


Abstract
Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, was metabolized by human liver microsomes to 5-hydroxy-, 6-hydroxy-, and N-deisopropyl-fluvastatin. Total metabolite formation was biphasic with apparentKm values of 0.2 to 0.7 and 7.9 to 50 μM and intrinsic metabolic clearance rates of 1.4 to 4 and 0.3 to 1.5 ml/h/mg microsomal protein for the high and lowKm components, respectively. Several enzymes, but mainly CYP2C9, catalyzed fluvastatin metabolism. Only CYP2C9 inhibitors such as sulfaphenazole inhibited the formation of both 6-hydroxy- and N-deisopropyl-fluvastatin. 5-Hydroxy-fluvastatin formation was reduced by compounds that are inhibitors of CYP2C9, CYP3A, or CYP2C8. Fluvastatin in turn inhibited CYP2C9-catalyzed tolbutamide and diclofenac hydroxylation withKi values of 0.3 and 0.5 μM, respectively. For CYP2C8-catalyzed 6α-hydroxy-paclitaxel formation the IC50 was 20 μM and for CYP1A2, CYP2C19, and CYP3A catalyzed reactions, no IC50 could be determined up to 100 μM fluvastatin. All three fluvastatin metabolites were also formed by recombinant CYP2C9, whereas CYP1A1, CYP2C8, CYP2D6, and CYP3A4 produced only 5-hydroxy-fluvastatin. Km values were ∼1, 2.8, and 7.1 μM for CYP2C9, CYP2C8, and CYP3A, respectively. No difference in fluvastatin metabolism was found between the CYP2C9R144 and CYP2C9C144 alleles, suggesting the absence of polymorphic fluvastatin metabolism by these alleles. CYP1A2, CYP2A6, CYP2B6, CYP2C19, CYP2E1, and CYP3A5 did not produce detectable amounts of any metabolite. This data indicates that several human cytochrome P-450 enzymes metabolize fluvastatin with CYP2C9 contributing 50–80%. Any coadministered drug would therefore only partially reduce the metabolic clearance of fluvastatin; therefore, the likelihood for serious metabolic drug interactions is expected to be minimal.
Fluvastatin (Lescol) is a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA)1reductase inhibitor which consistently lowers low density lipoprotein cholesterol levels by 20 to 30% at a daily dose of 20 to 40 mg (Levy et al., 1993; Peters et al., 1993;Davidson, 1994). Like most HMG-CoA reductase inhibitors, fluvastatin has a low incidence rate (<0.5%) of musculoskeletal side effects such as myopathy and rhabdomyolysis. These side effects, however, increase for certain HMG-CoA reductase inhibitors with increased systemic concentrations of the HMG-CoA reductase inhibitors when coadministered with other drugs. Lovastatin plasma exposure (area under the plasma concentration time curve), for example, increases 20-fold when coadministered with cyclosporine A (Olbricht et al., 1997) and the incidence of musculoskeletal side effects increases up to 28% (Tobert, 1988). In combination with itraconazole, lovastatin concentrations increase 10-fold (Kantola et al., 1997); this combination has been associated with rhabdomyolysis (Lees and Lees, 1995). Most of these interactions have been attributed to the inhibition of CYP3A, which is the major enzyme metabolizing most HMG-CoA reductase inhibitors including lovastatin, simvastatin, atorvastatin, and cerivastatin (Wang et al., 1991; Prueksaritanont et al., 1997; Boberg et al., 1997; Physicians’ Desk Reference, 1998). Additional factors besides metabolism may also contribute to the observed drug interactions. For example, lovastatin has recently been shown to be a substrate for P-glycoprotein (Dimitroulakos and Yeger, 1996) and increased lovastatin concentrations could in part be due to a decrease of biliary clearance after P-glycoprotein inhibition. A similar mechanism might also explain increased (5–23-fold) plasma concentrations of pravastatin in the presence of cyclosporine A (Regazzi et al., 1993), because pravastatin is thought not to be metabolized by CYP3A.
