


Abstract
To evaluate effects of multiple dosing of ketoconazole (KTZ) on hepatic CYP3A, the pharmacokinetics of intravenous midazolam (MDZ, 0.5 mg/kg) before and during multiple dosing of KTZ were investigated in beagle dogs. KTZ tablets were given orally to dogs (n = 4) for 30 days (200 mg b.i.d.). With coadministration of KTZ, t1/2β of MDZ were significantly increased both on day 1 (2-fold) and on day 30 (3-fold). Total body clearance (CLtot) of MDZ declined gradually during the first 5 days after the start of KTZ treatment, and thereafter CLtot appeared to reach a plateau phase (one-fourth), depending on plasma KTZ concentrations. The effects of KTZ on the biotransformation of MDZ were also investigated using dog liver microsomes (n = 5). TheKi values of KTZ for MDZ 1′-hydroxylation and 4-hydroxylation were 0.0237 and 0.111 μM, respectively, indicating that KTZ extensively inhibits hepatic CYP3A activity in dogs. CLtot values estimated from in vitroKi values corrected by unbound fraction of KTZ and unbound concentrations of the drug in plasma were consistent with in vivo CLtot of MDZ. The results in this study suggest that KTZ treatment is necessary until plasma concentrations of the drug reach a steady state to evaluate the effect of multiple dosing of the drug on hepatic CYP3A in vivo. In addition, it is suggested thatKi values corrected by unbound fraction of KTZ and unbound concentrations of the drug in plasma enable precise in vitro-in vivo scaling.
In human liver and small intestine, cytochrome P-450 3A (CYP3A) is the most important subfamily among the cytochrome P-450 superfamily. CYP3A catalyzes the biotransformation of a wide variety of exogenous and endogenous substances (Guengerich, 1999) and plays a significant role in the metabolism of about half of the available drugs (Guengerich, 1995). Because of the large number of drugs metabolized by CYP3A, the potential for drug-drug interactions to occur is substantial (Dresser et al., 2000). Drug-drug interactions may cause serious adverse effects in clinical practices. For example, case reports of drug-drug interactions resulting in adverse effects have been published for felodipine and erythromycin (Bailey et al., 1996), lovastatin and itraconazole (Lees and Lees, 1995), and cisapride and diltiazem (Thomas et al., 1998).
The antimycotic agent ketoconazole (KTZ1) is one of the potent CYP3A inhibitors (Albengres et al., 1998; Lomaestro and Piatek, 1998). Its inhibitory effects on in vitro metabolic activity of CYP3A using liver microsomes have been well studied by many investigators. They have demonstrated that KTZ is the most potent CYP3A inhibitor among all the CYP3A inhibitors tested (Newton et al., 1995;von Moltke et al., 1996; Wang et al., 1999). It has also been reported that KTZ causes clinically relevant interactions with different CYP3A substrates, including cyclosporine (Gomez et al., 1995), tacrolimus (Floren et al., 1997), and terfenadine (Honig et al., 1993). However, only few studies have been performed on the change of the decrease in CYP3A metabolic activity when KTZ is given by multiple dosing over a long term (Venkatakrishnan et al., 2000). KTZ has to be given chronically in clinical cases.
In the present study, we examined in dogs the effects of multiple oral dosing of KTZ on in vivo hepatic CYP3A activity by determining the intravenous pharmacokinetics of midazolam (MDZ), a classical probe for CYP3A activity (Thummel et al., 1994a,b). Moreover, we investigated the effects of KTZ on the biotransformation of MDZ using dog liver microsomes to quantify the inhibitory effects of KTZ on CYP3A metabolic activity and to examine whether the in vivo drug-drug interaction was quantitatively predictable even in the situation that the interacting drug was administered over a long term. We selected beagle dogs as an animal model because the animals have been extensively used to investigate metabolism of xenobiotics both in vivo and in vitro and the plasma concentration-time profile of KTZ after oral administration in dogs is similar to that in humans, although body weight-standardized dose is different between the species (Baxter et al., 1986).
Experimental Procedures
Materials.
KTZ was purchased as a tablet (Nizoral) from Janssen Pharmaceutica (Titusville, NJ) and as a reagent from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). MDZ and the metabolites, 1′-OH MDZ and 4-OH MDZ, were obtained from Daiichi Pure Chemicals Co., Ltd. (Tokyo, Japan) and a MDZ injectable solution (Dormicam) was from Yamanouchi Pharmaceutical Co. (Tokyo, Japan). Diazepam was obtained as a reagent from Sigma (St. Louis, MO). All other chemicals used as reagents were of analytical and HPLC grade.
Animals.
Nine beagle dogs (male, 1-year-old) weighing 10 to 15 kg were obtained from CSK Research Park Co. Ltd. (Nagano, Japan). Four of nine dogs were used for in vivo studies; five were used for in vitro studies. The dogs were allowed access to water ad libitum and were given food twice a day (8 AM and 8 PM).
In Vivo Studies.
Study design
Four beagle dogs were used to investigate the effects of multiple dosing of KTZ on hepatic CYP3A activity. KTZ was given orally to the dogs 1 h after feeding for 30 days (200 mg/body b.i.d.; 9 AM and 9 PM). MDZ (0.5 mg/kg) was intravenously administered 7 days before the beginning of multiple dosing of KTZ (day 0 as control) and 1 h after KTZ administration on the morning of days 1, 2, 3, 5, 8, 12, 19, and 30 after the beginning of the KTZ treatment to evaluate hepatic CYP3A activity in vivo by means of total body clearance (CLtot) of MDZ (Thummel et al., 1994a,b).
