


Abstract
The absorption, excretion, and metabolism of the endothelin receptor antagonist bosentan was investigated in healthy male subjects by administration of 14C-labeled compound. Four subjects received a single oral dose of 500 mg of bosentan (3.7 MBq), and four other subjects received a single i.v. dose of 250 mg of bosentan (3.7 MBq). Radioactivity and concentrations of bosentan and its metabolites were measured in plasma, urine, and feces samples. More than 97% of drug-related material was recovered on average within 3.5 days after oral dosing and within 5 days after i.v. dosing. More than 90% of radioactivity was found in feces after both oral and i.v. dosing. Most of the radioactivity in urine and feces represented bosentan and three metabolites. Ro 48-5033, the major metabolite in plasma, urine, and feces, is the result of hydroxylation at the t-butyl group of bosentan. The two other metabolites Ro 47-8634 and Ro 64-1056 represent minor metabolite species. Ro 47-8634 is the product ofO-demethylation of the phenolic methyl ester, and Ro 64-1056 is generated by both demethylation and hydroxylation. The radioactivity in plasma could almost entirely be attributed to bosentan and the two metabolites Ro 48-5033 and Ro 47-8634, whereby both metabolites exhibited much lower plasma levels than bosentan. Hepatic metabolism followed by biliary excretion of the metabolites apparently represents the major pathway of elimination for bosentan in humans.
Endothelin-1, a 21-amino acid peptide, is the most potent and longest-acting vasoconstrictor known to date. Since its first description and isolation in 1988 (Yanagisawa et al., 1988), evidence has been collected to show that the endothelin system plays a significant role in the pathogenesis of a variety of diseases, including hypertension, heart failure, renal failure, and pulmonary diseases (Noll et al., 1996). The potential of various endothelin antagonists as therapeutic interventions in some of these indications has been investigated in a number of small clinical trials (Webb et al., 1998).
Bosentan is a potent nonpeptide endothelin-receptor antagonist (Clozel et al., 1994) whose vasodilatory effects were shown in patients with hypertension (Krum et al., 1998) and chronic heart failure (Kiowski et al., 1995; Suetsch et al., 1998). The effects on systemic and pulmonary hemodynamic parameters in patients with chronic heart failure were very promising after both acute i.v. doses (Kiowski et al., 1995) and 14 days of oral therapy (Suetsch et al., 1998). Therefore, bosentan is currently under development for the treatment of patients with chronic heart failure. Single- and multiple-dose pharmacokinetics of bosentan in healthy volunteers have been described previously (Weber et al., 1996; C.W., R. Schmitt, H. Birnboeck, G.H., and H. Eggers, submitted for publication). The drug exhibits an oral bioavailability of approximately 50%, which decreased at doses above 500 mg. Bosentan has a low systemic plasma clearance of 7 to 8 liters/h and a moderate volume of distribution of 0.2 liters/kg. Its half-life in humans was between 5 and 8 h. In animals (rat and dog), the drug was eliminated mainly through hepatic metabolism followed by biliary excretion of the metabolites (Ubeaud et al., 1995). In human subjects, almost no unchanged drug was excreted into urine (Weber et al., 1996; C.W., R. Schmitt, H. Birnboeck, G.H., and H. Eggers, submitted for publication). Therefore, metabolism and renal or biliary excretion of metabolites appeared to be the major pathway of its elimination.
The clinical study described here was performed to investigate the rate and route of elimination of bosentan in human subjects to identify the metabolites produced and to investigate the rate and route of excretion of these metabolites.
Materials and Methods
Radiolabeled Drug.
[14C]Bosentan (Fig.1) was synthesized at F. Hoffmann-La Roche Ltd. (Basel, Switzerland). The oral [14C]bosentan suspension showed a specific activity of 7.4 MBq/g, and the i.v. [14C]bosentan solution had a specific activity of 14.8 MBq/g. The radiochemical purity of14C-labeled bosentan was >98%, as determined by HPLC.

