


Abstract
Cytochrome P-4503A, CYP2B, and P-450 reductase are induced by glucocorticoids, antiglucocorticoids such as pregnenolone 16α-carbonitrile, and drugs such as rifampin and phenobarbital. Although the pregnane X receptor is reported to mediate steroid and drug activation of CYP3A via a conserved cis-element in CYP3A genes, discrepancies exist between the induction of the endogenous CYP3A genes and the activation of the pregnane X receptor. It is a formal possibility that the glucocorticoid receptor may account for some of these discrepancies. To determine the requirement in vivo of the glucocorticoid receptor in expression of CYP3A and CYP2B, we compared the induction of these proteins in the livers of normal mice and mice with a targeted mutation in the glucocorticoid receptor. Mice lacking the glucocorticoid receptor show no difference in constitutive hepatic expression of CYP3A but show a decrease in the level of CYP2B. Glucocorticoid receptor-deficient mice challenged with either dexamethasone or pregnenolone 16α-carbonitrile failed to induce CYP2B proteins, whereas CYP2B was readily induced in (+/+) mice. In contrast, CYP3A and P-450 reductase proteins were induced by either inducer in wild-type and glucocorticoid receptor-null mice. Similarly, rifampin induced CYP3A in either wild-type or glucocorticoid receptor-null mice. Despite reports that rifampin is a nonsteroidal ligand for the human glucocorticoid receptor, rifampin failed to induce tyrosine aminotransferase in mice regardless of glucocorticoid receptor genotype, and rifampin did not compete for ligand binding to either mouse or human glucocorticoid receptor. Phenobarbital induced CYP3A, CYP2B, and P-450 reductase in all mice, but the amplitude of induction was diminished 37% in glucocorticoid receptor-null mice. Thus, there are distinctly different essential requirements of CYP3A, CYP2B, and P-450 reductase genes for the glucocorticoid receptor in their induction by steroids and drugs.
Glucocorticoids (both agonists and antagonists), rifampin, and phenobarbital induce cytochromes P-450, particularly CYP3A11 and 3A13 in mice and CYP3A4 in humans in vivo (Watkins et al., 1985; Wrighton et al., 1985b; Yanagimoto et al., 1997) and in their cultured hepatocytes (Schuetz and Guzelian, 1984; Strom et al., 1996). Rat CYP3A1 is unresponsive to rifampin (Wrighton et al., 1985b). Many of these same compounds induce rat, mouse, and human CYP2B proteins (Strom et al., 1996; Honkakoski and Negishi, 1998). In humans, these inductions lead to clinically important drug-drug interactions when glucocorticoids, phenobarbital, or rifampin is administered concurrently with medications (e.g., cyclosporin A, contraceptives, and so on) that are normally metabolized by these CYPs. Understanding the molecular mechanisms leading to drug and steroid induction of CYP3A and CYP2B, and particularly whether these cytochromes share identical regulatory mechanisms, ultimately leads to better models for screening and predicting drug interactions.
In probing the mechanism of glucocorticoid induction of these cytochromes, an unresolved issue has been the possible direct or indirect involvement of the glucocorticoid receptor in CYP3A induction. Although glucocorticoids induce the expression of many genes by ligand-mediated activation of the glucocorticoid receptor, we determined that glucocorticoid and, paradoxically, antiglucocorticoid induction of CYP3A reflected a nonclassic glucocorticoid-receptor induction process (Schuetz and Guzelian, 1984). Several recent reports have identified a unique nuclear hormone receptor, the pregnane X receptor (PXR)1(Kliewer et al., 1998; Lehmann et al., 1998; also referred to as hSXR,Blumberg et al., 1998), which appears to mediate steroid and drug induction of CYP3A. Mouse and human PXRs are ligands activated by high concentrations of glucocorticoid agonists (dexamethasone) and phenobarbital and in a species-specific manner by the glucocorticoid antagonist (pregnenolone 16α-carbonitrile; PCN) and by rifampin (Kliewer et al., 1998; Lehmann et al., 1998). Ligand-stimulated PXR transcriptionally activates hormone response elements found in the ratCYP3A1 (Kliewer et al., 1998) and human CYP3A4genes (Blumberg et al., 1998; Lehmann et al., 1998), elements that are conserved in the mouse CYP3A11 gene (Toide et al., 1997).
However, closer inspection of these and other reports suggests that the PXR may not totally account for drug and steroid induction of CYP3A, and therefore, the possibility exists that the glucocorticoid receptor is also required for the induction of this cytochrome. First, dexamethasone, which is an efficacious inducer of rat, mouse (Wrighton et al., 1985b; Yanagimoto et al., 1997), and human CYP3A (Watkins et al., 1989), was only a weak activator of mouse (Kliewer et al., 1998) and human (Bertilsson et al., 1998; Blumberg et al., 1998) PXRs. Indeed, Bertilsson et al. (1998) found no activation of human PXR by dexamethasone, concluding that there must be additional mechanisms for its induction of CYP3A4. Second, none of the four reports on PXR have used the authentic CYP3A4 5′-flanking sequences (which contain putative glucocorticoid receptor binding elements; Ogg et al., 1999); instead, only reporters with multiple copies of the PXR binding motif were used. Therefore, it cannot be concluded with certainty that PXR is the singular transcription factor required for steroid and drug induction of CYP3A. Indeed, it was recently reported (Pereira et al., 1998) that a functional glucocorticoid response element (GRE) bound glucocorticoid receptor in the rat CYP3A1 gene, and it was suggested that cooperation of the upstream GRE and downstream elements (e.g., PXR element) may be required for the maximal response of CYP3A to glucocorticoids. Similarly, cotransfection of glucocorticoid receptor with the CYP3A4 promoter in HepG2 cells demonstrated a requirement of glucocorticoid receptor for CYP3A induction by steroids and drugs (Ogg et al., 1999). Third, the antiglucocorticoid RU486 [a mouse (Kliewer et al., 1998) and human (Lehmann et al., 1998) PXR activator] paradoxically blocks dexamethasone transcriptional activation of the rat CYP3A1 and human CYP3A4 promoters (Burger et al., 1992; Ogg et al., 1999), again suggesting a role for the glucocorticoid receptor in dexamethasone induction of CYP3A. Finally, rifampin is a good inducer of mouse CYP3A (Wrighton et al., 1985b; Yanagimoto et al., 1997), yet rifampin is reported to be a weak activator of mouse PXR, suggesting there may be an alternate mechanism for rifampin induction of CYP3A in the mouse. However, rifampin was recently demonstrated to be a nonsteroidal ligand for the human glucocorticoid receptor (Calleja et al., 1998). Therefore, the possibility existed that the glucocorticoid receptor played a role in steroid and rifampin induction of CYP3A.