Blood sampling.
Blood samples (2.5 ml) were collected at 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, and 2 h (on days 0, 1, 2, and 3) or at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, and 4 h (on days 8, 12, 19, and 30) after MDZ injection for measurement of plasma MDZ concentrations. According to the previous study (Baxter et al., 1986) and our preliminary study on pharmacokinetics of KTZ in beagle dogs after single orally administration of KTZ (data not shown), blood samples (1 ml) were obtained at 3 and 12 h after KTZ administration for the determination of peak and trough concentrations of KTZ in plasma. Plasma was separated from whole blood by centrifugation and stored at −80°C before HPLC analysis.
In Vitro Studies.
Preparation of dog liver microsomes
Five beagle dogs were euthanized by intravenous pentobarbital injection (25 mg/kg) to obtain liver samples. The microsomal fractions were prepared as described by van der Hoeven and Coon (1974). The obtained samples were frozen at −80°C until used. The protein levels and cytochrome P-450 contents were determined as described by Lowry et al. (1951) and Omura and Sato (1964), respectively.
Enzyme kinetic analysis.
The kinetic and inhibition studies for MDZ in dog liver microsomes were performed on the incubation condition described as follows: 0.23 ml of incubation mixture containing dog liver microsomes (approximately 0.5 mg/ml) and an NADPH generating system [50 mM phosphate buffer (pH 7.4), 0.5 mM NADP, 5 mM glucose 6-phosphate, 0.4 U glucose-6-phosphate dehydrogenase, and 5 mM MgCl2] were preincubated at 37°C for 5 min. Varying quantities of MDZ in 1% methanol solution, to yield final incubate concentrations ranging from 6.14 to 368 μM, were added to a series of incubation tubes. For inhibition studies, incubations were also performed with coaddition of KTZ in ethanol solution (final concentration 4.7, 9.4, and 18.8 μM) because preliminary studies showed that the KTZ concentrations were more suitable than the others for estimating inhibition constant (Ki). Formation of metabolites was linear with respect to incubation time (0–10 min) and microsomal protein concentration (0–0.5 mg/ml). The reactions were initiated by adding 10 μl of MDZ solution and 10 μl of ethanol (without KTZ) or KTZ solution. The final reaction volume was 250 μl. After incubation at 37°C for 10 min, the enzyme reactions were terminated with 100 μl of acetonitrile and placed on ice. After centrifugation at 3000g for 5 min, the resulting supernatant was immediately applied to HPLC system.
Microsomal protein binding of MDZ and KTZ.
To obtain unbound concentrations of MDZ and KTZ in the assay system, the binding of the drugs to microsomal protein was determined. One milliliter of the mixture as the same in enzyme kinetic analysis except NADP was incubated at 37°C for 30 min and transferred to a device of ultrafiltration kit (Centrifree; Amicon, Beverly, MA), followed by centrifugation at 2000g for 5 min. The resulting ultrafiltrate was immediately analyzed to obtain unbound concentrations of MDZ and KTZ.
The binding percentages were 35.8 ± 4.1% (CV = 11.5%) at 3.07 μM and 36.4 ± 3.7% (CV = 10.2%) at 30.7 μM for MDZ; 71.0 ± 3.4% (CV = 4.8%) at 18.8 μM for KTZ, respectively (n = 4). The inter-day CV values ranged from 2.0 to 15.3% at 3.07 μM and from 3.0 to 13.6% at 30.7 μM for MDZ and from 1.4 to 8.0% at 18.8 μM for KTZ, respectively (during 3 days, four determinations for each day).
Plasma protein binding of KTZ.
The unbound fraction of KTZ in plasma was determined by an ultrafiltration method to perform in vitro-in vivo scaling, as described under Data Analysis. Blood was collected from the dogs used in in vitro study when the liver was obtained from the animals. The separated plasma was pooled and used for plasma protein binding study. Ten microliters of KTZ solution (2.5 mg/ml in ethanol) was added to 990 μl of plasma. The mixture was transferred to a device of ultrafiltration kit (Centrifree; Amicon) and centrifuged at 2000g for 5 min. The resulting ultrafiltrate was immediately injected into an analytical column (described below). The binding percentages of KTZ in plasma were 96.8 ± 0.7% (CV = 0.7%,n = 4). The inter-day CV values ranged from 0.7 to 0.9% (during 3 days, four determinations for each day).
HPLC Analysis.