For fluvastatin, relatively few cases of musculoskeletal side effects have been reported, even when administered in combination with other drugs (Peters et al., 1993; Peters, 1996; Plosker and Wagstaff, 1996). This may be attributed to its favorable biopharmaceutical profile. Fluvastatin is completely absorbed from the intestinal tract. Systemic exposure, however, is limited due to first-pass metabolism with maximal plasma concentrations of 0.35 μM after a 40-mg (0.1-mMol) oral dose. Fluvastatin is almost exclusively eliminated via metabolism, mainly hydroxylation, at the 5- and 6-position of the indole moiety andN-deisopropylation. Only the hydroxylated metabolites retain some HMG-CoA reductase inhibitory activity, yet they are not found in the systemic circulation (Tse et al., 1992; Dain et al., 1993).
Based on these pharmacokinetic characteristics, transport proteins such as P-glycoprotein should not be involved in the disposition of fluvastatin. Drug interactions affecting fluvastatin disposition should therefore be limited to metabolic interactions. However, although fluvastatin has been reported to inhibit CYP3A competitively at high (200 μM) concentrations (Ikeda et al., 1997), CYP3A does not appear to be the major enzyme eliminating fluvastatin. CYP3A inhibitors do not affect fluvastatin to the same extent as the other HMG-CoA reductase inhibitors. Fluvastatin area under the plasma concentration time curve has been increased only 1.9-fold in the presence of cyclosporine A (Goldberg and Roth, 1996) and fluvastatin plasma concentrations have been unchanged when given together with itraconazole (Kantola et al., 1997). At much lower concentrations (∼0.2 μM), fluvastatin has been reported to also competitively inhibit CYP2C9 (Transon et al., 1996).
The objective of this study was to further define the enzymes involved in fluvastatin elimination using in vitro techniques. Furthermore, the reciprocal effects on the biotransformation of fluvastatin and potentially coadministered drugs were assessed to provide guidance for clinical use.
Materials and Methods
Chemicals.
[14C]Fluvastatin (2 GBq/mmol) (R*,S*-(±)-7-[3-(4-fluorophenyl)-1-(methylethyl)-[3-14C] 1H-indol-2-yl]-3,5-dihydroxy-6-heptenoic acid monosodium salt) was obtained from Novartis Pharmaceuticals Corporation (East Hanover, NJ). The purity of the labeled fluvastatin was 94.5% by HPLC. [3H]Glibenclamide (1.35 GBq/mmol, cyclohexol-3,3-3H) was obtained from DuPont-NEN (Boston, MA). [14C]Paclitaxel (2.3 GBq/mmol, 2-benzoyl ring-UL-14C) and [14C]phenacetin (0.5 GBq/mmol, phenacetin-ring-UL-14C) were obtained from Sigma (St. Louis, MO). [14C]Tolbutamide (2 GBq/mmol), [14C]S-mephenytoin (2.2 GBq/mmol), and [3H]cyclosporine A (296 GBq/mmol) were obtained from Amersham (Little Chalfont, UK).
Unlabeled diclofenac, 4′-hydroxy-diclofenac, valsartan, cyclosporine A, isradipine, and fluvastatin metabolites (5-hydroxy-, 6-hydroxy- andN-deisopropyl- fluvastatin) were obtained from Novartis. Janssen Biotech NV (Olen, Belgium) provided itraconazole. Furafylline and ketoconazole were purchased from Ultrafine Chemicals (Manchester, UK). Chlorzoxazone, chlorpropamide, sulfaphenazole, quinidine, erythromycin, sulfinpyrazone, ethinyl estradiol, glyburide, troleandomycin, nifedipine, dextromethorphan, phenacetin, clofibrate, paclitaxel, sparteine, and tolbutamide were purchased from Sigma. Glibornuride and mibefradil were a gift from Hoffman-La Roche (Basel, Switzerland), lovastatin was obtained from Merck (West Point, PA), and pravastatin from Bristol-Myers Squibb (Princeton, NJ). All other reagents were obtained from commercial sources and were of the highest quality available.
Biologicals.
Human liver tissue which could not be used for transplantation was obtained as either pieces or microsomes from the International Institute for the Advancement of Medicine (Exton, PA), GGM-002; from Vitron Inc. (Tucson, AZ), HL44, HL45, HL46; and from the Novartis liver bank, M8 (Ball et al., 1992). Microsomes from livers GGM-002 and M8 were prepared in house by differential centrifugation as described previously (Ball et al., 1992). Microsomal protein was determined by the Bradford method (Bradford, 1976) and the cytochrome P-450 (P-450) content was determined from the carbon monoxide spectra according to the method of Omura and Sato (1964). Total P-450 content was 0.44, 0.23, 0.26, 0.36, and 0.29 nmol P-450/mg for microsomal GGM-002, HL44, HL45, HL46, and M8, respectively.