There also is evidence that the classic glucocorticoid receptor may participate in regulation of the CYP2B1/2 genes. For example, the mouse CYP2B10 and rat CYP2B1/2 (Stoltz et al., 1998) genes contain GREs, and the rat CYP2B2 GRE is functionally activated by glucocorticoids (Jaiswal et al., 1990). There are indications as well that the glucocorticoid receptor is required for maximal phenobarbital induction of CYP2B10 in mice and of CYP2B2 in rats (Shaw et al., 1993; Honkakoski and Negishi, 1998), and a putative GRE has been identified as part of a phenobarbital response unit in the 5′-flanking sequences of the rat CYP2B2 gene (Stoltz et al., 1998).
To determine whether the induction of these structurally dissimilar gene products by steroids and drugs requires the glucocorticoid receptor, we analyzed the induction of these CYP genes in glucocorticoid receptor-null mice. The majority of mutant mice carrying disruption of the glucocorticoid receptor gene die within minutes of birth due to atalectasis of the lungs (Cole et al., 1995), with 12% of the homozygous mutants surviving. The glucocorticoid receptor-null mice have impaired activation of many hepatic enzymes, including gluconeogenic enzymes and tyrosine aminotransferase (TAT), which require the glucocorticoid receptor for glucocorticoid activation (Cole et al., 1995). By using adult glucocorticoid receptor-null mice, we have found that there is an absolute requirement of the glucocorticoid receptor for steroid induction of CYP2B, but the receptor is not obligatory for glucocorticoid, antiglucocorticoid, or rifampin induction of CYP3A or P-450 reductase.
Experimental Procedures
Materials.
Mouse 3T3-L1 cells were obtained from American Type Culture Collection (Rockville, MD). Dulbecco's modified Eagle's medium with no phenol red (DMEM) and the nonradioactive steroids aldosterone, dexamethasone, and rifampin were obtained from Sigma Chemical Co. (St. Louis, MO). FBS was obtained from CSL (Melbourne, Australia). The highly specific synthetic glucocorticoid and progesterone receptor antagonist RU38486 (17β-hydroxy-11β-4-dimethylamino-phenyl-17α-1-propynl-estra-4,9-dien-3-one) was a generous gift from Roussel-UCLAF (Paris, France). [3H]Aldosterone (60–80 Ci/mmol) was obtained from New England Nuclear (Boston, MA). [3H]Dexamethasone (60–80 Ci/mmol) was obtained from Amersham (Buckinghamshire, UK).
Mice.
All experiments with glucocorticoid receptor (+/+) and (−/−) mice were carried out using littermates of C57BL6-129/J (+/−) heterozygote intercrosses (Cole et al., 1995) and represented the 12% survivors of the (−/−) offspring. Glucocorticoid receptor genotype of all littermates was determined by Southern blotting of tail DNAs. Mice ranged in age from 7 weeks to ∼1 year. Three mice (representing males and females) of (+/+) genotype and three mice of (−/−) genotype were used for each drug treatment. For drug induction studies, mice were age matched as closely as possible. Mice received daily i.p. injections of 50 mg/kg dexamethasone or PCN (Upjohn Co.) or rifampin for 2 days and were sacrificed on day 3. PCN was administered in water containing 2% Tween 80. Dexamethasone was administered in corn oil. Rifampin was administered in water containing 10% DMSO. Sodium phenobarbital in water was administered daily i.p. (75 mg/kg) for 3 days, and the mice were sacrificed on day 4.
RNA Isolation and Northern Blot Analysis.
Total RNA was isolated from mouse livers and analyzed by Northern blot with CYP3A11 cDNA (Yanagimoto et al., 1997). Blots were analyzed using a PhosphorImager or autoradiographed, and band intensities were quantified with Image Quant software or densitometry.
Immunoblot Analysis.