MDZ concentrations were analyzed by HPLC with UV detector as previously described (Carrillo et al., 1998) using diazepam as an internal standard. For plasma concentrations, thawed plasma (1 ml) was placed in 10-ml test tubes basified with glycine buffer (0.75 M, pH = 9). All tubes were spiked with 50 μl of internal standard containing 0.64 nmol of diazepam. The samples were extracted with 4 ml of diethyl ether, and the upper organic layer was transferred to clean conical tubes. The solvent was evaporated to dryness under a stream of nitrogen. The remaining solid was reconstituted with 200 μl of mobile phase, and 50 μl of the solution was injected to HPLC. For unbound concentration in microsomal solution, the ultrafiltrate was directly applied to HPLC. The mobile phase consisted of 100 mM acetate buffer (pH 4.7 with acetic acid), acetonitrile, and methanol (53:41.4:5.6, v/v/v); the flow rate was 1.0 ml/min. The analytical column was a reversed-phase TSK-gel ODS-120T 250 × 4.6-mm i.d. (TOSOH Co., Tokyo, Japan). Column effluent was monitored by UV absorbance at 254 nm. The recovery of MDZ was 79.3 ± 8.7% at 100 ng/ml and 80.2 ± 9.5% at 1000 ng/ml (n = 4). The quantification limit for MDZ was 2.5 ng/ml, and the interassay coefficient of variation was 5.5 to 11.2% at 100 ng/ml and 5.1 to 6.4% at 1000 ng/ml (during 3 days, four determinations for each day). The calibration curve was linear over a concentration range of 2.5 to 2500 ng/ml (r2 = 0.99).
KTZ concentrations were determined by HPLC with fluorescence detector as previously described (Yuen and Peh, 1998) with a slight modification. For concentrations in plasma, an aliquot of 250 μl was deproteinized with 3 volumes of acetonitrile, followed by centrifugation at 10,000g for 5 min, and the resulting supernatant was applied to HPLC system. For unbound concentrations in plasma or microsomal solution, the ultrafiltrate was directly applied. The mobile phase consisted of 50 mM Tris buffer (pH 6.0 with 2 N hydrochloric acid) and acetonitrile (60:40, v/v); the flow rate was 1.5 ml/min. The analytical column was a reversed-phase phenyl column (Radial-Pak Cartridge, 100 × 5-mm i.d.; Waters Associates, Inc., Milford, MA). The recovery of KTZ was 98.5 ± 3.5% at 100 