


Abstract
Delavirdine mesylate (U-90152T) is a highly specific nonnucleoside reverse transcriptase inhibitor currently under development for the treatment of AIDS. The excretion, disposition, and metabolism of delavirdine were investigated in Sprague-Dawley rats after oral administration of [14C]delavirdine mesylate at single doses ranging from 10 to 250 mg/kg and multiple doses ranging from 20 to 250 mg/kg/day. Excretion studies showed that feces was the major route of elimination, delavirdine was well absorbed (>80%) after a 10 mg/kg single dose, and excretion was dose-dependent. The metabolism of delavirdine in the rat was extensive. The following metabolites were identified (% of dose in rats given 10 and 100 mg/kg, respectively): 6′-hydroxy delavirdine (7.1% and 15.6%) and its glucuronide (12.2% and 6.2%) and sulfate (5.5% and 3.2%) conjugates, despyridinyl delavirdine (12.1% and 11.7%) and its conjugate (13.0% and 11.7%), desalkyl delavirdine (16.5% and 13.4%), and itsN-sulfamate, 6′- and 4′-sulfate conjugates (2.9% and 3.9%). Cleavage of the amide bond in delavirdine to giveN-isopropylpyridinepiperazine and indole carboxylic acid constituted a minor pathway. Degradation of 6′-hydroxy delavirdine generated despyridinyl delavirdine and the pyridine-ring opened MET-14. The metabolic pathway of delavirdine involvedN-desalkylation, pyridine ring hydroxylation, pyridine ring cleavage, and amide bond cleavage.
Delavirdine mesylate (U-90152T; fig. 1) is a potent and selective nonnucleoside inhibitor of HIV-11 reverse transcriptase (1), an enzyme that catalyzes the conversion of genomic viral RNA into proviral DNA. The process of reverse transcription is essential to HIV replication (2-4). This enzyme has therefore been selected as a target for the development of therapeutics for the treatment of HIV infection. Examples include the nucleoside analogs 3′-azido-3′-deoxythymidine (zidovudine) (5, 6), 2′,3′-dideoxyinosine (didanosine) (7-9), 2′,3′-dideoxycytidine (zalcitabine) (10), and 2′,3′-didehydro-3′-deoxythymidine (stavudine) (11). However, the toxicity associated with these drugs (12) due to inhibition of cellular DNA polymerases (6, 8, 12, 13), together with the emergence of resistant strains of the virus (14-17), have focused efforts on developing specific nonnucleoside reverse transcriptase inhibitors. Several classes of compounds that have been reported to inhibit HIV-1 reverse transcriptase include the thiobenzimidazolones (18), dipyridodiazepinones (e.g. nevirapine) (19), pyridinones (e.g.l-697,661) (20), TSAO compounds (21), HEPT compounds (22), α-anilinophenylacetamide (e.g. loviride) (23), pyrimidobenzothiazepines (24), thiadiazole derivatives (25), and benzophenone derivatives (26). In vitro experiments have shown delavirdine mesylate to be a potent inhibitor of laboratory strains and clinical isolates of HIV-1 in various cell culture systems (1, 27-31), exhibiting no significant inhibition of cellular polymerases α and β (1). The potent inhibition of replication by delavirdine was comparable with the antiviral activity of nucleoside or other nonnucleoside reverse transcriptase inhibitors (1, 27). Delavirdine mesylate is currently under phase III clinical evaluations for the treatment of AIDS. This study describes the routes and extent of excretion, the disposition of delavirdine and its metabolites, and the isolation and identification of the metabolites of delavirdine after oral administration of [14C]delavirdine mesylate to male and female Sprague-Dawley rats.

Structure of delavirdine mesylate, including the positions of the 14C labels.
* [14C-carboxamide]delavirdine mesylate; # [2-14C-pyridine]delavirdine mesylate.
Materials and Methods
Chemicals.
All chemicals used in this study were of analytical grade. Solvents were Burdick & Jackson high purity grade (Burdick & Jackson, Muskegon, MI). Water was distilled and purified through a Milli-Q reagent water system (Millipore Corp., Bedford, MA). Ultima Gold (Packard Instruments, Meriden, CT) was used for LSC. Carbo-sorb E (Packard) and Permafluor E (Packard) were used for combustion of samples. Ultima-Flo M (Packard) was used as scintillant for flow-through detection. Sulfatase (EC 3.1.6.1, type VI from Aerobacter aerogenes), β-glucuronidase (EC 3.2.1.31 from Helix pomatia, type H-5), and Trizma buffer (Tris-HCl, 0.2 M, pH 7.4) were obtained from Sigma Chemical Co. (St. Louis, MO). Methanol-d4 (99.96% D) was obtained from Cambridge Isotope Laboratories (Cambridge, MA).
Delavirdine mesylate, [14C-carboxamide]delavirdine mesylate, [2-13C-pyridine]delavirdine mesylate, [2-14C-pyridine]delavirdine mesylate, desalkyl delavirdine (U-96183), despyridinyl delavirdine (U-102466),N-isopropylpyridinepiperazine (U-88703), and indole carboxylic acid (U-96364) were synthesized by Pharmacia & Upjohn, Inc. (for structures, see fig. 7). The specific activities of the radiolabeled test substances were 34.7 μCi/mg and 57.8 μCi/mg for [14C-carboxamide]delavirdine mesylate and 79.58 μCi/mg for [2-14C-pyridine]delavirdine mesylate, with radiochemical purities of >99% by HPLC. Chemical purities of the nonradiolabeled and [2-13C-pyridine]delavirdine mesylate were >97.6% and 96.5%, respectively. The structure and positions of the 14C labels are shown in fig. 1.

Metabolic pathway of delavirdine in rats.
Animals.
Sprague-Dawley rats were obtained from Charles River Laboratories (Portage, MI). For the biliary excretion and metabolism study, male rats (328–341 g, 10–11 weeks) were surgically implanted with bile duct and duodenum cannulas and placed in Nalgene metabolism cages equipped with urine and feces separators using harnesses connected to swivels. Sodium taurocholate was infused at a rate of 1 ml/hr at a concentration of 7.68 mg/ml as replacement for the loss of bile salts. For the excretion and metabolism study, male (238–260 g, 8.5 weeks) and female (207–231 g, 10.5 weeks) rats were surgically implanted with superior vena cava cannulas and housed in metabolism cages. Female rats (211–249 g, 9.5 weeks) housed in holding (rats killed at 1 and 4 hr) or metabolism (rats euthanized at 12 and 24 hr) cages were used for the metabolism study with [2-14C-pyridine]delavirdine mesylate. Environmental conditions were maintained as follows: lights, 12 hr on/12 hr off; relative humidity, 50 ± 10%; temperature, 68°–75°F; and ventilation, 18–20 room air changes per hour. Rats were provided with food (Certified Rodent Diet 5002C, PMI Feeds, Inc., St. Louis, MO) and water ad libitum. Rats were fasted ∼16 hr before and 4 hr after radiolabeled dose administration. Animals in this study were cared for and used in accordance with The Guide for the Care and Use of Laboratory Animals, DHEW Publication (National Institutes of Health) 85-23, 1985 and with The Animal Welfare Act Regulation 9 CFR 3, August 15, 1989 and as modified on March 19, 1991.
Instrumentation.
Samples were oxidized in a Packard model 307 sample oxidizer (Packard Instrument Co., Meriden, CT) equipped with a Packard Oximate 80 robotics system. A Packard Tri-Carb Liquid Scintillation Analyzer model 1900 CA or model 1900TR was used for radioactivity counting. Feces were homogenized using an Omni 2000 variable speed homogenizer (Omni International, Waterbury, CT) or a Stomacher Blender (Tekmar Co., Cincinnati, OH). A Turbo Vap LV evaporator (Zymark Corp., Hopkinton, MA) or a Speed Vac concentrator model AS260 (Savant Instruments, Inc., Farmingdale, NY) was used to concentrate samples.
Chromatography.
The HPLC system consisted of a Perkin-Elmer Series 410 quaternary pump (Perkin-Elmer Corp., Norwalk, CT); a Perkin-Elmer ISS 200 LC sample processor; a Perkin-Elmer LC-235 or LC-235C diode array detector; a Radiomatic model A-280, A-515, or A-525 flow-through detector (Packard Instrument) containing a 500-μl liquid cell and a Foxy 200 fraction collector (Isco, Inc., Lincoln, NE).