Mouse liver microsomes were prepared (Wrighton et al., 1985b), and 10 or 20 μg of protein was analyzed on 10% slab polyacrylamide gels, electrophoresed, and immunoblotted with monoclonal anti-rat CYP3A1 Ig8 (Hostetler et al., 1987), polyclonal goat anti-rat CYP3A1 antibodies (Hostetler et al., 1987), or polyclonal goat anti-rat CYP2B antibodies (raised against purified rat CYP3A and CYP2B proteins) previously developed by Dr. S. Wrighton (Eli Lilly & Co., Indianapolis, IN) and described (Wrighton et al., 1985a; Hostetler et al., 1989). Monoclonal antibodies against rat CYP1A (CD2, 3, and 5) and rat CYP2B1 (monoclonal antibodies BE28.2 and BE26.2) were previously obtained from Dr. Paul Thomas (Rutgers University, Piscataway, NJ). Monoclonal H-8 antibody against rat CYP2B (from Dr. Milton Adesnik, Albert Einstein University, NY) was used previously in our laboratory (Schuetz et al., 1986). Sheep anti-TAT was obtained from Dr. Robert Ivarie (University of Georgia, Athens, GA) (Schuetz and Guzelian, 1984). Rabbit anti-rat CYP4A (from Dr. Richard Okita, Washington State University, Pullman, WA) and anti-rat P-450 reductase (from Dr. Ken Thummel, University of Washington, Seattle, WA) were generously provided. All primary antibodies were followed by appropriate secondary antibodies coupled with peroxidase and developed with the enhanced chemiluminescence detection system (Amersham).
Competition Binding Studies.
Steroid binding to receptors was performed with a whole-cell binding assay. 3T3-L1 cells were plated at a density of 1 to 2 × 105 cells/35-mm well and grown to confluence with DMEM supplemented with 10% FBS. At confluence, the cells were washed twice with DMEM without serum to remove endogenous steroids and serum proteins. Then, 2.5 ml of fresh medium without serum was added, and the cells were incubated for 30 min at 37°C. Cells were then incubated in a total volume of 2.5 ml with either: 1) 2 nM [3H]aldosterone plus 1 μM RU38486 and increasing concentrations (0–1000 nM) of nonradioactive aldosterone or rifampin, or 2) 2 nM [3H]dexamethasone and increasing concentrations (0–1000 nM) of nonradioactive dexamethasone or rifampin. After incubation for 60 min at 37°C, the cells were washed twice with isotonic saline, scraped, and counted in a scintillation counter.
Human brain tumor tissue (100 mg) was homogenized in ice-cold buffer consisting of 8.5 mM Na2HPO4 · 7H2O, 1.5 mM KH2PO4, 10 mM Na2MoO4 · 2H2O, 20% (v/v) glycerol, and 2 mM monothiolglycerol, pH 7.4, and homogenates were centrifuged at 150,000g for 60 min at 4°C to yield cytosol. Cytosol protein levels (2–4 mg/ml) were determined with the Bradford assay. For competition studies, cytosols (100 μl) were incubated for 16 to 18 h at 4°C with 2 mM tracer, [3H]dexamethasone (100 μl), and increasing concentrations (0–10,000 nM) of nonradioactive dexamethasone or rifampicin (100 μl). The total incubation volume was 300 μl. Bound and free steroids were separated by the addition of 300 μl of an ice-cold suspension of hydroxyapatite (15%, w/v) in 50 mM tris(hydroxymethylaminomethane) and 10 mM KH2PO4, pH 7.2. After incubation for 20 min at 4°C with intermittent shaking, tubes were centrifuged (1000g, 5 min), the supernatant was aspirated, and the pellet was washed three times with ice-cold buffer (8.5 mM Na2HPO4 · 7H2O, 1.5 mM KH2PO4, and 10 mM Na2MoO4 · 2H2O, pH 7.2). Washed hydroxyapatite pellets were resuspended in 2 ml of ethanol at room temperature for 15 min and centrifuged, and the supernatant was taken for liquid scintillation spectrometry.
Results
We first examined the expression of CYP3A mRNA in livers of untreated male and female mice. Northern blot analysis of total RNA from male or female glucocorticoid receptor (+/+) and (−/−) mice probed with a CYP3A cDNA showed they expressed similar levels of CYP3A mRNA regardless of glucocorticoid receptor genotype (Fig.1). Analysis of poly(A)+ RNA from female mice confirmed that there was no effect of glucocorticoid receptor genotype on constitutive expression of CYP3A mRNA (not shown).