ng/ml and 99.1 ± 1.9% at 1000 ng/ml (n = 4). The quantification limit for KTZ was 10 ng/ml, and the interassay coefficient of variation was 3.4 to 4.7% at 100 ng/ml and 1.3 to 2.1% at 1000 ng/ml (during 3 days, four determinations for each day). The calibration curve was linear over a concentration range of 0.05 to 50 μg/ml (r2 = 0.99).
Concentrations of 1′-OH MDZ and 4-OH MDZ in the in vitro assay system were determined by HPLC, as previously described (von Moltke et al., 1996), using diazepam as an internal standard. The reaction solutions terminated with 100 μl of acetonitrile were centrifuged at 10,000g for 5 min, and the resulting supernatant was immediately applied to HPLC system. The peak area was linearly related to the concentration within the tested range of 10 to 1000 ng/ml for 1′-OH MDZ (r2 = 0.99) and 4-OH MDZ (r2 = 0.98). The recovery of 1′-OH MDZ and 4-OH MDZ were 101 ± 3 and 101 ± 6% at 1000 ng/ml, respectively (n = 4). The interassay CV values were 2.4 to 4.9% for 1′-OH MDZ and 3.7 to 7.6% for 4-OH MDZ at 1000 ng/ml (during 3 days, four determinations for each day).
Data Analysis.
Pharmacokinetic parameters after intravenous MDZ administrations
Plasma concentration-time curves after MDZ injection were fitted to the following biexponential equation using the nonlinear least-squares regression (Yamaoka et al., 1981).
The kinetic parameters for metabolism of MDZ and inhibition by KTZ in dog liver microsomes. As previously reported in humans and mouse (von Moltke et al., 1996; Warrington et al., 2000), formation of 4-OH MDZ from MDZ without KTZ was consistent with single-enzyme Michaelis-Menten kinetics, and the formation with KTZ was consistent with single-enzyme Michaelis-Menten kinetics and competitive inhibition (Fig. 3A). Accordingly, the following equations were fitted to the observed data using the nonlinear least-squares regression (Yamaoka et al., 1981) to estimate kinetic parameters for 4-hydroxylation of MDZ and inhibition by KTZ in dog liver microsomes:
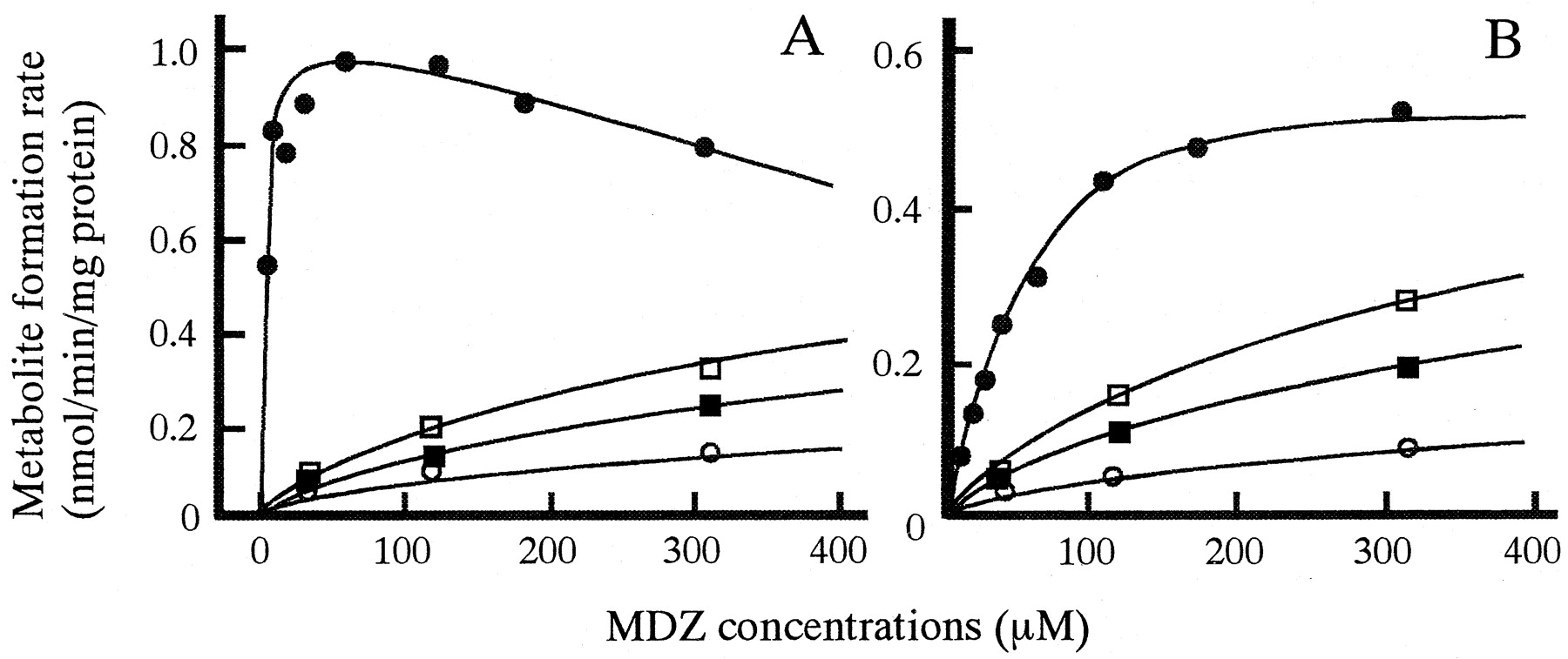
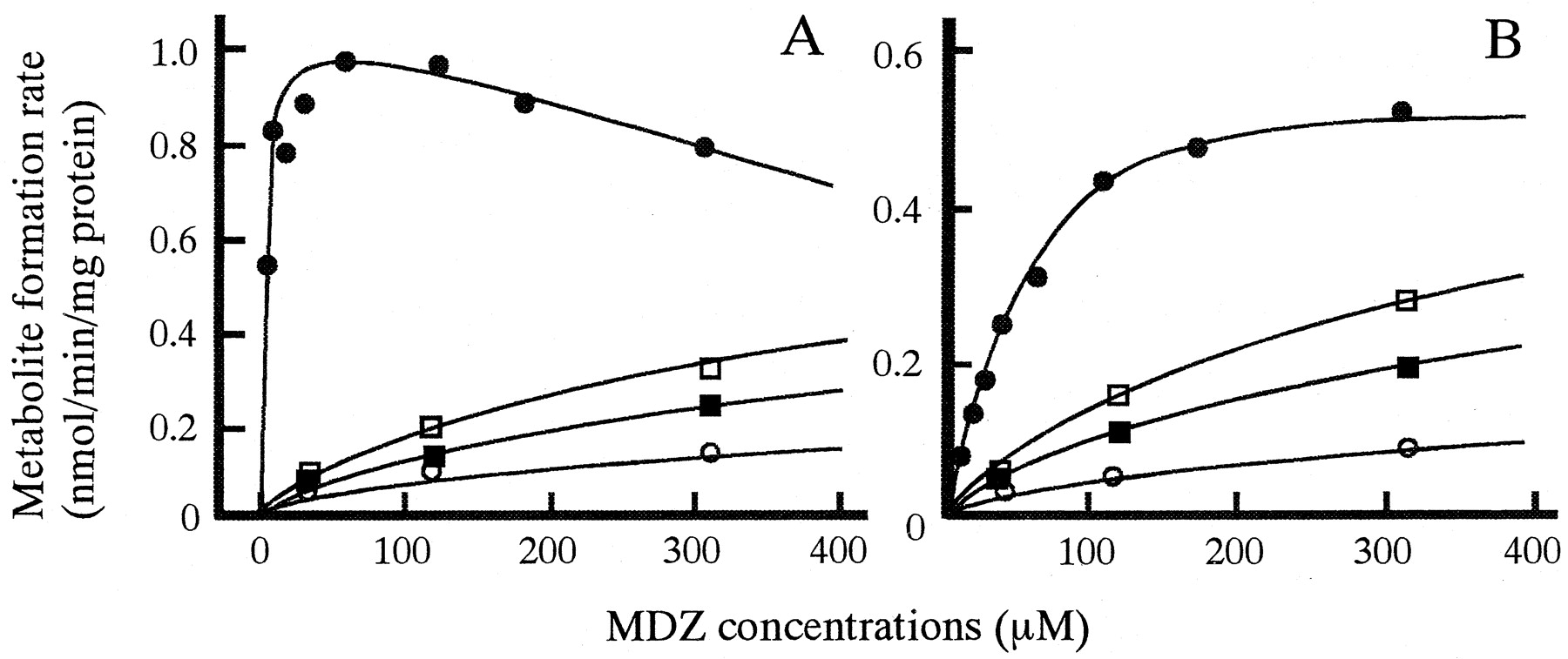
In vitro biotransformation of MDZ to 1′-OH MDZ (A) and 4-OH MDZ (B) and inhibition by KTZ in microsomes prepared from a representative dog liver.