Chemical structure of14C-labeled bosentan (4-tert.-butyl-N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2,2′-bipyrimidine-4-yl]benzenesulfonamide); *position of 14C label
Subjects.
Eight healthy young male subjects participated in this study. The study was approved by the Independent Ethics Committee in Assen, the Netherlands. Written informed consent was obtained from each subject before enrollment. Physical examinations were performed, and medical histories, routine laboratory tests, ECGs, and vital signs were recorded before and after the course of drug treatment. Subjects with clinically relevant deviation from normal or with any major illness within 1 month before the screening examination or smoking more than 10 cigarettes a day were not included. Subjects who worked regularly with ionizing radiation or radioactive material and subjects who received any ionizing radiation or radioisotope for research, for diagnostic reasons, or for therapy within 12 months before study start were excluded. In addition, only subjects with frequent bowel movements (≥ once per 48 h) were included. During the study, no concomitant medication was allowed with the exception of medications to treat adverse events. The subjects were not allowed to drink alcoholic or xanthine-containing beverages or smoke during the entire study period. Subjects stayed in the clinic from the evening before dosing until the last safety assessment was performed.
Study Design.
This was an open-label, randomized, parallel group study. After eligibility screening, four subjects received a single oral dose of 500 mg of 14C-labeled bosentan (3.7 MBq), and another four subjects received a single i.v. dose of 250 mg14C-labeled bosentan (3.7 MBq) as an infusion of 15-min duration. The oral dose was administered as 100 ml oral suspension of bosentan in water. For the i.v. dosing, 125 ml of a 4.2% glucose solution containing 250 mg of bosentan was used. Each dose was given within 30 min following a standardized breakfast. Blood samples were collected from a forearm vein via an i.v. catheter before dosing, at 5, 15, 30, 45, and 60 min and 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, and 36 h postdose, and every 12 h thereafter until radioactivity measured in urine and feces samples had returned to less than three times that of background levels or until at least 95% of the total radioactivity had been recovered in urine and feces, whatever was shorter. Urine was quantitatively collected during predefined time windows, i.e., 0 to 4, 4 to 8, 8 to 12, 12 to 16, 16 to 24, 24 to 36 h postdose, and every 12 h thereafter. All feces samples passed by the subjects were collected. The minimum number of collections was four samples. Twelve lead ECGs were recorded before dosing and after all blood, urine, and feces samples had been collected. Vital signs (blood pressure and heart rate) were measured at the same time points as ECGs were recorded and, in addition, before and at 10 h following dosing as well as every morning thereafter until sample collection was completed. Clinical laboratory tests were performed during screening, just before dosing, and after completion of sample collection.
Sample Collection.
Blood samples for measurement of radioactivity and plasma concentrations of bosentan and its metabolites were collected into Vacutainers (Becton Dickinson & Co., Lincoln Park, NJ), containing EDTA as anticoagulant and centrifuged at 4°C. Plasma was separated and stored at −20°C until assayed. Urine was collected into glass containers and kept at 4°C until the end of each collection interval. At the end of each collection interval, the volume was measured, and portions were stored at −20°C until analysis. All feces produced during the study were collected directly into plastic containers, weighted, and stored at −20°C until analysis.
Measurement of Radioactivity.