SPE was performed using Extract-Clean columns (Alltech Associates, Inc., Deerfield, IL) containing 500 mg of C8 or C18 packing. Columns were washed with ∼3 ml acetonitrile, then equilibrated with 6 ml of 0.1 M ammonium acetate buffer (pH 4) before use. A Supelco Visiprep vacuum manifold (Supelco, Inc., Bellefonte, PA) was used for SPE.
MS.
PCI-PB-LC/MS was performed using a Finnigan 4021 quadruple mass spectrometer (Finnigan MAT, San Jose, CA) equipped with a PB interface (Thermabeam, Extrel Corp., Pittsburgh, PA; interface built at Pharmacia & Upjohn, Inc.). The instrument was operated in the PCI mode using ammonia as reagent gas. Electron energy was set at 70 eV. Interface temperatures were controlled by Vestec thermospray electronics (Vestec, Houston, TX). Data were acquired and reduced by a Vector II data system (Teknivent Corp., Maryland, MO). HPLC analyses were performed on a system consisting of a Perkin-Elmer ISS 100 sample processor (Perkin-Elmer Corp.) interfaced to a Perkin-Elmer Series 410 quaternary pump with a Perkin-Elmer SEC-4 solvent environment control and a Waters 490MS UV detector (Waters, Milford, MA). Samples were analyzed on a YMC 5 μm-basic 4.6 mm i.d. × 25 cm column (YMC, Inc., Wilmington, NC) connected to a YMC 5 μm-basic 4.0 mm i.d. × 2.3 cm guard cartridge. The mobile phase consisted of elution at 0.5 ml/min with a 20-min linear gradient from 10% A/90% D to 60% A/40% D, followed by 20-min 60% A/40% D, then a 10-min linear gradient to 80% A/20% D; where A = acetonitrile, D = 0.1 M ammonium acetate (pH 4). Alternatively, samples were isocratically eluted at 0.5 ml/min with 35% acetonitrile/2% isopropyl alcohol/63% 0.1 M ammonium acetate (pH 4).
ESI mass spectra were obtained on a Finnigan-MAT TSQ 7000 triple quadruple mass spectrometer (Finnigan-MAT) directly coupled to the HPLC system via a Finnigan atmospheric pressure ionization source. Data were collected on a DEC 3000 model 300× computer running OSF/1 version 2.0 as operating system. The mass spectrometer was controlled using Instrument Control Language version 8.0, and data were processed using Interactive Chemical Information System version 8.1.1 software. Argon was used as collision gas at a pressure of 2.2 mtorr and collision energy of −30 eV. The spray voltage was set to 4 kV, and the capillary was operated at 250°C. The HPLC system consisted of a Hewlett-Packard 1050 Series pump and autosampler (Hewlett-Packard, Naperville, IL), a Thar two-position valve actuator (Thar Designs, Inc., Pittsburgh, PA), and a YMC 5 μm-basic 4.6 mm i.d. × 25 cm column connected to a YMC 5 μm-basic 4.0 mm i.d. × 2.3 cm guard cartridge. The mobile phase was the same as for PB-LC/MS.
APCI mass spectra were obtained using a Finnigan-MAT TSQ 7000 triple quadrupole mass spectrometer (Finnigan-MAT). Samples were analyzed using the same conditions described for ESI. The APCI was operated with a vaporizer heater at 400°C, a corona of 5 μA, a capillary temperature of 200°C, and a N2 sheath gas of 80 psi.
FAB mass spectra were obtained on a VG AutospecQ hybrid mass spectrometer (VG Analytical, Division of Fisons, Manchester, UK), using an accelerating voltage of 8 kV, argon as collision gas, and acidified glycerol as matrix. Data were acquired using VG Analytical’s Opus software package version 2.0 FX.
NMR Spectroscopy.
1H NMR spectra were recorded at 500 MHz using a Bruker AMX-500 spectrometer (Bruker Instruments, Inc., Billerica, MA). Data were processed on a Bruker X-32 computer using Bruker UXNMR software version 920801. Samples were dissolved in ∼300–500 μl methanol-d4 in a 3 mm i.d. Teflon insert (WILMAD, Buena, NJ); the teflon insert was placed in a 5 mm NMR Tube (WILMAD). Spectra were recorded at a temperature of 303°K as free induction decays of 32K complex points. The residual CHD2OH pentet was used as reference at 3.30 ppm. 2D 1H COSY spectra were recorded in the magnitude mode using 1K by 1K data table with 256 increments in F1 and no zero-filling in F2. Unshifted sinebell windows were applied before transformation. 2D inverse1H-13C HETCOR phase sensitive spectra (32) were recorded with a relaxation delay of 1 sec, GARP decoupling of13C during acquisition (33), the time-proportional phase increment method (34) to achieve quadrature detection in F1, 4 dummy scans, a 2K by 1K complex point data table, and 128 increments in F1 with no zero-filling in F2. Sinebell-squared window functions shifted π/2 rad were applied in both F2 and F1.
1H NMR spectra were recorded at 400 MHz using a Bruker AMX-400 spectrometer (Bruker Instruments), a 3 mm 1H/BB inverse probe (Nalorac Cryogenics Corp., Martinez, CA) with a 90° pulse, and a pulse length of 12.0 μsec. Samples were dissolved in ∼115 μl methanol-d4 in a Wilmad 328PP 3 mm NMR tube (WILMAD). Spectra were recorded at 300°K as free induction decays of 4K complex points.
13C NMR spectra were recorded at 75.5 MHz 13C observed and 300.13 MHz for 1H WALTZ-16 decoupling using a Bruker AM-300 spectrometer. The sample was dissolved in ∼120 μl methanol-d4 in a 5 mm o.d. Shigemi microcell (Shigemi Co., Ltd., Tokyo, Japan). Spectra were recorded at 21°C, 90° observe pulse, 5.4 μsec pulse length, 3.28 sec pulse repetition period, and 90° 1H pulses of 96 μsec decoupling power. Methanol-d4 was used as reference at 49.0 ppm.
UV Spectrometry.
UV spectra were obtained on a Perkin-Elmer LC-235 or LC-235C diode array detector. Effluent was monitored at 295 and 255 nm, using a 5 nm bandwidth, 16 sec peakwidth, and a scanning rate of 1 scan every 5 to 20 sec. Peak purity information was obtained by comparison of the absorbance of each and every defined wavelength in 5 nm increments of the UV spectrum taken on the leading edge with the corresponding absorbance of the spectrum taken on the trailing edge (35). This comparison was determined at ∼20% peak height. A given peak was taken as homogeneous if the peak purity index was 1.5 or lower.
Dosing and Sample Collection.
Three separate studies were conducted: a single- and multiple-dose biliary excretion and metabolism study with [14C-carboxamide]delavirdine mesylate, a multiple-dose excretion and metabolism study with [14C-carboxamide]delavirdine mesylate, and a single-dose metabolism study with [2-13C/14C-pyridine]delavirdine mesylate.
Blood samples were stored on ice and centrifuged to separate the plasma. Bile, urine, and feces samples were collected at room temperature, immediately analyzed, or stored at −20°C until further analysis.
Single- and Multiple-Dose Biliary Excretion and Metabolism [14C-Carboxamide]Delavirdine Mesylate Study.
Delavirdine mesylate and [14C-carboxamide]delavirdine mesylate were dissolved in 80% propylene glycol/20% water containing 3 μl methanesulfonic acid per milliliter to final concentrations of 4 and 40 mg/ml. Three male rats received single oral doses of ∼10 mg/kg and 180 μCi/kg, and three male rats were administered single oral doses of ∼100 mg/kg and 180 μCi/kg. One additional male rat was administered 100 mg/kg/day (given twice a day: one dose in the morning and a second dose 8 hr later) and 46 μCi/kg/day for 4.5 days. Doses were administered by oral gavage using a dosing syringe attached to an animal feeding needle. In the single-dose study, bile was collected over the time periods 0–1, 1–2, 2–4, 4–6, 6–8, 8–12, 12–24, 24–48, and 48–72 hr; urine and feces were collected over the time periods 0–12, 12–24, 24–48, and 48–72 hr. In the multiple-dose study, bile was collected over the time period 0–24, 24–48, 48–72, 72–88, 88–89, 89–90, 90–92, 92–96, 96–112, 112–136, and 136–145 hr after the first dose; urine and feces were collected over the time periods 0–24, 24–48, 48–72, 72–88, 88–112, 112–136, and 136–145 hr after the first dose.