MDZ was incubated without KTZ (closed circles) or with 4.7 μM KTZ (open squares), 9.4 μM KTZ (closed squares), or 18.8 μM KTZ (open circles). Lines were calculated by the nonlinear least-squares regression analysis.
The pattern of 1′-OH MDZ formation from MDZ was consistent with Michaelis-Menten kinetics with uncompetitive substrate inhibition, and the inhibition by KTZ showed competitive inhibition (von Moltke et al., 1996; Warrington et al., 2000) (Fig. 3B). The following equations were fitted to 1′-OH MDZ data points:
The prediction of inhibitory effect of KTZ on in vivo clearance of MDZ.
The decreasing ratio of intrinsic clearance both for 1′-hydroxylation and for 4-hydroxylation were estimated from in vitro data using the following equation:
We attempted to determine whether the in vivo inhibitory effect of KTZ on hepatic CYP3A activity was predictable in quantity from in vitro data. CLtot of MDZ predicted from in vitro data (CLtot in vitro) was calculated usingVmax, Km, andKi represented in Table 2 and plasma unbound concentrations of KTZ. The following assumptions were used in this calculation: 1) the elimination of MDZ from body is only by metabolism, and transformations from MDZ account for only 1′- and 4-hydroxylation because the unchanged drug was not detected in urine 0 to 6 h after intravenous administration of the drug to dogs and because the sum of concentrations of MDZ, 1′-OH MDZ, and 4-OH MDZ 1 h after incubation in vitro was almost consistent with MDZ concentration before incubation in preliminary study; 2) the unbound concentrations of KTZ in liver are equal to those in plasma; and 3) the plasma unbound fraction for MDZ is not affected by KTZ. First, the product of plasma unbound fraction and CLint for MDZ (fu × CLint) was calculated by substituting CLtot on day 0 obtained from in vivo studies to the following equation:
Kinetic parameters for the biotransformation of MDZ and inhibition by KTZ in dog microsomes
Statistical analysis.
Differences in the pharmacokinetic parameters before and with KTZ were analyzed by use of the paired Student's t test and were regarded as statistically significant when p values were below 0.05.
Results
Effects of Multiple Oral Dosing of KTZ on the Pharmacokinetics of i.v. MDZ in Dogs.
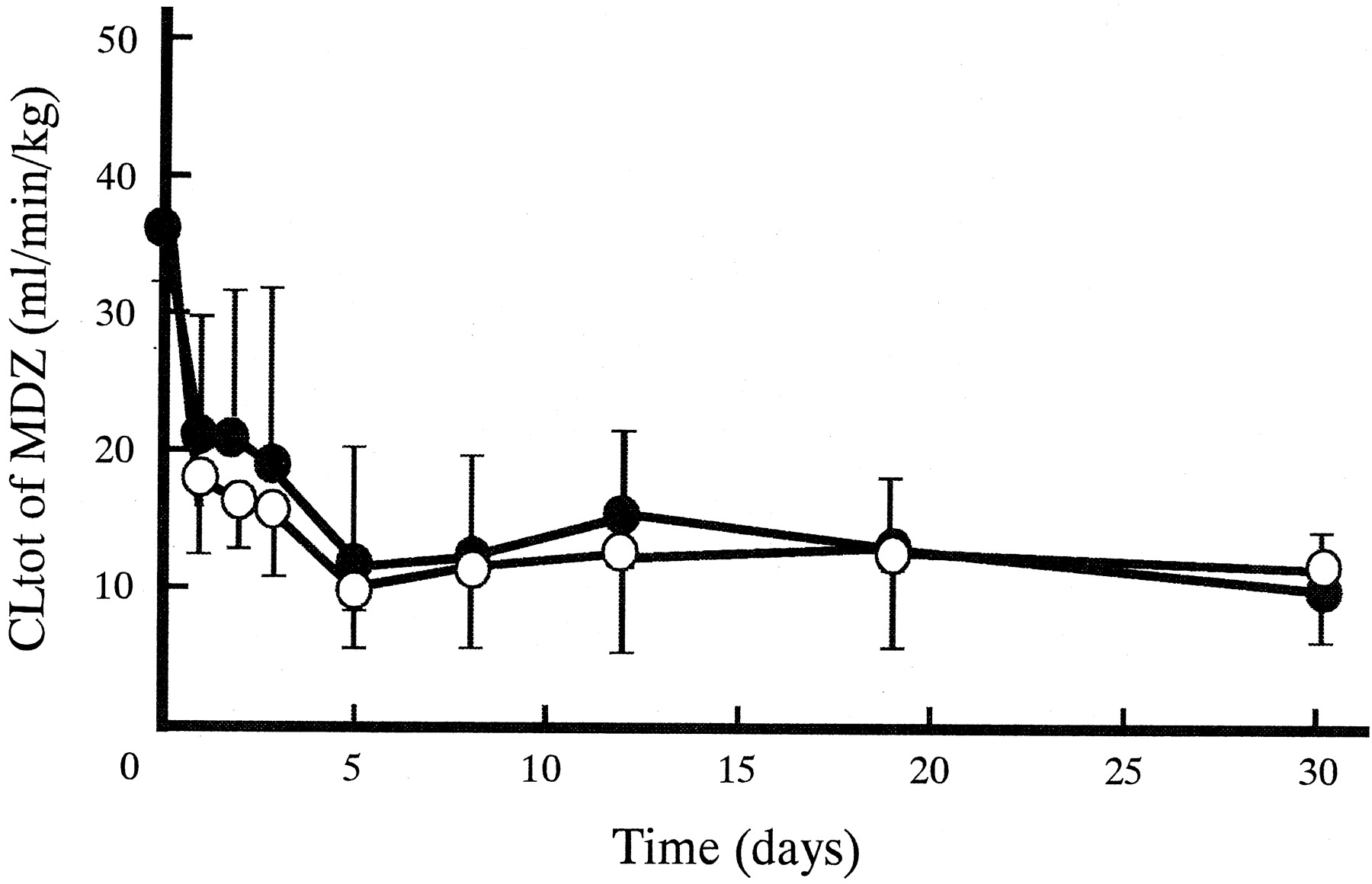
Figure 1 shows plasma concentration-time profiles of MDZ after intravenous administrations on day 0 (without KTZ), 1 (corresponding to one single dosing of KTZ), and 30 (after the last dose of KTZ). Plasma MDZ concentrations increased by oral KTZ administration. The extent on day 30 was much larger than that on day 1. The pharmacokinetic parameters for MDZ on day 0, 1, and 30 are represented in Table 1. With coadministration of KTZ, the t1/2β for MDZ significantly increased both on day 1 (2-fold) and on day 30 (3-fold). Statistically significant decreases of CLtot were observed both on day 1 (one-half) and on day 30 (one-fourth). Compared with the parameters on day 1,t1/2β on day 30 was 2-fold larger whereas CLtot was halved. Vdss for MDZ did not change. Figure 2 shows the changes in CLtot for MDZ after the start of multiple oral dosing of KTZ. CLtot for MDZ declined gradually during the first 5 days, and thereafter it appeared to reach a plateau phase at about 10 ml/min/kg. During the experiment, significant decreases in CLtot were observed.