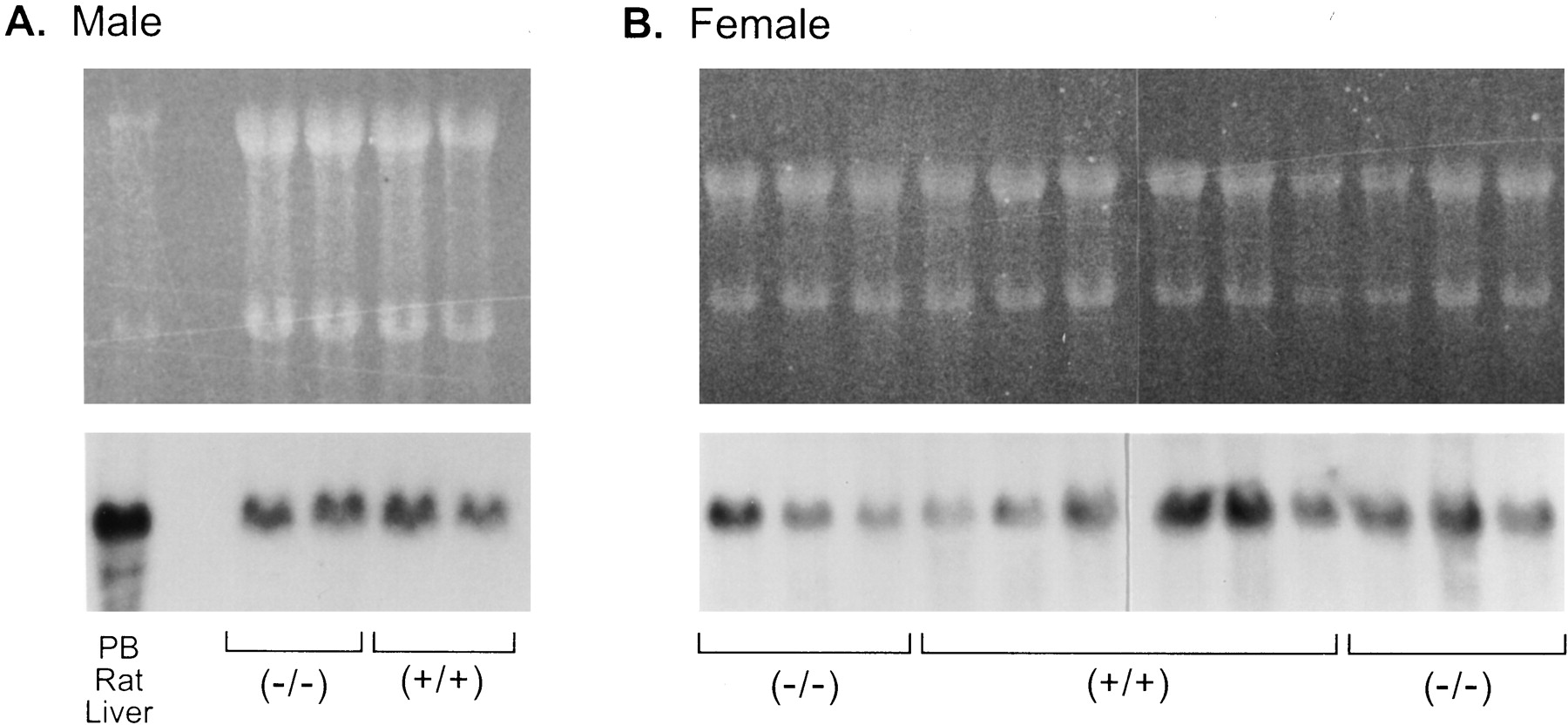
Expression of CYP3A mRNAs in the livers of glucocorticoid receptor (+/+) and (−/−) mice.
Total RNA (20 μg) from the livers of individual glucocorticoid receptor (+/+) and (−/−) male mice (∼1 year old) (A) and female mice (7 weeks to 1 year old) (B) was analyzed by Northern blotting followed by ethidium bromide staining (top) and using a CYP3A1 cDNA (bottom) (Wrighton et al., 1985b). Total RNA (10 μg) from the liver of a rat treated with sodium phenobarbital (PB Rat Liver) was run as a positive control.
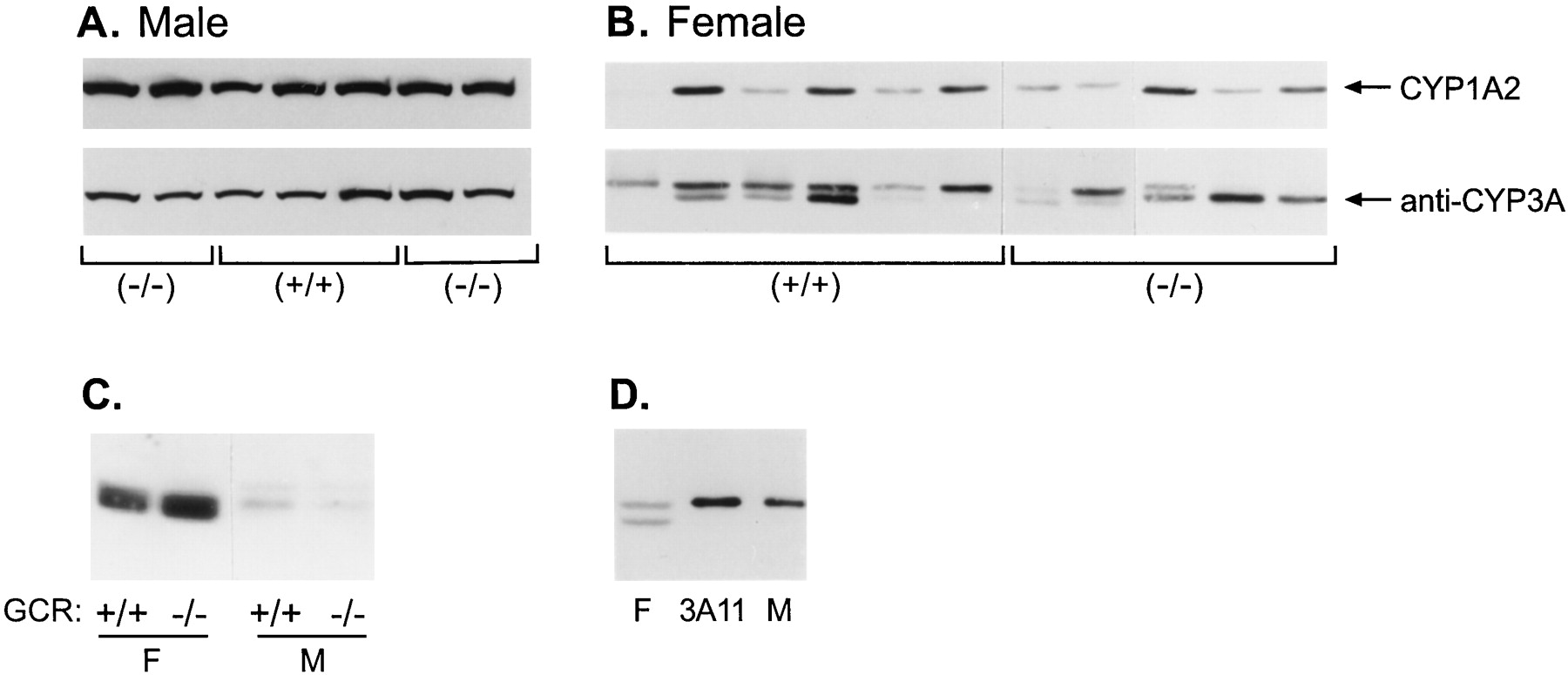
We next investigated the expression of hepatic cytochromes P-450 using antibodies that cross-react with CYP1A1 and CYP1A2. Only CYP1A2 is constitutively expressed in mouse liver, and its expression was similar in glucocorticoid receptor (+/+) and (−/−) mice regardless of gender (Fig. 2, A and B). Expression of the multiple hepatic proteins immunoreactive with monoclonal anti-CYP3A1 (Fig. 2, A and B) or with polyclonal anti-CYP3A1 (not shown) was unaffected by the presence of the glucocorticoid receptor in either male or female mice. Expression of CYP3A proteins was variable among both glucocorticoid receptor (+/+) and (−/−) females and could be due to age and mixed genetic background differences among the mice. However, because there was no uniform effect of glucocorticoid genotype on CYP3A protein, we conclude that the glucocorticoid receptor does not regulate constitutive CYP3A expression. Figure 2C illustrates the dramatic gender difference in the hepatic expression of CYP3A proteins in untreated male and female mice used in this study, consistent with the sexually dimorphic expression of many CYPs in certain strains of mice (Teglund et al., 1998; Park et al., 1999).

