxicological investigation of the metabolite itself is warranted. In most cases, animals will have greater exposures to metabolites than humans (by virtue of the higher doses used in animal toxicology studies relative to clinical doses). However, to demonstrate this, a bioanalytical method is needed for each human metabolite of interest. Generating and validating assays for multiple metabolites can be a large resource burden and result in delays in drug development (Humphreys and Unger, 2006). Because it is only necessary to demonstrate that animals have greater exposure levels than human, but not how much greater, a fully validated good laboratory practice (GLP) bioanalytical method to quantitate the metabolite is excessive. In this report, we have described an abbreviated but robust approach to demonstrate the relative animal/human exposures to a metabolite.
In this methodology, it has been demonstrated that the animal versus human concentration ratios determined from LC-MS/MS analyte/IS peak area ratios can adequately demonstrate whether the ratio of nominal concentrations in animal versus human is greater than unity within an acceptable error range. Of importance, this can be accomplished even in the absence of authentic standards of the metabolites, biosynthesized metabolites, or standards generated using radiometric or NMR calibration. In this strategy, the key bioanalytical mass spectrometric issues encountered when one is using response ratios for metabolite exposure assessment have been addressed, and solutions are provided: 1) the matrix difference across species was eliminated by mixing animal plasma samples with control human plasma and vice versa; 2) quantitative methods using MRM transitions were developed using MIM_EPI scans in the prepared unknown samples; 3) EPI scans triggered by MRM and MIM transitions were used to provide the confirmation of metabolite identification in unknown samples; 4) linearity of the instrument response was tested and established using serial dilution of the unknown samples in the mixed plasma matrix; and 5) selectivity for isomeric metabolites with similar mass spectra was further optimized through chromatographic separation.
The hybrid quadrupole linear ion trap spectrometer was selected for this methodology because it maintains the wide dynamic range of triple quadrupole instruments and fast scanning capability of linear trap instruments. MRM transitions themselves are not specific enough to determine metabolites in unknown samples without synthetic standards. EPI scans are necessary to confirm metabolite identification by comparing the spectra with product ion scans previously generated from metabolite profiles using other instrumentation, e.g., LTQ, Q-TOF, or Orbitrap spectrometers. The scanning mode in quadrupole yields a poor duty cycle; thus, the typical triple quadrupole mass spectrometer is not adequate for use of this strategy. Linear ion trap instruments have fast scanning capability, but the dynamic range is narrow because of space charge effects in the trap, and the low energy collisions produced by in-trap resonance excitation often require multiple stages of fragmentation (MSn) to provide the same coverage of the fragments generated in the triple quadrupole instruments (Hager and Yves Le Blanc, 2003). The ideal mass spectrometer characteristics for the execution of this strategy are a wide dynamic range, fast scanning capability, and high mass resolution for metabolite identification in unknown samples. Further advances in mass spectrometry may simplify the method development and analysis described here as well as increase the confidence level in the measurements.
The statistical analysis indicated that the concentration ratios in animal versus human experimentally determined from the mass spectrometric peak area ratios correlated well with the ratios of the actual concentrations of the analytes in animal versus human. From these data, the nominal concentration of the metabolites in animal plasma could be determined to be greater than that in human plasma with 95 or 99% confidence, if the animal/human ratio determined by this methodology is ≥1.83 or ≥2.07, respectively, for a singlicate measurement (n = 1), and the confidence level can be increased by averaging the results of additional independent measurements. Although the average ratio and error margins were improved by repeating the analysis, increasing the number of measurements beyond three did not appreciably improve the confidence limit. Pooling of the samples across pharmacokinetic/toxicokinetic sampling times using a mathematical algorithm can be done to generate a single sample for each species that reflects the exposure (Hamilton et al., 1981; Hop et al., 1998). Thus, ascertaining the metabolite exposure ratio between laboratory animals and humans can be done by analyzing as few as three samples (i.e., blank human + blank animal plasma as a control, pooled human plasma sample diluted with blank animal plasma, and pooled animal sample diluted with blank human plasma). Greater numbers of samples can be analyzed depending on the number of species and dose levels as well as on whether greater confidence in the data are desired by analysis of replicates. This methodology can provide the animal/human concentration ratios for all selected metabolites in one overnight batch analysis. In applying this methodology strategically to meet the needs of MIST, a majority of metabolites will never require synthesis, assay development and validation, or direct administration to animals if the ratio of the mass spectrometric peak area ratio is higher than the minimal ratio, e.g., ≥2.0 with 99% confidence. Instead, only a handful of metabolites may need to be investigated further if their animal/human concentration ratios are less than unity. This possibility offers tremendous resource and time savings in drug development. However, one key limitation of the method is that the stability of individual metabolites may not be fully established. Nevertheless, replicate analysis of the sample covering storage duration could indicate whether an analyte has a stability issue, i.e., the ratio would change significantly upon assay repetition. For metabolite types known to possess stability issues, (e.g., acyl glucuronide conjugates), stabilization of the metabolites in plasma samples should be undertaken when samples are collected.