Plasma MDZ concentration-time profiles after intravenous administration (0.5 mg/kg) on day 0 (open squares), 1 (closed circles), and 30 (open circles) after multiple oral dosing of KTZ (200 mg b.i.d.).
Each point represents the mean ± S.D. (n = 4). Lines were calculated by the nonlinear least-squares regression analysis. ∗, significant differences between the parameters on day 0 and the others by the paired Student's t test (p < 0.05). ‡, significant differences between the parameters on day 1 and 30 by the paired Student'st test (p < 0.05).
Effects of multiple oral dosing of KTZ (200mg b.i.d.) on pharmacokinetic parameters of MDZ (0.5mg/kg) after intravenous administrations to beagle dogs

Effect of multiple oral dosing of KTZ (200 mg b.i.d.) on CLtot for MDZ (0.5 mg/kg i.v.) in dogs.
Each point represents the mean ± S.D. (n = 4). Significant differences between the value of CLtot on day 0 and the others were observed by the paired Student'st test (∗, p < 0.05).
The Profiles and Kinetic Parameters for the Biotransformation of MDZ and the Inhibition by KTZ in Dog Liver Microsomes.
Figure 3 shows the profiles for 1′-hydroxylation and 4-hydroxylation of MDZ and the inhibition by KTZ in microsomes prepared from a representative dog liver. KTZ competitively inhibited both pathways of MDZ metabolism. Table2 shows the kinetic parameters for biotransformation of MDZ and the inhibition by KTZ in dog liver microsomes. The mean value of Km for formation of 1′-OH MDZ was 4.84 μM, with a high affinity. TheKs (substrate inhibition constant) values averaged 443 μM, and ranged from 275 to 704 μM. The mean value ofKm for formation of 4-OH MDZ was approximately 5-fold larger than that of 1′-OH. These results show that CLint for 1′-hydroxylation accounted for more than 90% of the sum of both pathways. TheKi values for both pathways were approximately 100-fold lower than those ofKm, indicating that KTZ possesses potent inhibitory effects on both pathways of MDZ metabolism in dogs.
The Prediction of Inhibitory Effect of KTZ on in Vivo Clearance of MDZ.
Average concentrations of KTZ in plasma on day 30 ranged from 3.64 to 19.1 μg/ml (Fig. 4). Using these values and unbound fractions of KTZ (fu = 0.032), plasma unbound concentrations of KTZ were obtained (0.219–1.15 μM). The ratio of intrinsic clearance on day 30 after the beginning of multiple dosing of KTZ (CLint′) to that without KTZ (CLint) calculated from eq. 6 are represented in Table 3. CLint for formation from MDZ to 1′-OH MDZ decreased to about 10- to 50-fold by KTZ, whereas that for 4-OH MDZ decreased to about 3- to 10-fold. Therefore, the total intrinsic clearance of MDZ metabolism decreased to about 9- to 40-fold by KTZ. The profiles of CLtotof MDZ predicted from in vitro data (CLtot in vitro) and the observed CLtot in vivo (CLtot in vivo) are shown in Fig.5. The plot of the CLtot in vitro values against CLtot in vivo values is also represented in Fig. 6. Both Figs. 5 and 6 shows that the CLtot in vivo and CLtot in vitro were almost comparable.

Plasma KTZ peak concentrations in each dog after multiple oral dosing of KTZ (200 mg b.i.d.).
Open circles, dog 1; open squares, dog 2; closed squares, dog 3; closed circles, dog 4.