Human lymphoblast-expressed human CYP1A1 (53 pmol P-450/mg protein), CYP1A2 (140 pmol P-450/mg protein), CYP2A6 (140 pmol P-450/mg protein), CYP2B6 (86 pmol P-450/mg protein), CYP2C8 (28 pmol P-450/mg protein), CYP2C9C144 (15 pmol P-450/mg protein), CYP2C9R144 (47 pmol P-450/mg protein), CYP2C19 (14 pmol P-450/mg protein), CYP2D6 (42 pmol P-450/mg protein), CYP2E1 (150 pmol P-450/mg protein), CYP3A4 (65 pmol P-450/mg protein), and baculovirus-expressed human CYP2C8 (370 pmol P-450/mg protein), CYP2C9R144 (500 pmol P-450/mg protein), CYP2C9C144 (480 pmol P-450/mg protein), and CYP3A4 (230 pmol P-450/mg protein) were purchased from Gentest Corp. (Woburn, MA).
Metabolism.
Fluvastatin, diclofenac, and glibornuride were incubated with human liver microsomes in a final volume of 500 μl of 0.1 M phosphate buffer at pH 7.4 in the absence or presence of different inhibitors. Substrate and inhibitor (0–100 μM) were added in dimethyl sulfoxide not to exceed 1% v/v. Metabolism was initiated by the addition of 0.2 mM β-NADPH and the NADPH regenerating system to give final concentrations of 1 mM NADP+, 5 mM isocitrate, 5 mM MgCl2, and 1 U isocitrate dehydrogenase. For fluvastatin, the reactions were stopped with half the volume of cold methanol and for diclofenac with 125 μl of cold acetonitrile. Typical incubation conditions such as substrate concentration, incubation time, and microsomal protein content were as follows: fluvastatin (0.04–20 μM, 25–30 min, 200 μg), diclofenac (5–100 μM, 10 min, 100 μg), and glibornuride (0.025–1.5 mM, 40 min, 500 μg). Human liver microsomal incubations with all other substrates were performed essentially as described previously (Fischer et al., 1998). For fluvastatin (50 μM) incubations with lymphoblast-expressed human P-450 (1 mg), the incubation time was 60 min.
HPLC Analysis.
The incubation media was separated from the denatured protein by centrifugation at ∼40,000g for 15 min using an Avanti centrifuge (Beckman Instruments, Palo Alto, CA). The pellets of the glyburide and glibornuride samples were then extracted once with 100 μl of methanol, and the extract was combined with the primary supernatant. Aliquots of the supernatant were directly analyzed by HPLC on either a HP1090 (Hewlett Packard, Waldbronn, Germany) or an Alliance (Waters, Milford, MA) system. Fluvastatin and its metabolites were separated by gradient HPLC (LC 18-DB, 5-μm particle size, 20 × 4.6 mm and 250 × 4.6 mm; Supelco Inc., Bellefonte, PA) with 10 mM ammonium acetate (pH 7.4) and methanol at a total flow of 1 ml/min at 50°C. Methanol was held constant at 0% for 3 min and then increased linearly to reach 35% at 33 min and then to 100% at 80 min. Glibornuride was chromatographed at 40°C (LC 18, 5-μm particle size, 20 × 2.1 mm and 100 × 2.1 mm; Brownlee, San Jose, CA) at a total flow of 0.4 ml/min. The mobile phases were 10 mM ammonium acetate (pH 4.3) and acetonitrile. The proportion of acetonitrile was increased linearly from 0 to 60% during 40 min. Acetonitrile reached 80% at 45 min and was held constant until 50 min. Mephenytoin was chromatographed at 25°C (C18 Genesis, 4 μm, 250 × 3.1 mm; Jones Chromatography Inc., Lakewood, CO). The mobile phases were 20 mM ammonium acetate (pH 7.4) and acetonitrile with a total flow rate of 0.6 ml/min. The proportion of acetonitrile was increased linearly from 0 to 100% during 30 min. Diclofenac was chromatographed at 40°C (LC 18-DB, 5-μm particle size, 30 × 4.6 mm and 250 × 4.6 mm) at a total flow of 1 ml/min. The mobile phase consisted of 100 mM sodium phosphate (80%) and acetonitrile (20%) containing triethylamine (0.02%). Fluvastatin, glyburide, and their metabolites were detected by radioactivity monitoring using β-Ram (IN/US Systems Inc., Tampa, FL) and glibornuride and alternatively glyburide by UV detection at 230 and 238 nm, respectively. Diclofenac and its 4′-hydroxy-metabolite were monitored by electrochemical detection using a Coulochem II (ESA, Inc., Bedford, MA). All other compounds were analyzed essentially as described by Fischer et al. (1998). All compounds and metabolites were identified by their HPLC retention times and compared to chromatograms of reference compounds. Fluvastatin metabolites were additionally identified by liquid chromatography/mass spectrometry (LC/MS) analysis.