Radioactivity was determined in the infusion solutions, the oral suspensions, and in plasma, urine, and feces by liquid scintillation counting with external standardization (Tri-Carb 1900 TR liquid scintillation analyzer; Canberra Packard Instruments Co., Downers Grove, IL). Four hundred-microliter aliquots of the infusion solutions were mixed with 10 ml of liquid scintillation fluid (Ultima Gold, Canberra Packard AG, Zürich, Switzerland), and 100-μl aliquots of the oral medication were mixed with 200 μl of dimethyl sulfoxide and 10 ml of Ultima Gold for liquid scintillation counting. In addition, the remainders in each drinking bottle used for oral dosing were determined by rinsing with 55 ml of dimethyl sulfoxide and liquid scintillation counting of a 1-ml aliquot in 10 ml of Ultima Gold. The radioactivities found in the infusion solutions, the oral suspension samples, and the remainders of the drinking bottles were used to accurately calculate the dose that was administered to each subject. One-milliliter aliquots of plasma samples were mixed with 5 ml of Pico Fluor 30 (Packard, Groningen, the Netherlands), and 1-ml aliquots of urine samples were mixed with 10 ml of Pico Aqua (Packard) for scintillation counting. All feces samples collected within 24-h intervals, calculated from the time of drug administration, were pooled. They were mixed with arabic gum solution (ratio of approximately 1:4, w/w) and homogenized for approximately 2 min using an Ultra-Turrax mixer type T25 with a shaft type IKA 25GM (Janke and Kunkel GmbH, Staufen, Germany). Two accurately weighed aliquots of the homogenate (within the order of magnitude of 400 mg) were combusted using a biological oxidizer (OX 500, Zinsser Analytic GmbH, Frankfurt, Germany). The produced [14C]O2 was directly trapped in the scintillation cocktail (15 ml of Pico Solve C-300, Packard) and analyzed by liquid scintillation counting. The results were corrected for the combustion recovery. The mean of two determinations was used for the pharmacokinetic evaluations. For plasma, urine, and feces samples, the limit of quantification was defined as twice the 14C radioactivity in a blank sample.
Bosentan and Metabolite Assay.
Plasma concentrations of bosentan and its metabolites Ro 47-8634 and Ro 48-5033 were determined by a specific liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS1) method with a limit of quantification of 2.0 ng/ml (Lausecker et al., 1998). The performance of the analytical method was monitored by simultaneous analysis of independently prepared quality control samples of various concentrations. The interassay precision was always ≤5.5% for bosentan, ≤6.9% for Ro 47-8634, and ≤14.7% for Ro 48-5033. The interassay accuracy of the method ranged between 97.4 and 97.6% for bosentan, between 92.1 and 97.9% for Ro 47-8634, and between 101.6 and 106.7% for Ro 48-5033.