The extent of decrease in in vivo CLint for MDZ metabolism predicted from in vitro data

Comparison of CLtot for MDZ calculated from in vitro data (CLtot in vitro, open circles) with that observed in vivo (CLtot in vivo, closed circles) after the start of multiple oral administration of KTZ (200 mg b.i.d.) in dogs (n = 4).
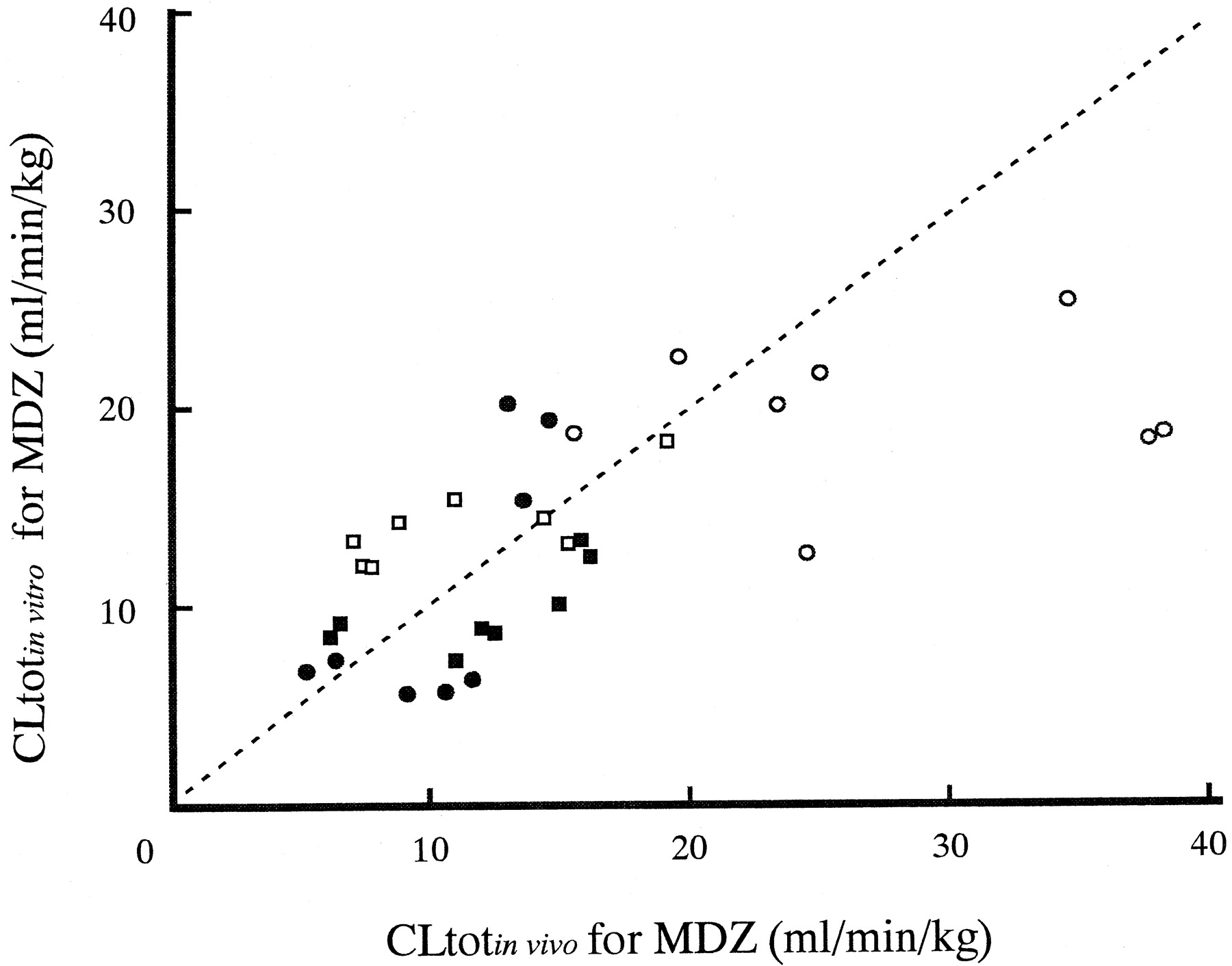
Correlation of CLtot for MDZ between observed in vivo (CLtot in vivo) and that calculated from in vitro data (CLtot in vitro) in dogs.
The dotted line indicates a 1:1 correspondence. Open circles, dog 1; open squares, dog 2; closed squares, dog 3; closed circles, dog 4.
Discussion
The purpose of this study was to investigate the effects of multiple oral dosing of KTZ on hepatic CYP3A activity because KTZ is chronically administered to patients. MDZ was intravenously administered to assess hepatic CYP3A activity (Thummel et al., 1994a,b). Moreover, it was tested by inhibition study using liver microsomes to determine whether the drug-drug interaction was quantitatively predictable even in the situation that the interacting drug was administered over a long term.
Plasma concentration-time profiles of i.v. MDZ in dogs were markedly affected by coadministration of KTZ (Fig. 1). KTZ caused increases in AUC after intravenous MDZ administrations (2-fold on day 1; 3- to 4-fold on day 30). Also, KTZ significantly changedt1/2β and CLtot of MDZ in contrast to Vdss (Table 1). These observations are consistent with those reported in a previous study with humans (Tsunoda et al., 1999). These results indicate that KTZ inhibits hepatic CYP3A activity in dogs and in humans.