Expression of CYP1A2 and CYP3A proteins in the livers of glucocorticoid receptor (GCR) (+/+) and (−/−) mice.
Microsomal protein (20 μg) of from the livers of individual glucocorticoid receptor (+/+) and (−/−) male (A) and female (B) mice was analyzed by immunoblotting using either monoclonal antibody for rat CYP1A1/2 or monoclonal antibody against rat CYP3A1. All of the males were approximately 1 year old. Lanes left to right, the ages of the females were 6 weeks, 2.5 months, 7 weeks, 1 year, 6 months, 7 weeks, 7 weeks, 7 weeks, 7 months, 7 months, and 10 months. C, microsomal protein (10 μg) from female (F) and male (M) glucocorticoid receptor (+/+) and (−/−) mice were compared for the level of immunoreactive CYP3A proteins with anti-CYP3A1 on the same gel. D, liver microsomal protein (2 μg) from a female (F) mouse, 1 pmol of purified mouse liver CYP3A11 isolated from a phenobarbital-treated mouse (Bornheim and Correia, 1990), and 10 μg of microsomes from a male (M) mouse were immunoblotted with monoclonal anti-CYP3A1 IgG. All samples were analyzed on 10% polyacrylamide gels and immunoblotted as described inExperimental Procedures.
We next attempted to identify which of the mouse liver proteins immunoreactive with anti-CYP3A antibodies was CYP3A11. To date, two highly related mouse CYP3A cDNAs expressed in adult mouse liver have been cloned, CYP3A11 (Yanagimoto et al., 1992) andCYP3A13 (Yanagimoto et al., 1994) (accession nos. X60452 andX63023, respectively), but only CYP3A11 has been purified (Bornheim and Correia, 1990). We previously determined that there are four proteins in mouse liver that immunoreact with CYP3A antibodies (Schuetz et al., 1996). On SDS-polyacrylamide gel electrophoresis, one CYP3A doublet selectively immunoreacts with monoclonal anti-CYP3A1, and the other doublet (faster migrating) immunoreacts with monoclonal anti-CYP3A4 (Schuetz et al., 1996). We observed these same four proteins in more than 14 inbred strains of female mice (E.G.S., unpublished observation). Monoclonal anti-CYP3A1 (Fig. 2D) and polyclonal anti-CYP3A1 antibody (not shown) each recognized purified CYP3A11 as the same single immunoreactive protein on immunoblots. Immunoreactive CYP3A11 comigrated with the immunoreactive protein in male liver microsomes and with the higher-molecular-weight protein in female mouse liver microsomes (Fig. 2D). Mixing experiments of CYP3A11 together with the mouse liver microsomes revealed comigration of CYP3A11 with the upper band in females (not shown). It is possible CYP3A13 is the lower-molecular-weight anti-CYP3A1 protein in mouse liver (Yanagimoto et al., 1997).
Dexamethasone, PCN, and rifampin are known inducers of mouse liver CYP3A proteins and mRNAs (Wrighton et al., 1985b; Yanagimoto et al., 1997). The treatment of male mice with dexamethasone, PCN, or rifampin induced CYP3A proteins in both glucocorticoid receptor (+/+) and (−/−) genotypes (Figs. 3 and4). Although the total amount of induced CYP3A protein within treatment groups was similar, regardless of glucocorticoid receptor genotype [e.g., PCN (+/+) and (−/−) had similar amounts of immunoreactive CYP3A protein], given the heterogeneity in the control levels of CYP3A and without full dose-response analysis, we cannot say with certainty that the proteins are induced to exactly the same extent. However, it is clear that the glucocorticoid receptor is not essential for these agents to induce CYP3A. Because dexamethasone and PCN are inducers of mouse and rat liver CYP2B (Hardwick et al., 1983; Honkakoski and Negishi, 1998), we next determined whether the glucocorticoid receptor had a role in mouse CYP2B induction. Although some investigators find CYP2B10 is the higher-molecular-weight species on immunoblots (Jarukamjorn et al., 1999), we lack authentic purified proteins to confirm the identity of the actual immunoreactive CYP2B proteins and refer hereafter to the upper immunoreactive band as CYP2B because in male mice only the higher-molecular-weight species was significantly induced by dexamethasone and PCN (Figs. 3 and5) and phenobarbital (Figs. 5 and6), which is consistent with this being a CYP2B protein. Surprisingly, dexamethasone and PCN induced CYP2B only in glucocorticoid receptor (+/+) mice (Figs. 3 and 5). Moreover, basal expression of CYP2B (higher-molecular-weight protein) was significantly decreased in the glucocorticoid receptor (−/−) compared with (+/+) mice (Figs. 3 and 6). There was no induction by rifampin of CYP2B proteins in any of the mice. No attempt was made to measure enzyme activities for CYP2B because there have been no definitive studies to date correlating distinct CYP2B mouse liver proteins with microsomal testosterone hydroxylase activities in C57BL/6J and 129SvJ mice.