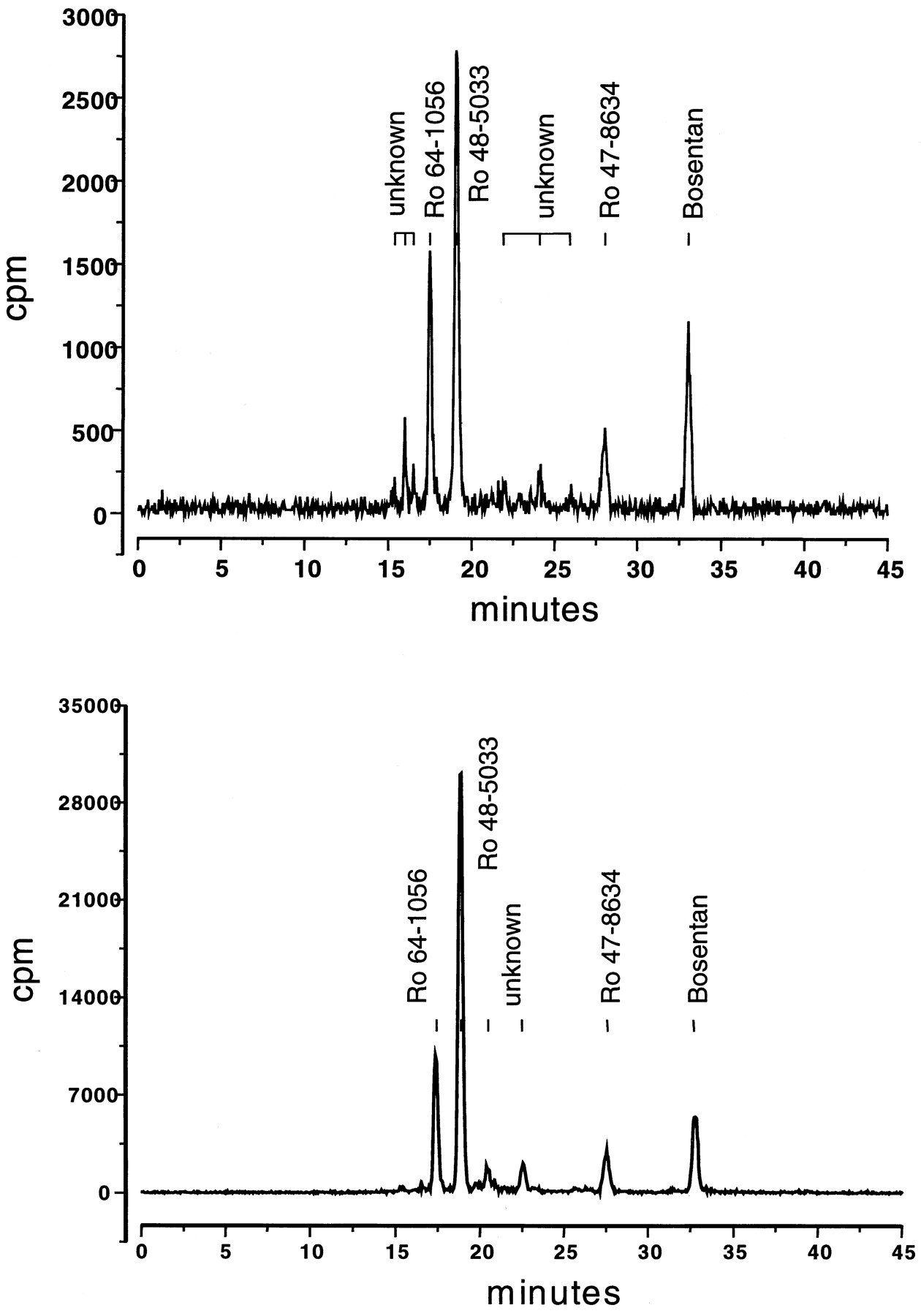
Urine and feces samples were analyzed by a radiometric HPLC assay. Urine samples (1.5-ml aliquots) were applied to a Superspher 100 column (250 × 4 mm; Merck, Darmstadt, Germany) and eluted with a linear gradient of 50 mM ammonium acetate buffer (pH 3.5) containing 1.3% tetrahydrofuran and acetonitrile containing 3% tetrahydrofuran over 40 min. Radioactivity in the eluate was determined by online radioactivity monitoring (Berthold LB 506 C-1 radioactivity monitor; EG & G Berthold, Bad Wildbad, Germany) (Fig. 2A). Fecal samples were diluted with 2 volumes of methanol, sonicated for 20 min (Bransonic 5; Branson, Bender and Hobein, Zurich, Switzerland), and centrifuged for 15 min at 10,000g (Sigma 2–15; Merck-ABS, Dietikon, Switzerland). The pellets were re-extracted with methanol and evaporated to dryness (Rotavapor 611; Buechi, Switzerland). The residue was reconstituted in 50 mM ammonium acetate (pH 3) and methanol (1:1, v/v), filtered (RC 15 cellulose filters; Knauer, Berlin, Germany), and analyzed by HPLC as before (Fig. 2B). The recovery of radioactivity in the extracts ranged from 85 to 105%. The different peaks in the HPLC chromatograms of urine and feces samples were identified by LC-MS/MS. The structure of the three metabolites has been identified earlier by LC-MS/MS and NMR using samples from in vitro experiments with human hepatic microsomes (Dr. Gerard Hopfgartner, Roche Basel, personal communication). They have been confirmed by synthesis of the reference compounds Ro 48-5033, Ro 47-8634, and Ro 64-1056. The fragmentation process of bosentan has been extensively investigated (Hopfgartner et al., 1996, 1998) allowing the screening of metabolites by means of LC-MS/MS.

Typical urinary (top) and fecal (bottom) metabolite pattern after i.v. administration of 250 mg of14C-labeled bosentan.
Pharmacokinetic Evaluation.
Pharmacokinetic evaluation was performed using model independent methods. The pharmacokinetic parameters calculated were peak plasma concentrations (Cmax), time to reachCmax (Tmax), area under the plasma concentration time curve from time zero to infinity (AUC0-∞), apparent terminal elimination half-life (T1/2), systemic plasma clearance (CL), and volume of distribution at steady-state (Vss).