Multiple-Dose Excretion and Metabolism [14C-Carboxamide]Delavirdine Mesylate Study.
Delavirdine mesylate and [14C-carboxamide]delavirdine mesylate were dissolved in 80% propylene glycol/20% water containing 3 μl methanesulfonic acid per milliliter to final concentrations of 10 and 125 mg/ml. Four male and four female rats received single oral radiolabeled doses of 10 mg/kg and 200 μCi/kg, followed by multiple oral nonradiolabeled doses of 20 mg/kg/day given as 10 mg/kg doses twice a day (multiple-dose regimen was started 24 hr after the radiolabeled dose; one dose in the morning and a second dose 8 hr later) for the next 8 days; then, a second radiolabeled dose of 10 mg/kg and 200 μCi/kg was administered. In addition, four male and four female rats received the same dosing regimen at single radiolabeled doses of 125 mg/kg and 200 μCi/kg and multiple nonradiolabeled doses of 250 mg/kg/day (125 mg/kg doses given twice a day). Urine was collected over the time periods 0–12, 12–24, and at 24-hr intervals up to 192 and 120 hr after the first and second radiolabeled doses, respectively. Feces were collected at 24-hr intervals up to 192 and 120 hr after the first and second radiolabeled doses, respectively. Blood samples (175- to 300-μl aliquots, 5.7 ml total volume of blood withdrawn for the study) were drawn from the venous cannula at scheduled times through the 48-hr period after each radiolabeled dose.
Single-Dose Metabolism [2-13C/14C-Pyridine]Delavirdine Mesylate Study.
Delavirdine mesylate and [2-14C-pyridine]delavirdine mesylate were dissolved in 50% propylene glycol/50% water containing 3 μl methanesulfonic acid per milliliter to a final concentration of 50 mg/ml. A second formulation containing equimolar amounts of delavirdine mesylate and [2-13C-pyridine]delavirdine mesylate, as well as [2-14C-pyridine]delavirdine mesylate, was prepared at a concentration of 50 mg/ml. Twelve female rats were administered single oral doses of ∼250 mg/kg and 500 μCi/kg; 9 of 12 rats received the [2-12C/14C-pyridine]delavirdine mesylate formulation and 3 of 12 rats were administered the [2-12C/13C/14C-pyridine]delavirdine mesylate formulation. Terminal blood was collected at 1, 4, 12, and 24 hr after dosing. Urine and feces were collected over the time periods 0–12 and 12–24 hr postdosing from the rats killed at 12 and 24 hr.
Sample Preparation and Total Radiocarbon Analysis.
Aliquots of urine ranging in volume from 200 to 500 μl (2–3 replicates) and 25–50 μl portions of plasma (1–2 replicates) were measured gravimetrically, mixed with 5–10 ml of Ultima Gold, and analyzed by LSC. A 25-μl portion of whole blood was measured gravimetrically (2 replicates) into combustion cones, dried at room temperature, combusted, and analyzed by LSC. A 25-μl aliquot of bile was measured gravimetrically (2 replicates), mixed with 0.5 ml of water, and 10 ml of Ultima Gold. Feces were homogenized with 2–5 volumes of water containing 2% acetic acid, 500-μl portions of the fecal homogenates (2–5 replicates) were weighed into combustion cones, dried at room temperature, combusted, and analyzed by LSC.
Feces and whole blood were combusted completely to carbon dioxide and water. The resulting 14CO2 was trapped in 9 ml Carbo-sorb E and diluted with 10 ml Permafluor E+scintillation cocktail. The 14C activity was then quantified by LSC. The combustion efficiency of the sample oxidizer was determined by comparing the radioactivity recovered from replicate oxidations of predose feces or whole blood fortified with a known amount of [14C-carboxamide] or [2-14C-pyridine]delavirdine mesylate to that obtained by direct fortification of the Carbo-sorb/Permafluor trapping solution with the same amount of [14C]delavirdine mesylate. The efficiency was calculated at the start of a series of oxidations, as well as intermittently to ensure that the oxidizer efficiency did not change substantially. Radioactivity was measured by LSC with 10 min counting or 2ς standard deviation. Count rates in cpm were corrected to dpm using quench curves generated from sealed quenched standards in either Ultima Gold or Carbo-sorb/Permafluor cocktails.
The amounts of radioactivity in urine, bile, and feces were expressed as a percentage of the administered dose. For each sample, the dpm value for each aliquot was divided by the aliquot weight and multiplied by the total sample weight to afford total dpm in the sample. The total dpm for the sample was converted to a percentage of the dose by dividing by the dpm administered. Values for feces were corrected for combustion efficiency.
The amounts of radioactivity in whole blood and plasma were expressed in μM-eq. For each sample, the dpm value for each aliquot was divided by the aliquot weight and further divided by the specific activity of the dosing formulation to afford ng-eq/g tissue. Values were converted to μM-eq assuming a density of 1.0 g/ml and using the molecular weight of delavirdine mesylate (552.68 ng/nmol). Values for whole blood were corrected for combustion efficiency.
HPLC Profiles of Plasma, Urine, Bile, and Feces Samples.
Plasma, urine, bile, and feces samples were profiled for parent drug and metabolites by HPLC with flow-through radiochemical detection. Chromatographic analyses were performed on a YMC 5 μm-basic 4.6 mm i.d. × 25 cm column using gradient elution at 1.0 ml/min with 10% A/90% D for 5 min, followed by a 35-min linear gradient to 60% A/40% D, then 60% A/40% D for 5 min [where A = acetonitrile, D = 0.1 M ammonium acetate buffer (pH 4)]. Alternatively, plasma samples were profiled using isocratic elution with 1.0 ml/min 34% acetonitrile/2% isopropyl alcohol/64% 0.1 M ammonium acetate buffer (pH 4). The HPLC effluent was mixed with Ultima-Flo M in a 1:3 ratio. Peaks of radioactivity were quantitated on the Radiomatic detector using peak area integration. Column recovery was determined by comparing the total radioactivity from the sum of the integrated peaks in the 14C chromatogram with total radioactivity. The radioactivity in a given chromatographic peak was expressed as a percentage of the total radioactivity eluted during the analysis. Values were not corrected for column recovery, because essentially all of the injected radioactivity was recovered from the column. The amounts of delavirdine and its metabolites in urine, feces, and bile were expressed as percentages of the administered dose by multiplying the percentage of the total radioactivity in a peak by the percentage of the dose in a given sample. Values for feces were corrected for extraction recovery.
A 50-μl aliquot of each plasma sample was mixed with 100 μl acetonitrile, followed by centrifugation at 14,000 rpm for 5 min at 4°C. A 100- to 125-μl portion of the supernatant was diluted 1:1 with 0.1 M ammonium acetate buffer (pH 4) or evaporated in vacuo and reconstituted in 200 μl 10% acetonitrile/90% 0.1 M ammonium acetate buffer (pH 4). A 75- to 150-μl aliquot of prepared plasma sample was analyzed by HPLC with flow-through radiochemical detection.
Urine samples were centrifuged to removed large particulates. A 200- to 400-μl portion of urine samples collected over the time periods 0–12, 12–24, and 24–48 hr were directly analyzed by HPLC with flow-through radiochemical detection using gradient elution (vide supra).
A 100-μl portion of bile samples collected up to 24 hr for rats given 10 mg/kg and up to 48 hr for rats administered 100 mg/kg was mixed with 100 μl acetonitrile, followed by centrifugation at 14,000 rpm for 5 min at 6°C. A 100-μl aliquot of the supernatant was diluted 1:1 with 0.1 M ammonium acetate buffer (pH 4), and a 100-μl aliquot was analyzed by HPLC with flow-through radiochemical detection using gradient elution (vide supra).
Fecal samples collected over the time periods 0–24 and 24–48 hr from the biliary excretion and metabolism study were processed as follows. A 2-g aliquot of fecal homogenate was weighed in a 15-ml centrifuge tube and sonicated for 5 min with 5 ml 90% acetonitrile/10% 0.1 M ammonium acetate buffer (pH 4). The mixture was centrifuged at 2,500 rpm for 15 min at 6°C. The extraction procedure was repeated an additional time; the supernatants were combined and weighed. The extraction recoveries of radioactivity from all samples of feces were 69.0 ± 6.0% (mean ± SD for 2 replicates/fecal sample for each of 3 rats) for rats given 10 mg/kg and 89.0 ± 3.1% for rats administered 100 mg/kg. The fecal extract was concentrated to one-half its original volume. A 50- to 100-μl portion of the concentrated fecal extract was analyzed by HPLC with flow-through detection using gradient elution (vide supra). The procedure was validated by extraction of 2-g aliquot of predose fecal homogenate fortified with [14C-carboxamide]delavirdine mesylate (94.7 ± 0.6% recovery). All fecal samples were analyzed in duplicate.