Induction of hepatic cytochromes P-450 in male glucocorticoid receptor (+/+) and (−/−) mice.
Male glucocorticoid receptor (+/+) and (−/−) mice were treated i.p. with 50 mg/kg dexamethasone (DEX), PCN, or rifampin (RIF) for 48 h, and 10 μg of liver microsomal protein was analyzed by immunoblotting with polyclonal rabbit anti-rat CYP4A or anti-rat P-450 reductase antibodies or polyclonal goat anti-rat CYP3A1 IgG or monoclonal anti-rat CYP2B IgG.

Induction of hepatic cytochromes P-4503A in male glucocorticoid receptor (+/+) and (−/−) mice.
Liver microsomes (5 μg) from individual treated and control (CT) male glucocorticoid receptor (+/+) and (−/−) mice (see legend to Fig.3) were analyzed by immunoblotting with two different lots of polyclonal goat anti-rat CYP3A1 (1 or 3) or monoclonal anti-rat CYP3A1 (2) and developed for a short (SH) or long (L) duration.

Induction of hepatic cytochromes P-4502B in male glucocorticoid receptor (+/+) and (−/−) mice.
Liver microsomes (5 μg) from individual male glucocorticoid receptor (+/+) and (−/−) mice (see legend to Fig. 3) or a control (CT) (+/+) mouse or a mouse treated with phenobarbital (PB) were analyzed by immunoblotting with monoclonal antibodies raised against rat CYP2B (anti-CYP2B 1, 2, and 3, monoclonal antibodies H8, BE28.2, and BE26.2, respectively) or polyclonal goat anti-rat CYP2B (4).

Phenobarbital induces cytochromes P-450 in glucocorticoid receptor knock-out mouse liver.
A, liver microsomal protein (10 μg) from untreated control or phenobarbital-treated male (unlabeled) and female (F) glucocorticoid receptor (+/+) and (−/−) mice were analyzed on 10% polyacrylamide gel electrophoresis followed by immunoblotting with either polyclonal anti-P-450 reductase, anti-CYP3A1, or monoclonal anti-CYP2B antibody. All mice were 6 to 7 weeks old. A longer exposure of the control microsomes developed with CYP2B IgG is added (lanes 1–4) to demonstrate the significant decrease in CYP2B (higher-molecular-weight protein) expression in untreated (control) glucocorticoid receptor (−/−) mice. B, liver microsomal protein (4–10 μg) from individual phenobarbital-treated glucocorticoid receptor (+/+) and (−/−) mice was immunoblotted as in (A) and quantified by densitometry, revealing that CYP2B and CYP3A were each induced in receptor (−/−) mice to only 63% of their induction in (+/+) mice (100%).
The researchers at numerous laboratories are routinely using antibodies raised against rat liver CYPs or other rat liver proteins to study regulation of the orthologous genes in mice (e.g., Park et al., 1999). Several lines of evidence together support the specificity of antibodies raised against rat CYPs recognizing the corresponding orthologous mouse gene products in this study. First, purified CYP3A11 immunoreacted with the CYP3A1 monoclonal and polyclonal IgGs (Fig. 2D), and these CYP3A antibodies have been previously shown to be specific for CYP3A proteins (Hostetler et al., 1987). Second, the deduced amino acid sequence of CYP3A11 is 87.3 and 84.9% identical with rat CYP3A1/2, respectively (Yanagimoto et al., 1992), and CYP3A13 shows 71% amino acid identity with CYP3A11 (Yanagimoto et al., 1994). By definition, the amino acid identity between P-450 gene families is ≤40%, making it most likely that the antibodies recognize mouse CYP3A (or CYP2B). Third, we used a panel of anti-CYP3A and anti-CYP2B monoclonal and polyclonal antibodies and compared the immunoblots of liver microsomes from the treated mice (Figs. 4 and 5). Two separate preparations of polyclonal anti-CYP3A1 IgG and a CYP3A1 monoclonal IgG gave the same pattern of immunoreactive proteins. Similarly, three unique monoclonal and one polyclonal IgG raised against rat CYP2B gave identical results, making it exceedingly likely that the higher-molecular-weight protein recognized by these four different anti-CYP2B antibodies is indeed a mouse CYP2B protein (tentatively identified as CYP2B10). The specific CYP2 family lower-molecular-weight immunoreactive proteins remain undefined. Fourth, the monoclonal IgGs had previously been raised against purified rat liver CYP2B and CYP3A proteins, and all of the polyclonal IgGs have previously been backabsorbed against microsomes from a 3-methylcholanthrene-treated male rat (Hostetler et al., 1987) to prevent promiscuous immunoreactivity with other CYP family members. In addition, other investigators (Park et al., 1999) who examined CYP proteins in mouse liver microsomes found similar patterns of bands immunoreacting with anti-rat CYP2B antibodies (Jarukamjorn et al., 1999; Park et al., 1999) and have identified CYP2B10 and CYP2B9 as the slower and faster migrating immunoreactive proteins, respectively. Finally, the regulatory patterns of the CYP3A and the higher-molecular-weight CYP2B protein in these mice conform to their patterns of induction by dexamethasone, rifampin, PCN, and phenobarbital that have been reported previously (Wrighton et al., 1985b; Yanagimoto et al., 1997; Honkakoski and Negishi, 1998; Jarukamjorn et al., 1999). In total, the antibodies used in this study identify CYP3A and CYP2B proteins.