Mass Spectrometric Analysis.
Liquid chromatography flow was split equally between the TSQ-7000 mass spectrometer (Finnigan MAT, San Jose, CA) and an INUS β-Ram radioactivity monitor. Single and tandem quadrupole experiments were performed in the negative ion mode with an electrospray interface. Capillary temperature was 225°C and gas pressures were set at four bars for nitrogen sheath gas and one bar for nitrogen auxiliary gas. For most experiments, the electron multiplier setting was 1600 V. For tandem MS experiments, the collision cell voltage was 25 eV and the argon cell pressure was 1.3 × 10−6bar.
Data Analysis.
Metabolic rates were calculated from mean substrate concentrations over the incubation period. IC50 values were determined graphically by plotting the percent of the control activity against the inhibitor concentration. Michaelis-Menten parametersKm and Vmaxwere determined by analysis of linearized plots as well as nonlinear curve fitting using Hyper.exe (Easterby, Department of Biochemistry, University of Liverpool, Liverpool, UK) and Fig.P (BIOSOFT, Cambridge, UK) with the following equation:
Results
Biotransformation Pathways.
Fluvastatin was metabolized in five different human liver microsomal preparations to three primary metabolites (Figs.1A and 2). The metabolites exhibiting retention times of 42 and 45 min were identified as 6-hydroxy- and 5-hydroxy-fluvastatin, respectively, and the metabolite eluting at 52 min was identified asN-deisopropyl-fluvastatin. The assignment of the metabolite structures was based on LC/MS analysis and on retention times compared with synthetic reference compounds. The LC/MS analysis indicated changes of the nominal mass compared to fluvastatin of M + 16 for 5-hydroxy- and 6-hydroxy-fluvastatin and of M − 42 forN-deisopropyl fluvastatin. Quantitatively, at 0.2 to 0.6 μM fluvastatin concentrations in human liver microsomes, 5-hydroxy-fluvastatin was the most abundant metabolite, followed by 6-hydroxy-fluvastatin and N-deisopropyl-fluvastatin, which were of similar importance. Among the five livers studied, 5-hydroxy-fluvastatin formation varied 2- to 29-fold compared to 6-hydroxy-fluvastatin (data not shown), whereas there was less variability between 6-hydroxy-fluvastatin andN-deisopropyl-fluvastatin formation (0.7–1.4-fold).