The change in CLtot of MDZ after the start of multiple oral dosing of KTZ was investigated in this study. CLtot of MDZ decreased to 30% during the first 5 days, and thereafter reached a plateau phase (Fig. 2). Peak concentrations of KTZ in plasma also reached a steady state at about 5 days after the start of KTZ administrations (Fig. 4). These results show that the extent of inhibition of in vivo CYP3A activity depends on plasma KTZ concentration, suggesting that its inhibition by oral KTZ administration may be accounted for only by competitive inhibition on hepatic CYP3A. Therefore, KTZ treatment may be necessary until plasma concentration of the drug reaches a steady state to evaluate the effect of multiple dosing of the drug on hepatic CYP3A in vivo in humans and in dogs.
Because data of the effect of multiple oral dosing of KTZ on pharmacokinetics of i.v. MDZ in humans are lacking, the result on day 1 in this study was compared with that in humans described by Tsunoda et al. (1999). Plasma KTZ concentrations on day 1 in this study were close to those in the human study. The single dose of KTZ decreased CLtot of MDZ to 20% in humans and to 50% in dogs (Fig. 2). However, this comparison is incorrect because CLtot does not reflect directly hepatic metabolic capacity, especially in the case of drugs that represent a high extraction ratio (Ito et al., 1998), and because CLtot of MDZ represents a high extraction in dogs and a low extraction in humans (Tsunoda et al., 1999).
To compare the effects of KTZ on the metabolic capacity for MDZ or hepatic CYP3A activities between humans and dogs CLint that directly represents metabolic capacity was calculated by substituting CLtot to eq. 7. In this calculation, 25.4 and 42.3 ml/min/kg were used as the values ofQ for humans and dogs, respectively (Boxenbaum, 1980). The calculation shows that KTZ decreases CLint for MDZ to 16% for humans and to 26% for dogs. For estimatingKi values from eq. 6, inhibitor concentrations (I) were calculated by multiplying plasma KTZ concentrations (about 3 μg/ml for humans; approximately 4.4 μg/ml for dogs) by plasma unbound fraction. Substituting the calculatedR values and I to eq. 6, it was estimated thatKi values of KTZ for MDZ metabolism in humans is about 10-fold smaller than in dogs. Therefore, it is suggested that KTZ inhibits hepatic CYP3A activity more extensively in humans than in dogs.
Moreover, to quantify the inhibitory effect of KTZ on hepatic CYP3A in dogs, in vitro studies using prepared dog liver microsomes were conducted in this study. The Ki values of KTZ were markedly small, indicating that KTZ strongly inhibits MDZ metabolism. Comparison of Ki values of KTZ in dogs with those in humans shows that KTZ inhibits hepatic CYP3A stronger in humans than in dogs (von Moltke et al., 1996), supporting the possibility that KTZ inhibits in vivo hepatic CYP3A activity in humans more extensively than in dogs.
As shown in Fig. 5, CLtot values estimated from in vitro data were consistent with in vivo CLtotof MDZ. This success may be attributed to the following: 1) we used unbound Ki values based on Michaelis-Menten theory but not Ki values calculated from total concentration of KTZ, and 2) we also used unbound concentrations of KTZ in plasma as I on the assumption that the drug entered hepatocytes only by passive diffusion because the drug has not been reported to be actively transported from plasma into hepatocytes.von Moltke et al. (1998) successfully predicted in vivo inhibition of MDZ clearance by KTZ using total plasma concentration andKi values calculated from total concentration of KTZ. This success may be due to the similarity of unbound fraction of KTZ in an assay system containing microsomal proteins to that in plasma. In many cases, however, the similarity seems to be rare. Therefore, plasma unbound concentration of inhibitor and Ki values corrected by unbound fraction of inhibitor may result in precise in vitro-in vivo scaling in case of inhibitor that may be not actively transported into the liver.
In summary, multiple oral dosing of KTZ to dogs gradually decreased the CLtot of MDZ during the first 5 days, and thereafter CLtot almost stabilized during experimental period. These changes depended on plasma KTZ concentration, suggesting that its inhibition by oral KTZ administration may be accounted only by competitive inhibition on hepatic CYP3A. Therefore, KTZ treatment may be necessary until plasma concentration of the drug reaches a steady state to evaluate the effect of multiple dosing of the drug on hepatic CYP3A in vivo in humans and dogs. In vitro-in vivo scaling of KTZ inhibition was examined usingKi values corrected by unbound fraction of KTZ and unbound concentrations of the drug in plasma. CLtot of MDZ estimated from in vitro data was consistent with in vivo CLtot. For precise in vitro-in vivo scaling also in humans, we recommend use ofKi values corrected by unbound fraction of KTZ and unbound concentrations of the drug in plasma but not total concentration.
Acknowledgments
We thank Dr. A. S. J. P. A. M. van Miert for helpful comments about the manuscript. We thank Shizuka Kishimoto and Hideki Kayaba for skillful technical assistance.
Footnotes
- Abbreviations used are::
- KTZ
- ketoconazole
- CLtot
- total body clearance
- MDZ
- midazolam
- 1′-OH MDZ
- 1′-hydroxymidazolam
- 4-OH MDZ
- 4-hydroxymidazolam
- Vdss
- volume of distribution at steady state
- t1/2β
- half-life in the β-phase
- Received February 7, 2001.
- Accepted July 18, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}