A schematic diagram illustrating a strategy to use this bioanalytical approach in drug development to address steady-state metabolite exposures in human versus toxicology species is shown in Fig. 7. This approach is best applied to samples from multiple-dose studies such that steady-state metabolite exposures are assessed. Undertaking this approach can be triggered by two types of observations: 1) an actual demonstration using radiometric methods that a metabolite is present in human circulation at 10% or more of total drug-related material in a single dose ADME study or 2) anticipation that a human metabolite will be present in circulation at 10% of drug-related material. In the latter instance, such a prediction could be attempted from in vitro metabolism data or from qualitative investigations of circulating metabolite profiles in samples from phase 1 clinical studies, in which relative metabolite abundances are estimated from peak intensities in HPLC-UV-MS chromatograms. The focus of such an estimation should be to provide a comprehensive list of identified/predicted metabolites with possible MIST implications, rather than attempting to generate an accurate estimate of percentage of drug-related material. However, it should be recognized that estimates of which human metabolites will comprise >10% of total circulating drug-related material from anything other than studies using radiolabeled material may sometimes be incorrect. A major metabolite could be overlooked or a metabolite could be aggressively pursued only later to be demonstrated as a minor metabolite when the radiolabeled human study is done.

A schematic diagram for using the ratio approach for cross-species relative metabolite exposure assessments in drug development. The findings that could trigger this approach are the observation of a metabolite that is 10% or more of the total drug-related material in human plasma in the human radiolabel study or other data from which it can be reasonably anticipated that a metabolite may be more than 10% of the total in human. When this occurs, plasma samples from humans and relevant toxicology species that have been dosed to steady-state are acquired, and these samples are pooled such that a single sample per species is created in which the parent drug and metabolite(s) will be present at average concentration. These samples are analyzed as described under Materials and Methods. If the animal/human ratio of mass spectrometric peak area response, corrected for internal standard, exceeds 2.0, then no further analysis is needed because it can be concluded that animals have a higher exposure than humans. The investigator may decide to replicate this analysis twice more to gain confidence in the conclusion. If the mean animal/human ratio is less than 2.0, then replicate analyses should be done, and if the mean animal/human ratio is still less than 2.0, investment in the development of a conventional bioanalytical approach is warranted to determine the actual concentration values in individual plasma samples. If this analysis reveals that the animal/human steady-state exposure ratio is less than unity, more work will be needed to provide assurance of the safety of the metabolite. This could include seeking an alternate safety evaluation species that has greater exposure to the metabolite or direct administration of the metabolite to animals, which will require a validated GLP bioanalytical (BA) method to measure area under the curve (AUC) for exposure comparison. NOAEL, no observable adverse effect level.
When the animal/human ratio of LC-MS/MS peak area ratio for a given metabolite is ≥2.0 (if analyzed just once), then it is safe to conclude that the animal species has exposure to the metabolite that is at least equal to or greater than that in humans with a high confidence (99%). If the initially determined ratio is less than 2.0 (n = 1), and the subsequent average ratio is still less than 2.0 after three repetitive measurements (n = 3), it is possible that the exposure in animals may be lower than that in humans, and further investigation will be needed to measure the exposure values using a standard bioanalytical approach. In this case, a quantitation method using a chemically or biologically synthesized standard of the metabolite is probably needed. Should the human exposure to the metabolite still exceed that of laboratory animal species used in safety testing, further work is warranted to provide assurance of safety of the metabolite (Atrakchi, 2009; Frederick and Obach, 2010). Then it is worthwhile to check whether the metabolite is indeed ≥10% of the total drug-related material from the radiolabeled ADME study. If so, an alternative species may be selected for toxicology studies such that a greater exposure to the metabolite will be attained, or the metabolite will be directly administered to animals in a toxicology study. For these studies, a validated GLP method will be needed to analyze plasma samples to measure the circulating concentrations for comparison of the toxicokinetics to the human pharmacokinetics of the metabolite.
In this report, we have described an abbreviated, yet robust, approach to demonstrate that one species has had a greater exposure to a metabolite than another. Using six drugs and their metabolites, it was statistically shown that a response ratio of 2.0 is enough to be able to conclude that the actual concentration ratio is at least unity. The method does not require an authentic standard of the metabolite(s) and uses only a few pooled plasma samples to provide the data. Thus, it should offer a facile and inexpensive approach to provide data needed to support understanding of the safety of metabolites of new drugs.
Acknowledgments.
We thank Christopher Holliman for his support and for his review and valuable editing of the article.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.034637.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- MIST
- Metabolites in Safety Testing
- LC
- liquid chromatography
- MS/MS
- mass spectrometry
- MIM
- multiple ion monitoring
- EPI
- enhanced product ion
- MRM
- multiple reaction monitoring
- HPLC
- high-performance liquid chromatography
- AZT
- zidovudine
- IS
- internal standard
- ADME
- absorption, distribution, metabolism, and excretion
- MS
- mass spectrometry
- CI
- confidence interval
- GLP
- good laboratory practice.

- Received May 21, 2010.
- Accepted September 16, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}