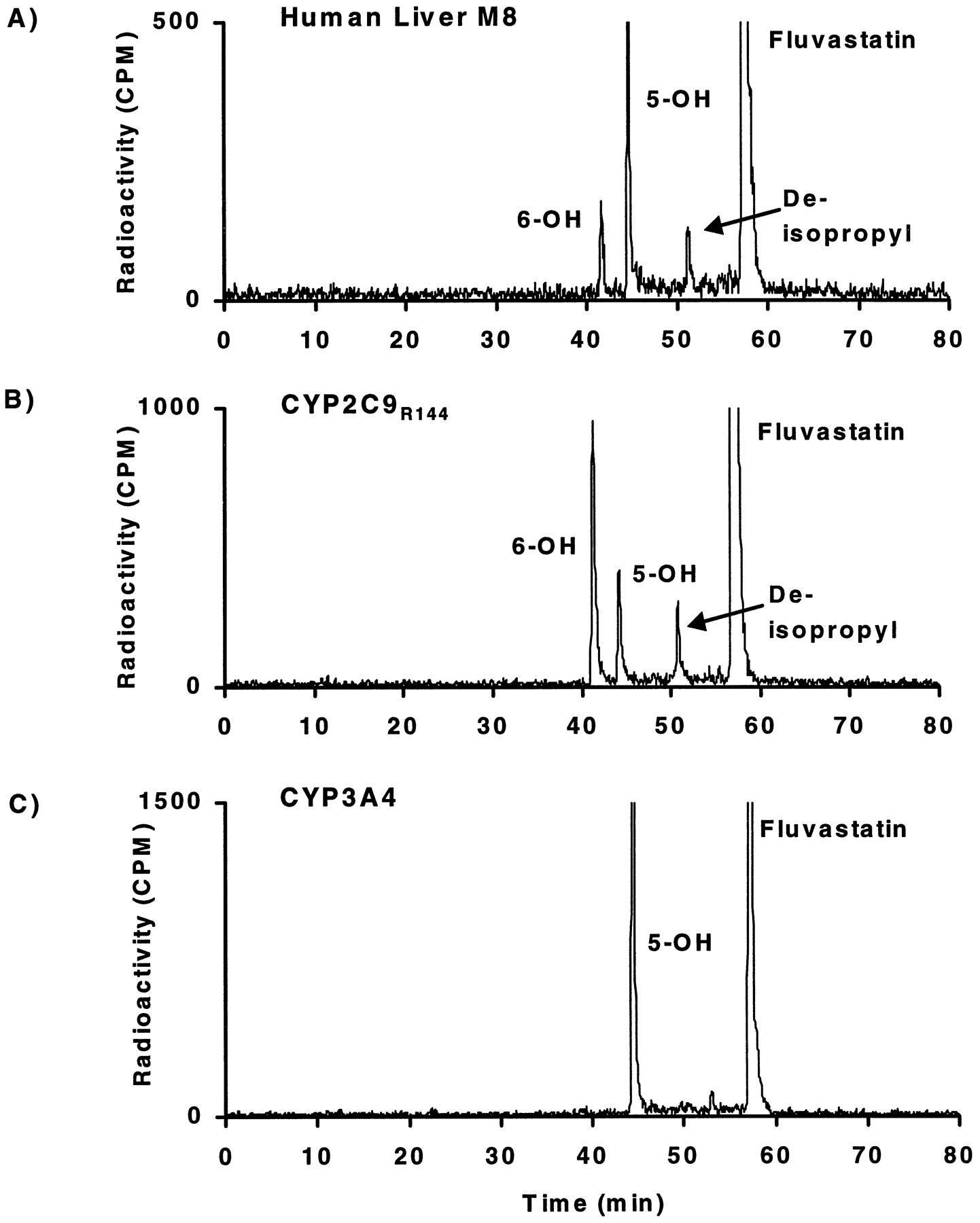
Fluvastatin metabolite profiles in human liver microsomes and individual CYP2C9 and CYP3A4.
Typical HPLC separation with radioactivity monitoring of fluvastatin and its metabolites formed in incubations of [14C]fluvastatin (0.6 μM) during 30 min with human liver microsomal protein (200 μg/ml) (A) or baculovirus insect cell-expressed CYP2C9R144 (B) and CYP3A4 (C) (1 mg cellular protein/ml).

Metabolite pathways of fluvastatin in human liver microsomes and human P-450s.
Individual human P-450 isoenzymes expressed in human lymphoblast cells or in insect cells also metabolized fluvastatin, but only CYP2C9 formed all three metabolites produced by human liver microsomes (Fig. 1B). Recombinant human CYP3A4 (Fig. 1C), CYP2C8, CYP2D6, and CYP1A1 (data not shown) formed only 5-hydroxy-fluvastatin. Quantitatively, for CYP2C9, 6-hydroxy-fluvastatin is the most abundant metabolite, followed by 5-hydroxy- and N-deisopropyl-fluvastatin. There was no difference in fluvastatin metabolite formation between the CYP2C9R144 allele and the CYP2C9C144 allele. In contrast, CYP1A2, CYP2A6, CYP2B6, CYP2C19, CYP2E1, and CYP3A5 did not produce detectable amounts of metabolites (data not shown).