Rat liver P-450 reductase is also induced by drug and steroid treatments (Simmons et al., 1987). We examined regulation of the orthologous mouse liver P-450 reductase in microsomes from these same mice. P-450 reductase was induced by dexamethasone, PCN, and rifampin regardless of glucocorticoid receptor genotype (Fig. 3). Further analysis of these same microsomes with antibodies against rat CYP4A proteins revealed multiple immunoreactive bands whose expression was unaffected by glucocorticoid receptor genotype. These results demonstrate that the glucocorticoid receptor is required for agonist and antagonist induction of CYP2B but not for steroidal and nonsteroidal induction of CYP3A and P-450 reductase proteins.
Because the constitutive hepatic expression of CYP3A and CYP2B was sexually dimorphic in these mice (i.e., females expressed significantly greater amounts of these cytochromes than males; Fig. 2C and not shown for CYP2B), it was impossible to compare CYP induction in microsomes from male and female mice on the same blots. Therefore, we next examined the induction of hepatic CYP3A and CYP2B in female glucocorticoid receptor (+/+) and (−/−) mice. The magnitude of rifampin induction of both CYP3A proteins in females was less robust than that in males due to the higher basal level of CYP3A in female mice (not shown) but supported the results in male mice that the glucocorticoid receptor is not required for induction of CYP3A. As in the male mice, CYP2B was not induced by rifampin in females regardless of glucocorticoid receptor genotype (not shown).
To determine the influence of glucocorticoid receptor genotype on CYP3A and CYP2B mRNAs, we examined hepatic mRNAs from male mice on Northern blots (Fig. 7). Hybridization with a CYP3A11 cDNA revealed induction of CYP3A mRNA by dexamethasone, PCN, and rifampin in glucocorticoid receptor (+/+) and (−/−) mice. The mRNA supported the protein results that the glucocorticoid receptor is not required for CYP3A to be induced by either dexamethasone, PCN, or rifampin. Using a CYP2B10 cDNA, we were unable to detect good hybridizable CYP2B mRNA in male livers (not shown), a finding consistent with observations by others of the low expression of CYP2B mRNAs in control and steroid-treated male mouse liver in vivo (Nemoto and Sakurai, 1995).

Northern blot analysis of CYP3A in the livers of glucocorticoid receptor (+/+) and (−/−) mice.
Total RNA from individual male glucocorticoid receptor (+/+) and (−/−) untreated control or dexamethasone (DEX)-, PCN-, or rifampin (RIF)-treated mice were analyzed by Northern blot analysis with aCYP3A11 cDNA or ethidium bromide (Et. Br.) staining.
Analysis of TAT protein in cytosols from these same livers showed that dexamethasone induced TAT only in glucocorticoid receptor (+/+) mice (Fig. 8), demonstrating the requirement of the glucocorticoid receptor for this induction (as has been seen in other glucocorticoid receptor-null mice; Reichardt et al., 1998) and confirming the absence of functional glucocorticoid receptor in the null mice (Fig. 8). The antiglucocorticoid PCN suppressed TAT protein but only in the livers of mice lacking the glucocorticoid receptor (Fig. 8). However, there was no apparent induction of TAT protein by rifampin in any of the mice (Fig. 8) regardless of glucocorticoid receptor genotype.

Induction of TAT in glucocorticoid receptor (+/+) and (−/−) mice.
Cytosol (50 μg) from the livers of control (CT) and PCN-, dexamethasone (DEX)-, or rifampin (RIF)-treated glucocorticoid receptor (+/+) and (−/−) mice were analyzed on 10% polyacrylamide gel electrophoresis and immunoblotted with anti-TAT antibody.
This result was surprising because a recent report suggested that rifampin is a high-affinity nonsteroidal ligand for the human glucocorticoid receptor in liver (Calleja et al., 1998). We therefore tested rifampin in a dose-dependent manner for its ability to compete 2 nM [3H]dexamethasone from glucocorticoid receptor in extracts from mouse 3T3-L1 cells (Fig.9A). Rifampin did not compete with [3H]dexamethasone for binding to GR even at a concentration of 1 μM. The IC50 value for dexamethasone binding was determined to be 8.5 nM. Similar results were found when we tested rifampin for competition of [3H]aldosterone binding to the mouse mineralocorticoid receptor in mouse 3T3-L1 cells and also found no ability to compete at concentrations of up to 1 μM, whereas the IC50 value for aldosterone binding to the mouse mineralocorticoid receptor was 2.4 nM (not shown). We also tested rifampin for competition of [3H]dexamethasone binding to the human glucocorticoid receptor (Fig. 9B) and also found no ability to compete at concentrations up to 10 μM. The IC50 value for dexamethasone binding to the human glucocorticoid receptor was 4.6 nM. This indicates that rifampin showed no physiological binding affinity for glucocorticoid receptor in either species tested.