Fecal samples collected over the time periods 0–12 and 12–24 hr from the 2-14C-pyridine metabolism study were combined and processed as before using a 5-g portion of fecal homogenate and 15 ml of 90% acetonitrile/10% 0.1 M ammonium acetate buffer (pH 4). The extraction recovery from the feces samples was 87.7%. A 100-μl portion of the fecal extract was diluted 1:3 (v:v) with 0.1 M ammonium acetate (pH 4), and a 200-μl aliquot was analyzed by HPLC with flow-through radiochemical detection using gradient elution (vide supra). The procedure was validated by extraction of a 8-g aliquot of predose fecal homogenate fortified with [2-14C-pyridine]delavirdine mesylate (96.3% recovery). Fecal samples were analyzed in duplicate.
Metabolite Isolation.
Metabolites were isolated from appropriate matrices as described herein. In general, at each purification step, an aliquot of each fraction was analyzed by LSC; from these data, aliquots of selected fractions were analyzed by HPLC with flow-through radiochemical detection. Fractions containing the same metabolite(s) were combined, weighed, and an aliquot was gravimetrically measured and analyzed by LSC. The amount of metabolite(s) in the fraction was estimated from the specific activity of the dosing formulation.
MET-2 (U-102466) was isolated as follows. Rat feces (11.6 g, 15.76 × 106 dpm, 11.0 mg of drug-related material) collected over the time period 24–88 hr from the multiple-dose biliary excretion and metabolism study were extracted 2 times with 30 ml of 90% acetonitrile/10% 0.1 M ammonium acetate buffer (pH 4) with sonication for 3 min. The fecal mixture was centrifuged at 2,300 rpm for 10 min at 6°C to obtain a supernatant containing ∼8.0 mg of drug-related material (11.54 × 106 dpm). A portion of the fecal extract was concentrated to dryness, dissolved in 1 ml 0.1 M ammonium acetate buffer (pH 4), and purified by C8 SPE. The SPE cartridge was stepwise eluted with 4 ml each 0%, 20%, 40%, 60%, 80%, and 100% acetonitrile in 0.1 M ammonium acetate buffer (pH 4). The 0% and 20% acetonitrile fractions were combined and concentrated to afford 1.2 mg of drug-related material (1.76 × 106dpm). Further HPLC purification with gradient elution [YMC 5 μm-basic 4.6 mm i.d. × 25 cm, 11 injections of 100 μl each, 1.0 ml/min, 5-min 10% A/90% D, 35-min linear gradient to 60% A/40% D, 5-min 60% A/40% D, A = acetonitrile, D = 0.1 M ammonium acetate buffer (pH 4)] gave 0.7 mg (1.05 × 106 dpm) of partially purified MET-2. A final HPLC purification with isocratic elution [YMC 5 μm-ODS-AQ 4.6 mm i.d. × 25 cm, 11 injections of 100 μl each, 0.5 ml/min, 7% acetonitrile/1% isopropyl alcohol/92% 0.1 M ammonium acetate buffer (pH 4)] afforded ∼0.5 mg (7.52 × 105 dpm) of purified MET-2.
MET-6 and MET-8 were isolated as follows. Rat bile (9.5 × 106 dpm, 6.6 mg of drug-related material) collected over the time period 72–88 hr from the multiple-dose biliary excretion and metabolism study was purified by semipreparative HPLC with gradient elution [YMC 5 μm-basic 10 mm i.d. × 25 cm, 35 injections of 400 μl each, 3.0 ml/min, 5-min 10% A/90% D, 35-min linear gradient to 60% A/40% D, 5-min 60% A/40% D, A = acetonitrile, D = 0.1 M ammonium acetate buffer (pH 4)] to afford partially purified MET-6 (1.44 × 106 dpm, 1.0 mg) and MET-8 (1.35 × 106 dpm, 0.9 mg). MET-6 was further purified by isocratic reversed-phase HPLC [YMC 5 μm-basic 4.6 mm i.d. × 25 cm, 20 injections of 100 μl each, 1.0 ml/min, 18% acetonitrile/2% isopropyl alcohol/80% 0.1 M ammonium acetate buffer (pH 4)] to afford ∼0.5 mg (7.18 × 105 dpm) of purified MET-6. Isocratic HPLC purification of MET-8 [YMC 5 μm-basic 4.6 mm i.d. × 25 cm, 20 injections of 100 μl each, 1.0 ml/min, 23% acetonitrile/2% isopropyl alcohol/75% 0.1 M ammonium acetate buffer (pH 4)] gave ∼0.5 mg (6.69 × 105 dpm) of partially purified MET-8.
MET-14 was isolated as follows. The remaining fecal homogenate (14.30 × 106 dpm, 3.2 mg of drug-related material) from a rat given [2-12C/13C/14C-pyridine]delavirdine mesylate collected over the time period 0–24 hr from the metabolism study was concentrated in vacuo. The residue was reconstituted in 15% acetonitrile/85% 0.1 M ammonium acetate buffer (pH 4) to afford 1.8 mg of drug-related material (7.96 × 106 dpm). The acetonitrile in the concentrated fecal extract was evaporated, the aqueous extract was loaded onto two C18 SPE columns, and the columns were eluted with 5 ml 10% acetonitrile/90% 0.1 M ammonium acetate buffer (pH 4), followed by 5 ml acetonitrile. The 10% acetonitrile fractions were combined (1.68 × 106 dpm, 0.38 mg) and purified by HPLC [YMC 5 μm-basic 4.6 mm i.d. × 25 cm, 46 injections of 200 μl each, 1.0 ml/min 10% acetonitrile/90% 0.1 M ammonium acetate buffer (pH 4)] to afford 0.09 mg (4.05 × 105 dpm) of partially purified MET-14. Further HPLC purification on a YMC 5 μm-basic 4.6 mm i.d. × 25 cm column isocratically eluted with 1.0 ml/min 4% THF/4% acetonitrile/92% 0.1 M ammonium acetate buffer (pH 4) gave 19 μg (9.51 × 104 dpm) of partially purified MET-14. Final purification on phenyl chromatography [YMC 5 μm-phenyl 4.6 mm i.d. × 25 cm, 3 injections of 100 μl each, 1.0 ml/min, 5% acetonitrile/5% methanol/90% 0.1 M ammonium acetate buffer (pH 4)] afforded 6 μg (2.52 × 104 dpm) of purified MET-14. Additional MET-14 was obtained by fortification of 12.5 g of predose fecal homogenate with [2-12C/13C/14C-pyridine]delavirdine mesylate (40.23 × 106 dpm, 9.1 mg of drug-related material). The fortified feces were extracted 2 times with 20 ml 90% acetonitrile/10% 0.1 M ammonium acetate buffer (pH 4) to obtain 7.9 mg of drug-related material (34.81 × 106 dpm). The fecal extract was concentrated in vacuo (23.84 × 106 dpm, 5.4 mg of drug-related material) and purified by SPE as described. The 10% acetonitrile fractions were combined (6.60 × 106 dpm, 1.50 mg) and further purified on a semipreparative HPLC column [YMC 5 μm-basic 10.0 mm i.d. × 25 cm, 37 injections of 200 μl each, 2.5 ml/min, 3% THF/3% acetonitrile/94% 0.1 M ammonium acetate buffer (pH 4)] to afford 0.21 mg (9.18 × 105 dpm) of partially purified MET-14. Final HPLC purification on phenyl chromatography [YMC 5 μm-phenyl 4.6 mm i.d. × 25 cm, 25 injections of 200 μl each, 1.0 ml/min, 5% acetonitrile/5% methanol/90% 0.1 M ammonium acetate buffer (pH 4)] gave 0.15 mg (6.42 × 105 dpm) of purified MET-14.
Metabolite Identification.