Intrinsic Metabolic Clearance.
The rate of fluvastatin metabolism was determined for three human liver microsomal preparations over a concentration range of 0.04 to 20 μM (Table 1) and for recombinant CYP2C8, CYP2C9, and CYP3A4 preparations (Table2). In human liver microsomal preparations, metabolism appeared biphasic. For the lower substrate concentration (0.04–0.6 μM), the Km was similar for all three livers (0.2–0.7 μM), whereas theVmax values ranged from 0.3 to 2.6 nmol/h/mg microsomal protein. At the higher concentrations (0.6–20 μM), Km values were 24 to 35 μM withVmax values ranging from 7.9 to 50 nmol/h/mg microsomal protein. This second phase contributed about 20 to 30% to the total intrinsic metabolic clearance of fluvastatin. For the formation of 6-hydroxy-fluvastatin andN-deisopropyl-fluvastatin (0.04–0.6 μM),Km values were 0.1 to 0.3 μM, which is similar to the high Km component for total metabolism. Vmax values for the formation of 6-hydroxy-fluvastatin and N-deisopropyl-fluvastatin were between 0.1 and 0.6 nmol/h/mg human liver microsomal protein.
Enzyme kinetic parameters for fluvastatin metabolism
Individual enzyme kinetic parameters for fluvastatin metabolism
Human enzymes CYP2C9R144 and CYP2C9C144 each exhibited aKm of ∼1 μM and theVmax values were also similar for both CYP2C9 alleles. The Km values observed for CYP2C8 and CYP3A4 were higher (i.e., 2.8 and 7.1 μM, respectively). The maximal velocities were extrapolated to human liver microsomes as described in Materials and Methods. The sum of the metabolic clearance estimates for recombinant CYP3A and CYP2C9 extrapolated for human liver microsomes was about 3-fold below the total intrinsic metabolic clearance estimates obtained from human liver microsomal incubations. Based on these calculations, the contribution of CYP2C9 and CYP3A to human liver fluvastatin metabolic clearance was similar.
Inhibition of Fluvastatin Metabolism.
Only compounds known to inhibit either CYP2C9 or CYP3A decreased fluvastatin metabolism (Table 3). Substrates and inhibitors of CYP2C9 such as diclofenac, warfarin, and sulfaphenazole inhibited all pathways of fluvastatin metabolism, but the largest effects were on the CYP2C9-catalyzed 6-hydroxy- andN-deisopropyl-fluvastatin formation. Typical CYP3A-specific inhibitors such as ketoconazole and troleandomycin were only inhibitory for 5-hydroxy-fluvastatin formation, which is in part formed by CYP3A. Compounds such as phenacetin and furafylline did not affect metabolism of fluvastatin. Both are known to inhibit CYP1A2. Similarly, substrates of CYP2D6 (quinidine, dextromethorphan, and sparteine) or CYP2E1 (chlorzoxazone) had no relevant effect on fluvastatin metabolism.
Inhibition of fluvastatin metabolism
Among the antihypertensive agents that might potentially be coadministered with fluvastatin, both nifedipine and isradipine were almost equally inhibitory of all three metabolites (IC50 ∼2–7 μM). Mibefradil only partially inhibited the formation of 5-hydroxy-fluvastatin, a metabolite that can be formed by several enzymes. Valsartan had no effect on fluvastatin metabolism. Among the antifungal agents, ketoconazole was the only compound that inhibited one of the metabolites, i.e., 5-hydroxy-fluvastatin formation. Itraconazole and terbinafine had little or no effect on any metabolite. The antihypercholesterolemic agents, lovastatin and clofibrate, inhibited both CYP2C9 and CYP3A (IC50 ∼5–10 μM), whereas pravastatin had no effect. The immunosuppressant cyclosporine A had only small inhibitory effect at high concentrations, which is consistent with clinical observations. Finally, the antidiabetic chlorpropamide had no effect on fluvastatin metabolism, whereas glyburide, and, to a lesser extent, tolbutamide were inhibitory of all fluvastatin metabolic pathways.
Effect of Fluvastatin on the Metabolism of other Drugs.