Cmax and Tmaxvalues were taken directly from the observed plasma concentration time data. The area under the curve was estimated using the linear trapezoidal rule up to the last measured concentration value. Extrapolation to infinity was performed by dividing the last measurable concentration by the apparent terminal elimination rate β. The terminal elimination rate β was estimated by performing standard unweighted log-linear least-squares regression analysis of the terminal phase. The T1/2 was calculated by dividing ln2 by β. Systemic plasma clearance after i.v. dosing (CL) was calculated as dose/AUC0-∞. The volume of distribution at steady state (Vss) was calculated as CL·(AUMC0-∞/AUC0-∞ −T1/2), whereby T is the duration of the i.v. infusion (Benet and Galeazzi, 1979).
Excretion of radioactivity into urine and feces was evaluated by calculation of cumulative amount of drug-related material excreted during the predefined time windows for urine collection and expressed in percent of dose administered. The total amount of bosentan, each metabolite, and unknown material into urine and feces was also calculated.
Results
Intravenous Administration.
On average, 98% of the dose was recovered in urine and feces (Table1 and Fig.3), more than 97% in three subjects and 93.1% in one subject. Most of the radioactivity was excreted in the feces, 92.9% of the dose on average. Only 5.2% were found in urine. Within the first 24 h, none or only negligible fecal excretion occurred, whereas renal excretion was almost complete at that time (see Fig. 3). Therefore, most (≥90%) of the radioactivity was excreted at later time points. Excretion was essentially complete 5 days after i.v. administration.
Total amounts of bosentan and drug-related material excreted into urine and feces following a single oral dose of 500 mg of 14C-labeled bosentan and following a single i.v. dose of 250 mg of14C-labeled bosentan

Cumulative excretion of radioactivity over time in urine and feces after oral administration of 500 mg of14C-labeled bosentan (A) and after i.v. administration of 250 mg of 14C-labeled bosentan (B) to four subjects each.
Most of the radioactivity in urine and feces, on average 1.9% of the dose in urine and 47.5% of the dose in feces, was attributable to the metabolite Ro 48-5033 (Table 1). The other two identified metabolites, Ro 64-1056 and Ro 47-8634, were found in lower quantities, each ≤1% of the dose in urine and less than 18% of the dose in feces. A number of minor metabolites, which have not been identified, were detected as peaks in the HPLC chromatograms (Fig. 2). They were apparent in greater quantity in feces than in urine and accounted for less than 9% of the total radioactivity. Approximately 10% of the radioactivity in feces and less than 1% of the radioactivity in urine were of unknown identity and could not be attributed to any peak in the HPLC chromatograms. Unchanged bosentan contributed little to the radioactivity in both urine and feces. Less than 1% of the dose was excreted as bosentan in urine and less than 4% in feces.
Total radioactivity in plasma could almost entirely be attributed to bosentan and the two metabolites Ro 48-5033 and Ro 47-8634 (see Fig.4). On average, the AUC0-∞ of bosentan and the two metabolites Ro 48-5033 and Ro 47-8634 accounted together for 95% of the AUC0-∞ of total radioactivity (estimated in nanomole equivalents of bosentan · h/ml using the molecular weight of bosentan for the calculation).

Mean plasma concentration time profiles of total radioactivity (microgram equivalents/liter), bosentan, the two metabolites Ro 47-8634 and Ro 48-5033, and the calculated sum of bosentan and the two metabolites after oral administration of 500 mg of 14C-labeled bosentan (A) and after i.v. administration of 250 mg of14C-labeled bosentan (B) to four subjects each.
The pharmacokinetic parameters of bosentan and the two major metabolites in plasma are summarized in Tables2 and 3, respectively. CL and Vss of bosentan amounted to 9.3 ± 4.2 liters/h and 24 ± 12 liters, respectively. The apparent terminal elimination half-life was 5.6 ± 2.3 h. The plasma concentrations of both metabolites never exceeded those of bosentan (Fig. 4). The half-life of the major metabolite Ro 48-5033 was 6.1 ± 0.8 h. The half-life of Ro 47-8634 was lower, namely 2.9 ± 0.9 h.