The remaining plasma samples from the excretion and metabolism study collected 1 hr after each radiolabeled dose and from a rat given [2-12C/13C/14C-pyridine]delavirdine mesylate collected 1 hr after radiolabeled administration in the metabolism study were mixed with 2 volumes of acetonitrile. After centrifugation at 14,000 rpm for 5 min at 4°C, the supernatant was concentrated to one-third its original volume or evaporated to dryness and reconstituted in 10% acetonitrile/90% 0.1 M ammonium acetate buffer (pH 4) or diluted 1:2 with 0.1 M ammonium acetate buffer (pH 4). Aliquots of the concentrated plasma samples were analyzed by PCI-PB-LC/MS and electron ionization-PB-LC/MS.
A 30-ml portion of urine collected over the time period 48–72 hr from the multiple-dose biliary excretion and metabolism study was lyophilized and reconstituted in ∼3 ml of 10% acetonitrile/90% 0.1 M ammonium acetate buffer (pH 4). An aliquot of the concentrated urine sample was analyzed by PCI-PB-LC/MS. A 30-ml portion of urine collected over the time period 0–12 hr from the multiple-dose excretion and metabolism study was similarly prepared and analyzed by PCI-PB-LC/MS. Urine collected over the time period 0–12 hr from a rat given [2-12C/13C/14C-pyridine]delavirdine mesylate in the metabolism study was analyzed directly by ESI-LC/MS.
Concentrated fecal extracts collected over the time period 112–136 hr from the multiple-dose biliary excretion and metabolism study and over the time period 0–24 hr from a rat given [2-12C/13C/14C-pyridine]delavirdine mesylate in the metabolism study were analyzed by PCI-PB-LC/MS and ESI-LC/MS, respectively.
The purified MET-2 was dissolved in 300–400 μl methanol-d4 and analyzed by 1D 1H NMR and 2D HETCOR NMR. A 50-μl portion of MET-2 in methanol-d4 was evaporated and reconstituted in 50 μl of 50% acetonitrile/50% 0.1 M ammonium acetate buffer (pH 4) for PCI-PB-LC/MS and FAB/MS analyses.
The purified MET-6 was dissolved in ∼300 μl methanol-d4 and analyzed by 1D 1H NMR and 2D COSY NMR.
The partially purified MET-8 was analyzed by 1D 1H NMR and 2D COSY NMR as described for MET-6. The remaining MET-8 was concentrated to a volume of ∼25 μl, diluted with 100 μl methanol, and analyzed by PCI-PB-LC/MS and FAB/MS.
The purified MET-14 was dissolved in ∼120 μl methanol-d4 and analyzed by 1H and13C NMR. The remaining MET-14 was concentrated in vacuo and reconstituted in 600 μl aqueous methanol for ESI/MS and APCI/MS analyses.
Enzyme Hydrolysis.
A 20- to 25-μl portion of bile collected over the time period 2–4 hr was mixed with 80–175 μl of 0.1 M sodium acetate buffer (pH 5) and 100 μl β-glucuronidase solution (500 units/ml in 0.2% saline). The mixture was incubated at 37°C for 1 hr. The enzyme incubation was quenched by addition of 50–200 μl acetonitrile, followed by centrifugation at 14,000 rpm for 5 min at 6°C. The supernatant was evaporated or diluted 1:1 with 0.1 M ammonium acetate buffer (pH 4). A 50-μl aliquot of the sample was analyzed by HPLC with flow-through radiochemical detection using gradient elution (vide supra). A control sample containing rat bile and sodium acetate buffer (pH 5) was similarly conducted.
A 25-μl portion of bile collected over the time period 2–4 hr was mixed with 175 μl of sulfatase solution (4 units/ml in Trizma buffer) and incubated at 37°C for 1 hr. A 50-μl aliquot of the enzyme incubation was immediately analyzed by HPLC with flow-through radiochemical detection using gradient elution (vide supra). A control sample containing rat bile and Trizma buffer was similarly conducted.
Results
Mass Balance.
The cumulative excretion of drug-related radioactivity after single and multiple oral dose administration is shown in fig. 2. After a 10 mg/kg/day dose on day 1, 80 ± 3% of the dose was excreted in feces, 15 ± 3% was recovered in urine, for a total recovery (including cagewash) of 95 ± 1%. After multiple doses (day 10) of 20 mg/kg/day, 83 ± 4% was recovered in feces, 14 ± 3% was excreted in urine, for a total recovery of 97 ± 2%. After a dose of 125 mg/kg/day on day 1, 86 ± 5% was recovered in feces, urine accounted for an additional 9 ± 3%, and the total recovery was 95 ± 3%. After a dose of 250 mg/kg/day on day 10, 87 ± 2% and 8 ± 2% were excreted in feces and urine, respectively, for a total recovery of 95 ± 1%. Most of the dose (>87%) was excreted within 48 hr after dosing.

Excretion of drug-related radioactivity in rats after single and multiple oral dose administration of [14C-carboxamide]delavirdine mesylate.
○, Feces male; •, feces female; ▿, urine male; ▾, urine female; □, total male; ▪, total female; ◊, bile male.
Excretion in bile duct-cannulated rats is also summarized in fig. 2. Biliary excretion was the major route of elimination of drug-related material in rats administered a single 10 mg/kg oral dose, accounting for 58 ± 10% of the dose. The remaining 19 ± 9% was recovered in feces and 19 ± 2% was excreted in urine, for a total recovery of 96 ± 2%. The major route of elimination in rats administered a single 100 mg/kg dose was fecal excretion (46 ± 4%), whereas bile represented 35 ± 3% and urine 14 ± 3% of the dose, for a total recovery of 95 ± 1%. Mean absorption were 80 ± 10% and 52 ± 4% in rats administered 10 and 100 mg/kg doses, respectively.
Plasma Concentrations and Metabolite Profiles.
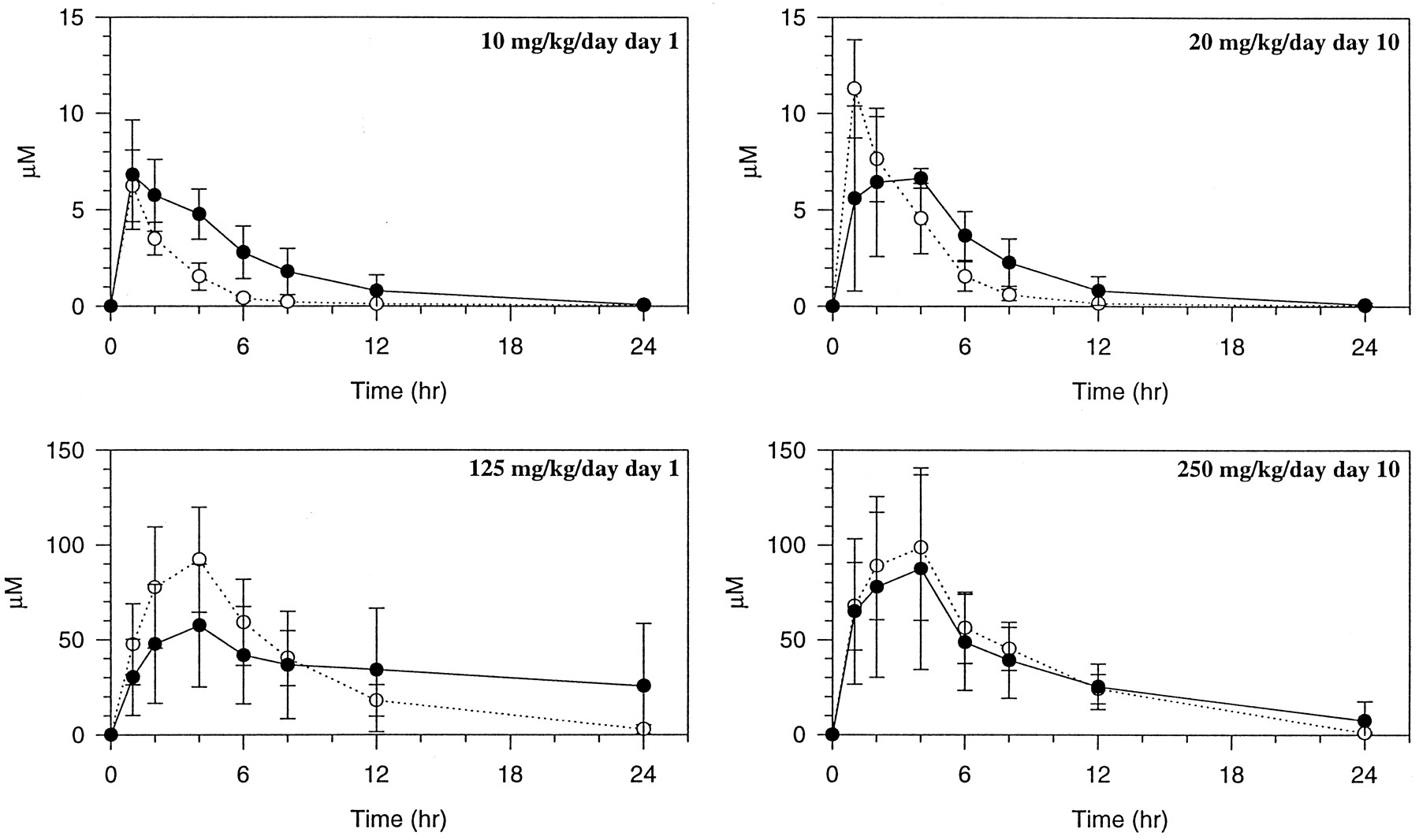
Plasma concentration-time profiles of drug-related material after multiple oral dose administration of nonradiolabeled delavirdine with single oral doses of [14C-carboxamide]delavirdine mesylate on days 1 and 10 are shown in fig. 3. Absorption was rapid as evidenced by mean tmaxvalues for drug-related material, which ranged from 1 to 4 hr, with the exception of 2 of 4 outlier female rats given 125 mg/kg/day on dose day 1.