Fluvastatin inhibited only the metabolism of compounds that are metabolized by CYP2C9 (Table 4). Fluvastatin was found to inhibit the metabolism of the CYP2C9 substrates diclofenac and tolbutamide. For tolbutamide 4-hydroxylation, which displayed monophasic Michaelis-Menten kinetics (Fig. 3), the maximal velocity (Vmax) and Michaelis-Menten constant (Km) were 17 nmol/h/mg and 160 μM, respectively. In the presence of fluvastatin (5 μM), there was an increase in Km to 1.3 mM and only a relative small decrease in Vmax as expected for a competitive inhibition. The mean inhibition constant (Ki) for three inhibitor concentrations was 0.3 μM. Diclofenac also exhibited monophasic Michaelis-Menten kinetics with a Km of 9.4 μM and aVmax of 170 nmol/h/mg.Km increased to 31 μM andVmax decreased to 95 nmol/h/mg in the presence of 1 μM fluvastatin. This is indicative of a mixed mode of inhibition with a mean Ki of 0.5 μM for three fluvastatin concentrations.
Effect of fluvastatin on the metabolism of characteristic cytochrome P-450 substrates and potentially coadministered compounds (IC50) in human liver microsomes

Lineweaver-Burk plots of tolbutamide 4-hydroxylation.
Human liver microsomal incubations of ∼0.7 to 100 μM tolbutamide were performed in the presence of 0, ▪; 0.1, •; 1, ▾; and 5 μM fluvastatin, ★.
Glyburide metabolism in human liver microsomes exhibited monophasic Michaelis-Menten kinetics, with aVmax of 33 nmol/h/mg protein and aKm of 8.4 μM. Several metabolites were detected, which correspond to hydroxylations at the phenylethyl moiety and at the cyclohexyl ring (Fischer et al., 1998). Fluvastatin selectively inhibited cyclohexyl hydroxylation of glyburide but had little effect on phenylethyl hydroxylation of glyburide. The inhibition of cyclohexyl hydroxylation of glyburide by fluvastatin was predominantly competitive as indicated by a 3-fold increase inKm from 9 to 27 μM with only a ∼30% decrease in Vmax from 320 to 230 nmol/h/mg for the highest 20 μM fluvastatin concentration. TheKi value was thus determined to be 5.9 ± 0.8 μM. Sulfaphenazole, a specific inhibitor of CYP2C9, also specifically inhibited glyburide cyclohexyl hydroxylation with an IC50 of ∼20 μM (data not shown). Glibornuride was metabolized to four unidentified metabolites in incubations with human liver microsomes. Metabolism followed monophasic Michaelis-Menten kinetics with a maximal velocity Vmax of 81 nmol/h/mg protein and a Km of 120 μM. In contrast with glyburide, the inhibition by fluvastatin of glibornuride metabolism was noncompetitive. The mean Kicalculated was 9.4 ± 2.1 μM. Sulfaphenazole also inhibited glibornuride metabolism with an IC50 of ∼10 μM (data not shown).
Discussion
The combined data indicate that fluvastatin is metabolized by multiple enzymes. Specifically, the metabolites that are primarily responsible for the elimination of fluvastatin (Dain et al., 1993) are formed by several enzymes: 5-hydroxy-fluvastatin by CYP2C9 and 6-hydroxy- and deisopropyl-fluvastatin mainly by CYP2C9, CYP3A4, and CYP2C8. The most relevant enzyme for in vivo metabolic clearance of fluvastatin is predicted to be CYP2C9, because it is the only one forming all three metabolites found in vivo. All other enzymes capable of metabolizing fluvastatin produced only the 5-hydroxy-fluvastatin metabolite. The relative contribution of 6-hydroxy- andN-deisopropyl-fluvastatin could therefore serve as an indicator for the contribution of CYP2C9 to fluvastatin elimination. In four healthy human volunteers, the contribution of CYP2C9 appeared to be ∼65% of fluvastatin elimination based on fecal metabolite profiles (Dain et al., 1993). CYP2C9 also appears to be responsible for the high Km component, which contributes 73 to 83% to total fluvastatin metabolism in human liver microsomes. The apparent Km for this component is similar to the Ki for the inhibition of CYP2C9 substrates, such as tolbutamide and diclofenac, by fluvastatin. Also specific CYP2C9 inhibitors, such as sulfaphenazole (Baldwin et al., 1995), strongly inhibit 6-hydroxy- andN-deisopropyl-fluvastatin formation but only inhibited to a limited extent 5-hydroxy-fluvastatin formation. In contrast, ketoconazole, a specific inhibitor of CYP3A (Baldwin et al., 1995), inhibited only 5-hydroxy-fluvastatin formation. Furthermore, the ketoconazole concentration required for a 50% inhibition of 5-hydroxy-fluvastatin formation was greater than would be expected for a reaction that is catalyzed only by CYP3A. This suggests the involvement of enzymes other than CYP3A in this pathway.