Pharmacokinetic parameters of bosentan following a single oral dose of 500 mg of 14C-labeled bosentan and following a single i.v. dose of 250 mg of 14C-labeled bosentan
Pharmacokinetic parameters of the metabolites Ro 48-5033 and Ro 47-8634 following a single oral dose of 500 mg of 14C-labeled bosentan and following a single i.v. dose of 250 mg 14C-labeled bosentan
Oral Administration.
After oral administration, 97.3% of the dose was recovered in urine and feces on average (Table 1 and Fig. 3), at least 94.5% in each individual. As following i.v. dosing, most of the radioactivity was found in the feces, 94.5% of the dose on average. Only 2.8% were found in urine. Excretion was essentially complete 3.5 days after oral dosing.
Most of the radioactivity in urine and feces, on average 1.1% of the dose in urine and 34.6% of the dose in feces, was attributable to the metabolite Ro 48-5033 (Table 1). The other two identified metabolites, Ro 64-1056 and Ro 47-8634, were found in lower quantities, their relative amounts being similar to that found after i.v. dosing. Less than 1% of the total radioactivity recovered in urine and less than 10% in feces were of unknown identity (minor metabolites). Unchanged bosentan was recovered to a higher extent in feces than after i.v. dosing, 30.2% of the dose as compared with 3.7% after i.v. administration. Renal excretion of bosentan was low, 0.1% of the dose was found in urine on average.
Most, but not all, of the radioactivity in plasma could be attributed to bosentan and the two metabolites Ro 48-5033 and Ro 47-8634. Especially at later time points (after 24 h), the total radioactivity was higher than expected based on the concentrations of bosentan and its two known metabolites (see Fig. 4). The AUC0-∞ of bosentan and the two metabolites, Ro 48-5033 and Ro 47-8634, accounted together for 79% of the AUC0-∞ of total radioactivity (estimated in nanomole equivalents of bosentan · h/ml using the molecular weight of bosentan for the calculation) on average.
Mean peak bosentan plasma concentrations of 3724 ± 791.6 μg/l (mean ± S.D.) were reached within 6.5 ± 3.8 h after dosing, at the same time when peak levels of radioactivity were measured. The plasma concentrations of both metabolites never exceeded those of bosentan (Fig. 4). Cmax of Ro 48-5033 was on average in the range of 11% of that of bosentan;Cmax of Ro 47-8634 was on average 5% of that of bosentan (Table 3). Peak plasma levels of Ro 47-8634 were reached always earlier than those of Ro 48-5033. The apparent terminal elimination half-life of bosentan was 7.3 ± 3.8 h. The half-life of the two metabolites, Ro 48-5033 and Ro 48-8634 was 10.3 ± 4.7 h and 3.6 ± 1.6 h, respectively.
Discussion
Based on the recovery of more than 95% of the total radioactivity administered in six subjects and more than 93% in two subjects, it can be concluded that the study was well conducted and the duration of sample collection was adequate. Excretion was almost complete in less than 5 days following i.v. dosing and in less than 3.5 days following oral dosing.