Plasma concentration-time profiles of drug-related material in rats after multiple oral dose administration of nonradiolabeled delavirdine mesylate with single oral doses of [14C-carboxamide]delavirdine mesylate on days 1 and 10.
○, Male; •, female.
A summary of the analyses of plasma samples for delavirdine and its metabolites by HPLC with radiochemical detection is presented in table1. Delavirdine was the major radioactive component in circulation after both single (dose day 1) and multiple (dose day 10) oral doses of [14C-carboxamide]delavirdine mesylate, and after a single dose of [2-14C-pyridine]delavirdine mesylate. The amounts of delavirdine in plasma ranged from 69.1 ± 5.4% in male rats given 10 mg/kg/day on dose day 1 to 98.7% in female rats administered a single dose of 250 mg/kg. MET-5 was also observed in circulation, ranging from 1.6 ± 2.0% in female rats given 250 mg/kg/day on dose day 10 to 27.5 ± 3.4% in male rats after 10 mg/kg/day on dose day 1. A gender-related difference was observed in rats given a 20 mg/kg/day dose, with a higher amount of MET-5 and a lower amount of delavirdine in male rats than in female rats.
Analyses of plasma samples for delavirdine (DLV) and its metabolites by HPLC with radiochemical detection1-a
HPLC Profiles of Urine, Bile, and Feces.
Radiolabeled components in urine, bile, and feces were analyzed for delavirdine and its metabolites by HPLC with flow-through radiochemical detection (table 2). The metabolism of delavirdine was extensive in the rat, with a small amount of the administered dose excreted as intact drug (<2% of the dose). Bile contained several components, of which MET-6 was the major metabolite. The major component in urine was MET-5; several minor metabolites were also observed in urine. Feces contained MET-1, MET-2, and MET-7 as major metabolites, with delavirdine and MET-5 as minor components. Representative chromatograms are shown in fig. 4.
Excretion of delavirdine (DLV) and its metabolites after single and multiple oral dose administration of [14C-carboxamide]delavirdine mesylate2-a

Representative HPLC radiochromatograms of rat plasma, urine, feces, and bile after single oral dose administration of [14C-carboxamide]delavirdine mesylate.
From top to bottom: plasma, urine, feces, and bile. DLV, delavirdine.
Metabolite Isolation and Identification.
The presence of delavirdine in plasma was indicated by HPLC retention time and UV spectrum comparisons to a synthetic standard, and was confirmed by LC/MS analysis. Analyses of plasma, bile, urine, and fecal samples from rats given [14C-carboxamide]delavirdine mesylate by CI/MS showed a protonated molecular ion for parent drug atm/z 457 (table 3). Loss of CH3SO2 gave an ion at m/z 379. Ions at m/z 221 and 137, resulting from cleavage of the carbonyl—piperazine bond and pyridine ring, respectively, were also observed. Cleavage of the carbonyl–indole linkage gave an ion atm/z 249. Likewise, of plasma, urine, and feces samples from a rat administered [2-12C/13C/14C-pyridine]delavirdine mesylate by ESI/MS showed protonated molecular ions for parent drug atm/z 457 and 458 and ions at m/z 221 and 222 (pyridinepiperazine fragment).
Summary of key MS fragmentation for delavirdine (DLV) and its metabolites
The major metabolite in plasma and urine, MET-5, had the same HPLC retention time and UV spectrum as a synthetic standard of desalkyl delavirdine (U-96183). The presence of this metabolite was confirmed by LC/MS. Analyses of plasma, bile, urine, and fecal samples from rats given [14C-carboxamide]delavirdine mesylate by CI/MS showed a protonated molecular ion for MET-5 at m/z 415, 42 amu lower than parent drug, indicative of loss of the isopropyl side chain (table 3). Loss of CH3SO2 gave an ion atm/z 337. Cleavage of the carbonyl—piperazine bond gave an ion at m/z 179, whereas cleavage of the carbonyl–indole linkage resulted in a fragment at m/z 207. Further confirmation of the presence of desalkyl delavirdine in urine and plasma was obtained by ESI/MS of samples obtained from a rat administered [2-12C/13C/14C-pyridine]delavirdine mesylate.
Two minor metabolites in urine, MET-10 and MET-12, were observed in rats treated with [14C-carboxamide]delavirdine mesylate and [2-14C-pyridine]delavirdine mesylate, respectively. These metabolites eluted at the same HPLC retention time as the synthetic standards U-96364 (MET-10) and U-88703 (MET-12), arising from cleavage of the carboxamide bond (for structures, see fig. 7). The presence of MET-10 in urine was confirmed by CI/MS, which showed a base peak at m/z 255 corresponding to the protonated molecular ion and a fragment at m/z 175 due to loss of CH3SO2 (table 3). The presence of MET-12 in plasma and urine was confirmed by ESI/MS (table 3). Protonated molecular ions were observed at m/z 221 and 222 (doublets 1 amu apart due to the presence of fragments containing 13C). Loss of isopropyl gave ions at m/z 178 and 179, and cleavage of the piperazine—pyridine bond gave ions at m/z 136 and 137.
MET-4 was observed as a minor metabolite in urine and feces. Phenyl chromatography resolved MET-4 into a mixture of three metabolites: MET-4a, MET-4b, and MET-4d. Analysis of feces from a rat administered [2-12C/13C/14C-pyridine]delavirdine mesylate by ESI/MS showed protonated molecular ions for MET-4a atm/z 495 and 496, 80 amu higher than desalkyl delavirdine, indicating addition of a sulfamate group (table 3). Loss of SO3H gave ions at m/z 415 and 416, whereas cleavage of the piperazine—pyridine bond gave an ion at m/z322. These data identified MET-4a as N-sulfamate desalkyl delavirdine. MET-4b and MET-4d were characterized as 6′-sulfate desalkyl delavirdine and 4′-sulfate desalkyl delavirdine based on HPLC retention time comparison to identified metabolites in the monkey and human (36).
The major metabolite in rat bile was MET-6, whereas MET-7 and MET-8 were observed as minor metabolites. Characterization of these metabolites was initially pursued by enzyme hydrolysis. Incubation of rat bile with β-glucuronidase (containing glucuronidase and sulfatase activities) hydrolyzed MET-6 to MET-7 and also cleaved MET-8 to MET-7. MET-7 on further standing at room temperature was converted to MET-2. Treatment of rat bile with sulfatase (containing no detectable β-glucuronidase activity) resulted in a decrease in MET-8 with concomitant increase in MET-7, whereas MET-6 remained unchanged. These data indicated that MET-6 and MET-8 were the glucuronic acid and sulfate conjugates of MET-7, respectively.