For the individual enzymes, the Km of recombinant CYP2C9 fluvastatin metabolism corresponded most closely to the higher Km component in human liver microsomes, whereas the Km of recombinant CYP3A for fluvastatin exhibits a lower Km, possibly reflecting the lower Km component observed in human liver. Both CYP2C9 and CYP3A would thus be predicted to be the major enzymes involved in fluvastatin metabolism.
Overall, fluvastatin appears to be metabolized in human liver by several enzymes, with CYP2C9 being the most important, followed by CYP3A4 and CYP2C8. There was no difference in fluvastatin intrinsic metabolic clearance for CYP2C9R144 and the variant allele CYP2C9C144 , which has been associated with a smaller warfarin maintenance dose in heterozygotes (Furuya et al., 1995). Based on the present data, no difference in fluvastatin clearance is expected in patients which express CYP2C9C144. The involvement of several enzymes in the metabolism of fluvastatin should minimize the effect, in case a coadministered compound inhibits one enzyme. Clinical observations confirm these findings. For example, neither CYP2C9 inhibitors/substrates such as the antidiabetics tolbutamide and glyburide, the anticoagulant warfarin, or the proton pump inhibitor omeprazole (Appel and Dingemanse, 1996) nor CYP3A inhibitors/substrates such as the immunosuppressive cyclosporine A or the antifungal itraconazole had significant clinical effects on fluvastatin. Furthermore, even a compound such as isradipine, which inhibits both CYP2C9- and CYP3A-mediated pathways of fluvastatin metabolism in vitro, is not expected to have a clinically significant effect, because isradipine plasma concentrations are >80-fold lower than the in vitro IC50 values.
Fluvastatin appears also to be safe with respect to its potential effect on other compounds. Fluvastatin was previously reported to inhibit CYP2C9 but not CYP3A and CYP2D6 in vitro (Transon et al., 1996). The present data confirm these findings and demonstrate that fluvastatin does not inhibit CYP1A2, CYP2C8, CYP2C19, or CYP3A. Only small effects on coadministered CYP2C9 substrates were also observed in vivo, in spite of the relatively potent inhibition of CYP2C9 in vitro. Compounds such as diclofenac, warfarin, tolbutamide, or glyburide were only minimally affected, with only Cmaxbeing increased (Transon et al., 1995; Appel and Dingemanse, 1996). This can be explained by the short terminal half-life of fluvastatin of 0.5 to 1 h, which would suggest that fluvastatin can inhibit CYP2C9 mainly during first-pass.
In summary, fluvastatin has a low potential for metabolic drug-drug interactions as compared to other HMG-CoA reductase inhibitors. Multiple enzymes are involved in the metabolism of fluvastatin, with CYP2C9 as the major one. Conversely, although fluvastatin is a potent inhibitor of CYP2C9, this effect is limited because of fluvastatin’s rapid systemic clearance. As a consequence, fluvastatin inhibits coadministered compounds’ metabolism only during first-pass.
Acknowledgments
We thank Dr. T. Ray and Dr. H. Andres for the synthesis of the radiolabeled fluvastatin, and Dr. C. Crespi for the helpful discussions.
Footnotes
-
Send reprint requests to: Dr. Volker Fischer, Novartis Institute for Biomedical Research, 59 Route 10, East Hanover, New Jersey 07936. E-mail: volker.fischer{at}pharma.novartis.com
- Abbreviations used are::
- HMG-CoA
- 3-hydroxy-3-methylglutaryl coenzyme A
- CYP or P-450
- cytochrome P-450
- LC/MS
- liquid chromatography/mass spectrometry
- Received August 20, 1998.
- Accepted November 16, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}