Renal excretion of bosentan and its metabolites is negligible; less than 10% of the total radioactivity was found in urine. The major portion of radioactivity was excreted into the feces in the form of metabolites. Contribution of gut toward metabolism of bosentan cannot be ruled out completely at present. However, because in rats and dogs the majority of drug-related material was found to be excreted in the bile and because clearance in humans could well be predicted by combining in vivo and in vitro (hepatocyte metabolism) data from a few animal species with the in vitro hepatocyte metabolic data in humans (Ubeaud et al., 1995), it is hypothesized that hepatic metabolism and biliary excretion of metabolites may also represent the major pathway of elimination for bosentan in humans. The larger amount of bosentan found in feces after oral dosing as compared with i.v. dosing (30.2% versus 3.7%) most probably reflects nonabsorbed drug material and additional first-pass material. A first-pass effect of maximally 20% was expected based on the systemic plasma clearance of bosentan and its blood/plasma distribution of 0.6 (C.W., R. Schmitt, H. Birnboeck, G.H., H. Eggers, submitted for publication). Based on the AUC data of bosentan in this study, an oral bioavailability of approximately 40% can be estimated. This figure is certainly only a rough estimate because of the low number of subjects studied per treatment group and the fact that this was not a crossover study. Overall, this value is in line with earlier findings of an oral bioavailability of 30 to 78% (individual data from Weber et al., 1996). Assuming that renal clearance relative to total clearance is similar following both oral and i.v. dosing, an oral bioavailability of bosentan of approximately 11% can be roughly estimated from the urine data in this study. The difference between urine-based and plasma (AUC)-based bioavailability figures (11% versus 40%) could point toward a higher metabolic clearance following oral dosing than following i.v. dosing, part of this probably being due to a first pass effect after oral dosing.
Most of the drug-related material in feces could be attributed to the metabolite Ro 48-5033, the major metabolite detected in plasma. The second major metabolite detected in urine and feces was Ro 64-1056, but its presence in plasma was not observed. Ro 47-8634 represented only a minor metabolite fraction in all matrices collected. Nearly all radioactivity measured after i.v. dosing in plasma could be attributed to bosentan and its two metabolites Ro 48-5033 and Ro 47-8634. Following oral dosing, however, at late time points (after 24 h postdose) the time course of radioactivity deviated from that of bosentan and its known metabolites and declined with an apparently longer half-life. Part of this difference may be due to the fact that concentrations of especially Ro 47-8634, but in some individuals also those of Ro 48-5033, were underestimated because they fell below their limit of quantification at these time points. It cannot be excluded that additional, not yet identified metabolites have been produced and are present in plasma in substantial quantities at late time points only. Because of the relatively low systemic concentrations of all three metabolites and because metabolism represents the major pathway of elimination for bosentan, it was concluded that the majority of metabolites produced in the hepatocytes were immediately excreted into bile without reaching the systemic circulation.
Based on this study, the metabolic pathway for bosentan in humans, as depicted in Fig. 5, is proposed. Ro 48-5033, the major metabolite, is the result of hydroxylation at thet-butyl group of bosentan; Ro 47-8634 is the product ofO-demethylation of the phenolic methylester. Ro 64-1056 results from the combination of both metabolic reactions.

Proposed metabolic pathway of bosentan in humans.
Because Ro 48-5033 exhibits an apparently longer half-life than bosentan, some accumulation of this metabolite in plasma might be expected during multiple dosing regimens. From the average half-life of 6 to 10 h, it seems unlikely that such an accumulation could lead to plasma levels of metabolites exceeding those of bosentan. The half-life of Ro 47-8634 may have been underestimated in most subjects because the concentrations fell below the limit of quantification of the analytical assay before the apparent terminal elimination phase was reached.
In summary, nearly all drug-related material was recovered within 3.5 (oral dosing) and 5 (i.v. dosing) days. Hepatic metabolism followed by biliary excretion of the metabolites most likely represents the major pathway of elimination for bosentan. Ro 48-5033 was the major metabolite detected in plasma, urine, and feces, but compared with bosentan it exhibited lower plasma concentrations. Two other metabolites, identified as Ro 47-8634 and Ro 64-1056, represent minor metabolite species.
Acknowledgment
We thank A. Karwoth-Graf for assistance in the pharmacokinetic evaluations.
Footnotes
-
Send reprint requests to: C.W., F. Hoffmann-La Roche Ltd., Dept. of Clinical Pharmacology, CH-4070 Basel, Switzerland. E-mail: cornelia.weber{at}roche.com
- Abbreviations used are::
- LC-MS/MS
- liquid chromatography-mass spectrometry/mass spectrometry
- AUC0-∞
- area under the plasma concentration time curve from time zero to infinity
- Received November 19, 1998.
- Accepted February 15, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}