Confirmation of MET-6 as a glucuronide was obtained by LC/MS analysis. The CI mass spectrum showed a protonated molecular ion for MET-6 atm/z 649, 192 amu higher than parent drug, indicative of addition of a hydroxy group followed by conjugation with glucuronic acid (table 3). Loss of the glucuronic acid moiety gave the base peak at m/z 473. Cleavage of the carbonyl—piperazine bond and the carbonyl–indole linkage gave ions at m/z 237 and 265, respectively, and indicated substitution on the right-hand side of the molecule (i.e. piperazine ring, pyridine ring, or isopropyl group). Loss of the isopropyl moiety from the ion at m/z 237 to generate an ion at m/z 194 indicated that the substituent was located on the pyridine or piperazine rings. Ions at m/z323 and 153 were observed and corresponded to fragments arising from cleavage of the pyridine–piperazine linkage. The ion at m/z153 was 16 amu higher than the corresponding ion for parent drug and established substitution on the pyridine ring. To determine the substitution position on the pyridine ring, MET-6 was isolated from rat bile. MET-6 showed a UV λmax at 300 nm, characteristic of the presence of an indole ring. The 1H NMR spectrum (500 MHz, methanol-d4; fig. 5, table4) showed the presence of six aromatic protons; four resonances were similar to corresponding resonances in the parent drug (fig. 5) and were assigned to protons on the indole ring (δ 7.56, 7.43, 7.17, and 6.84 ppm). A resonance for the pyridine H-6′ was not observed and the remaining two aromatic resonances for the H-4′ and H-5′ pyridine ring protons at δ 7.06 and 6.56 ppm, respectively, were shifted upfield compared with corresponding resonances in the parent drug (δ 7.50 and 7.37 ppm). In addition, the H-4′ and H-5′ protons in MET-6 appeared as doublets with a coupling constantJ4′-5′ of 8.3 Hz, whereas in the parent drug these protons were observed as doublets of doublets with coupling constants J4′-5′ of 8.3 Hz,J4′-6′ of 1.4 Hz, andJ5′-6′ of 5.7 Hz. These data established that MET-6 was substituted at the pyridine ring C-6′. Assignments were confirmed with a 2D COSY NMR experiment, which established homonuclear spin coupling between H-4 and H-6, H-7 and H-6, as well as between H-4′ and H-5′. Additional resonances in the 1H NMR spectrum of MET-6 corresponded to the glucuronic acid (δ 5.59, 3.77, and 3.51 ppm), the piperazine ring protons (δ 4.01 and 3.18 ppm), the isopropyl chain (δ 3.57 and 1.20 ppm), and the methyl sulfonate group (δ 2.89 ppm). A coupling constant between the anomeric H-1" and H-2" sugar protons confirmed the β-linkage of the glucuronic acid. A 2D COSY NMR confirmed these assignments with cross-peaks between H-1" and H-2", H-5" and H-4", the methine and methyl protons on the isopropyl chain, and between the piperazine protons. The NMR, MS, and enzyme hydrolysis data identified MET-6 as 6′-O-glucuronide delavirdine. The presence of small amounts (<1% of the radioactivity in plasma) of 6′-O-glucuronide in plasma was confirmed by HPLC retention time comparison and LC/MS analysis.

1H NMR spectra (500 MHz, methanol-d4) of the aromatic region of delavirdine and its metabolites.
From top to bottom: delavirdine, MET-6, MET-8, and MET-2.
1H NMR assignments for delavirdine and its metabolites4-a
Enzyme hydrolysis indicated that MET-8 contained a sulfate group. UV spectroscopy indicated the presence of an indole ring (λmax at 300 nm). Confirmation was pursued by LC/MS analysis. The CI mass spectrum of MET-8 showed a base peak atm/z 473, 16 amu higher than parent drug, indicative of loss of the sulfate group (table 3). Cleavage of the piperazine–pyridine linkage gave fragments at m/z 153 and 323 and indicated substitution on the pyridine ring. Cleavage of the carbonyl—piperazine bond and the carbonyl–indole linkage gave ions at m/z 237 and 265, respectively. Loss of CH3SO2 gave an ion at m/z 395. Confirmation of MET-8 as a sulfate conjugate was investigated by FAB/MS. However, loss of sulfate at m/z473, as well as cleavage of the carbonyl–piperazine linkage atm/z 237, were observed with no detectable molecular ion. The enzyme hydrolysis and MS data together indicated that MET-8 contained a sulfate group on the pyridine ring. To determine the position of substitution on the pyridine ring, MET-8 was partially purified. Despite the presence of impurities, 1H NMR spectroscopy was investigated, because endogenous bile components were not expected to be aromatic compounds and, as such, would not interfere with the aromatic resonances of the metabolite. The upfield region of the NMR spectrum of MET-8 from 1 to 4 ppm, as expected, showed the presence of impurities. However, the aromatic region of the spectrum was quite clean (fig. 5). Six aromatic protons were observed; four resonances were similar to corresponding resonances in the parent drug and were assigned to protons on the indole ring (δ 7.55, 7.43, 7.17, and 6.84 ppm; table 4). Similar to MET-6, a resonance for the pyridine H-6′ in MET-8 was not observed, and the remaining two aromatic resonances for the H-4′ and H-5′ pyridine ring protons at δ 7.06 and 6.99 ppm, respectively, were shifted upfield, compared with corresponding resonances in the parent drug (δ 7.50 and 7.37 ppm). The protons H-4′ and H-5′ in MET-8 appeared as doublets with a coupling constantJ4′-5′ of 8.4 Hz and established substitution at the pyridine C-6′. The NMR, MS, and enzyme hydrolysis data identified MET-8 as 6′-sulfate delavirdine.
Confirmation of MET-7 as 6′-hydroxy delavirdine was obtained by LC/MS analyses of rat plasma, bile, urine, and fecal samples. The CI mass spectra gave a protonated molecular ion for MET-7 at m/z473, 16 amu higher than parent drug, indicative of the presence of a hydroxyl group (table 3). Cleavage of the pyridine—piperazine bond gave ions at m/z 153 and 323. Comparison to the corresponding ions in the mass spectrum of parent drug (m/z137 and 323) indicated that MET-7 was hydroxylated at the pyridine ring. Attempts to purify MET-7 for determination of the position of substitution by NMR resulted in decomposition of MET-7 to MET-2. However, the MS data, together with the fact that enzyme hydrolysis of MET-6 and MET-8 generated MET-7, identified MET-7 as 6′-hydroxy delavirdine.
MET-2 was present as a minor metabolite in bile, urine, and feces of rats administered [14C-carboxamide]delavirdine mesylate. MET-2 was isolated from feces as a degradation product of MET-7, a major fecal component. The purified MET-2 showed a UV λmax at 296 nm, suggesting the presence of an indole ring. The 1H NMR spectrum (500 MHz, methanol-d4; fig. 5) showed the presence of four aromatic protons (δ 7.55, 7.42, 7.16, and 6.81 ppm), which were similar to corresponding resonances in the parent drug, and were assigned to the indole ring protons (table 4). In addition, eight piperazine protons (δ 3.88 and 2.99 ppm) and a methyl sulfonyl group (δ 2.88 ppm) were observed. Resonances for the pyridine ring and isopropyl chain were not observed. These data were consistent with a pyridine-cleaved structure. MS confirmed this assignment. The CI mass spectrum of MET-2 showed a base peak at m/z 323 corresponding to the protonated molecular ion and loss of CH3SO2 to give an ion at m/z 245. Likewise, the FAB mass spectrum of MET-2 showed ions at m/z323 and 87, corresponding to the protonated molecular ion and the fragment arising from cleavage of the carbonyl–piperazine linkage, respectively (table 3). The identification of MET-2 as despyridinyl delavirdine (U-102466) was further confirmed by comparisons of HPLC retention times and CI and FAB mass spectra with a synthetic standard.
As described previously, concentration of fecal extracts from rats given [14C-carboxamide]delavirdine mesylate resulted in the decomposition of MET-7 to MET-2. Degradation of MET-7 to MET-14 was observed in a similar experiment with rats administered [2-14C-pyridine]delavirdine mesylate. These results suggested that MET-14 was the pyridinol resulting from cleavage of the piperazine—pyridine bond in MET-7. MET-14 was initially isolated from feces of rats treated with [2-12C/13C/14C-pyridine]delavirdine mesylate. A small amount (6 μg) of purified MET-14 was obtained due to the difficulty of separating this metabolite from endogenous material. However, additional amounts (0.15 mg) of MET-14 were obtained through bacterial metabolism and degradation of predose feces fortified with [2-12C/13C/14C-pyridine]delavirdine mesylate. The purified MET-14 showed a UV λmax at 249 nm, a hypsochromic shift from the parent drug UV λmax at 295 nm, suggesting the absence of the indole ring. The 13C NMR spectrum (75.5 MHz, methanol-d4) showed the13C-2′ at 161.7 ppm, 16 ppm downfield from the C-2′ in delavirdine, consistent with an amide or carboxylic acid moiety and indicated a pyridine ring-opened structure. The 1H NMR spectrum (300 MHz, methanol-d4) showed a triplet at 5.8 ppm and indicated the presence of a vinyl proton coupled to a methylene with a coupling constant of 4.9 Hz. In addition, resonances for the N-isopropyl side chain were observed at 3.8 and 1.3 ppm, corresponding to the methine and methyl protons, respectively. Resonances for the aromatic region in the indole and pyridine rings (6.5 to 8 ppm), as well as the piperazine protons (3.4 and 4.1 ppm), were not observed and suggested a pyridine ring-opened structure. The1H and 13C NMR data supported the following structural features in MET-14: N-isopropyl, CH⋕CH2, and COOH or CONH2 at C-2′. These data, together with the fact that MET-14 resulted from degradation of 6′-hydroxy delavirdine, suggested that MET-14 was the pyridine ring-opened structure in fig. 6. Two possible structures for MET-14 were consistent with the data:13CONH2 at C-2′/12COOH at C-6′ or 13COOH at C-2′/12CONH2 at C-6′. Confirmation was investigated by MS. The ESI and APCI mass spectra of MET-14 showed protonated molecular ions at m/z184 and 185, 2 amu lower than expected, suggesting cyclization to the lactone in the mass spectrometer. Note that doublets 1 amu apart were observed due to isotopic contribution for fragments containing13C-2′. To resolve between the two possible structures for MET-14, MS/MS was investigated. The daughter ion mass spectra ofm/z 184 and 185 are shown in fig. 6. The daughter ion mass spectrum of m/z 184 showed ions at m/z 142, 114, 85, 68, and 43, whereas the daughter ion mass spectrum ofm/z 185 showed ions at m/z 143, 115, 86, 68, and 43. Therefore, ions differing by 1 amu (m/z 143, 142, 115, 114, 86, and 85) contained the C-2′ carbon, whereas ions atm/z 43 and 68 did not. Loss of isopropyl gave ions atm/z 142 and 143, and at m/z 43; further loss of12CO gave ions at m/z 114 and 115. The ions atm/z 85 and 86 can be rationalized by loss of12CH⋕O from the ions at m/z 114 and 115. These data were consistent with 13CONH2 at C-2′/12COOH at C-6′. The other structure,13COOH at C-2′/12CONH2 at C-6′, would be expected to lose 13CO from the ions atm/z 114 and 115 to give a fragment at m/z 86 or cleave NH212CH⋕O to yield ions atm/z 69 and 70. These spectroscopic data identified MET-14 as the pyridine ring-opened metabolite containing an amide group at C-2′.

ESI/MS/MS of MET-14.
(Top) Daughter ion mass spectrum of m/z 184. (Bottom) Daughter ion mass spectrum of m/z 185.
Discussion
After oral administration to rats, delavirdine was extensively metabolized before excretion. The major route of excretion wasvia the feces. Delavirdine was well absorbed (>80%) after a 10 mg/kg single dose. Absorption was reduced to 52% after a 100 mg/kg single dose. Excretion of drug-related material was dose-dependent; at higher doses and after multiple-dose administration, an increased percentage of the administered radioactivity was recovered in feces. Gender-related differences in excretion were not observed.
Delavirdine was metabolized by the rat to several metabolites. Unchanged delavirdine was not detected or was present in <0.5% of the dose in bile and urine, indicating rapid metabolism. Radioactivity in bile consisted of the following metabolites: MET-7 and its glucuronide (MET-6) and sulfate (MET-8) conjugates, MET-2 and its conjugate (MET-1), and MET-5 and its conjugates (MET-3 and MET-4). In urine, MET-5 was a major component; with MET-2 and its conjugate, MET-7, and conjugates of MET-5 as minor metabolites. In addition, MET-12 and MET-10 were detected as minor components in rat urine. Radioactivity in feces was mostly composed of MET-7 and MET-2 and its conjugate, with delavirdine and MET-5 as minor components. HPLC analysis of predose feces fortified with [14C-carboxamide]delavirdine suggested that bacterial metabolism of delavirdine may account for the presence of MET-7, which in turn degraded to MET-2 and the pyridine ring-opened MET-14.
1H NMR spectroscopy proved to be indispensable for the identification of metabolites, especially to ascertain the location of glucuronidation and sulfation. Comparison of the chemical shifts and the splitting of the aromatic signals between delavirdine and the isolated metabolites (fig. 5) proved to be extremely useful in assigning the structures of the metabolites, in view of the minute quantities isolated and in the absence of synthetic standards.
Figure 7 illustrates the scheme for the metabolism of delavirdine in the rat. The metabolism of delavirdine in the rat involves four pathways. First, N-desalkylation to MET-5 (U-96183), followed by conjugation with sulfate (MET-4a). Second, hydroxylation of MET-5 at the pyridine ring C-4′ (MET-15d) or C-6′ (MET-15b) and subsequent conjugation with sulfate give MET-4d and MET-4b, respectively. Third, hydroxylation of delavirdine at the pyridine ring C-6′ to MET-7 and subsequent conjugation with glucuronic acid or sulfate give MET-6 and MET-8, respectively, orN-desalkylation (MET-15b) and conjugation yield 6′-sulfate desalkyl delavirdine (MET-4b). Alternatively, the pyridine ring in MET-7 is cleaved to give MET-2 (U-102466) and the pyridine ring-opened MET-14. Further conjugation of MET-2 gives MET-1. A fourth minor pathway involves amide bond cleavage with release of MET-12 (U-88703) and MET-10 (U-96364).
Acknowledgments
The authors gratefully acknowledge B. W. Jones, L. R. Norris, R. J. Ouding, K. E. Rousch, and T. L. VandeGiessen for technical assistance with surgery, animal dosimetry, and sample collection. We also thank M. D. Koets for combustion of samples; R. S. P. Hsi, E. H. Chew, W. T. Stolle, and J. A. Easter for syntheses of the14C- and 13C-labeled drugs; D. L. Romero for synthesis of U-102466; and L. J. Larion for preparation of the vehicle in the dosing formulations.
Footnotes
-
Send reprint requests to: Dr. Mayland Chang, Drug Metabolism Research, Pharmacia & Upjohn, Inc., Kalamazoo, MI 49007.
- Abbreviations used are::
- HIV-1
- human immunodeficiency virus type 1
- HIV
- human immunodeficiency virus
- LSC
- liquid scintillation counting
- SPE
- solid-phase extraction
- PCI
- positive chemical ionization
- PB
- particle beam
- ESI
- electrospray ionization
- APCI
- atmospheric pressure chemical ionization
- FAB
- fast atom bombardment
- 2D
- two-dimensional
- COSY
- correlation spectroscopy
- HETCOR
- heteronuclear correlation
- GARP
- globally optimized alternating-phase rectangular pulses
- cpm
- counts per minute
- dpm
- disintegrations per minute
- THF
- tetrahydrofuran
- 1D
- one-dimensional
- CI
- chemical ionization
- metabolites—MET-2
- despyridinyl delavirdine (U-102466)
- MET-6
- 6′-O-glucuronide delavirdine
- MET-8
- 6′-sulfate delavirdine
- MET-14
- pyridine ring-opened metabolite
- MET-7
- 6′-hydroxy delavirdine
- MET-5
- desalkyl delavirdine
- MET-10
- indole carboxylic acid
- MET-12
- N-isopropylpyridinepiperazine
- MET-4
- mixture of MET-4a, MET-4b, and MET-4d
- MET-4a
- N-sulfamate desalkyl delavirdine
- MET-4b
- 6′-sulfate desalkyl delavirdine
- MET-4d
- 4′-sulfate desalkyl delavirdine
- TSAO
- 2′,5′-bis-(O-tert-butyldimethylsilyl)-3′-spiro-5"-(4"-amino-1",2"-oxathiole-2",2"-dioxide
- HEPT
- 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)-thymine
- Received July 7, 1996.
- Accepted November 18, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}