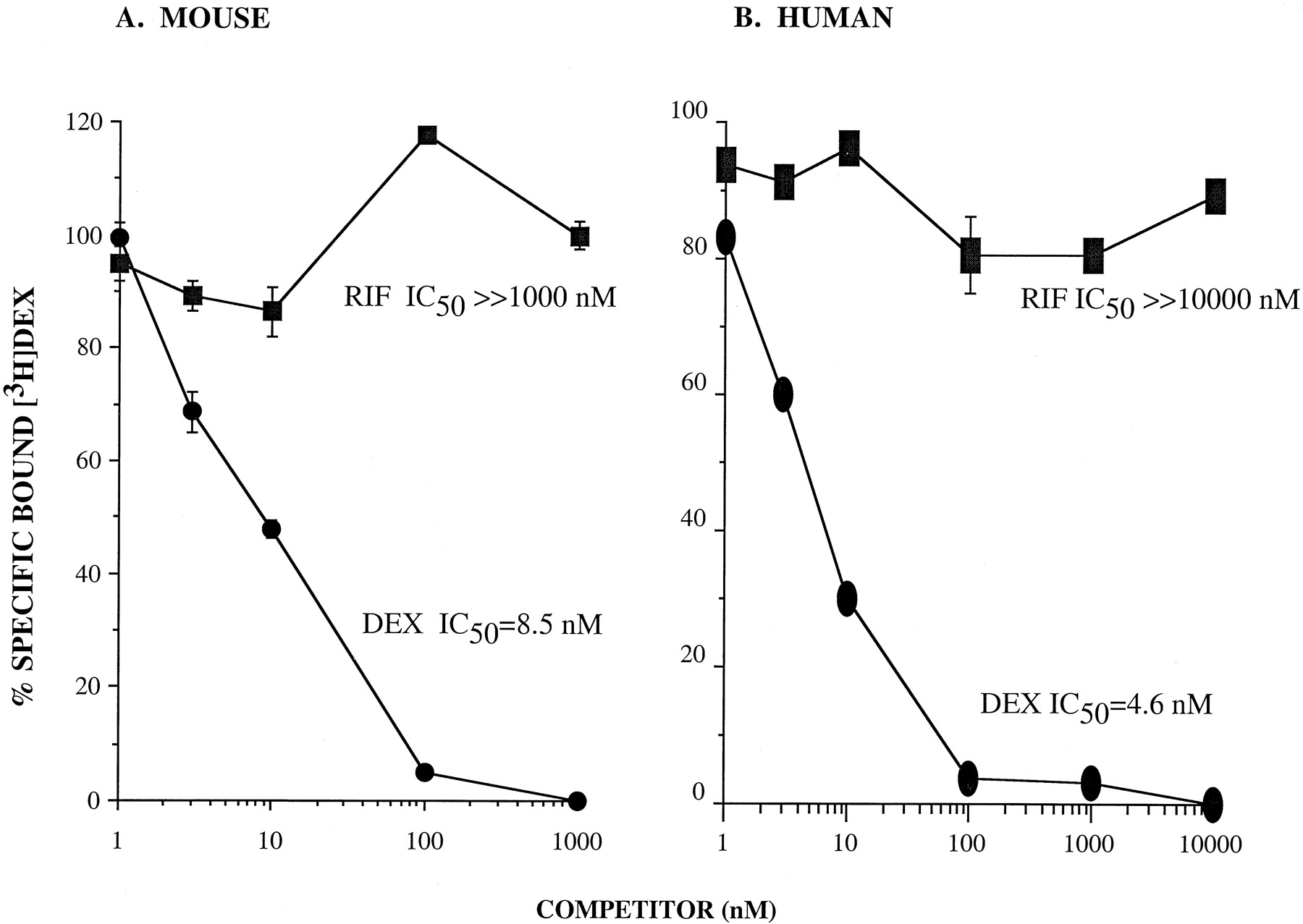
Rifampin cannot displace [3H]dexamethasone bound to mouse or human glucocorticoid receptor.
A, specificity of 2 nM [3H]dexamethasone binding in mouse 3T3-L1 cells: competition by dexamethasone (DEX, ●) and rifampin (RIF, ▪). The IC50 value, the concentration of cold competitor at which 50% of [3H]dexamethasone is competed off, is 8.5 nM (dexamethasone binding set at 100%). The IC50 value for rifampin was ≫1000 nM. B, specificity of 2 nM [3H]dexamethasone binding in human brain tumor tissue: competition by dexamethasone (DEX, ●) and rifampin (RIF, ▪). Dexamethasone, IC50 = 4.6 nM (dexamethasone binding set at 100%). Rifampin, IC50 ≫10,000 nM. Values (6 × 3.5-cm wells/point) are means ± S.E., except where value for S.E. is less than the size of symbol.
Because we and others have found that glucocorticoid treatment can affect the magnitude of phenobarbital induction of CYP (Kocarek et al., 1994; Sidhu and Omicinski, 1995), we used glucocorticoid receptor-null mice to determine whether the glucocorticoid receptor had a role in phenobarbital induction of CYPs. The glucocorticoid receptor was not obligatory for phenobarbital to induce CYP2B or CYP3A proteins (see Fig. 6). However, the magnitude of CYP2B and CYP3A induction by phenobarbital was attenuated in glucocorticoid receptor-null male mice, although it was not obvious in these immunoblots developed to detect CYP levels in untreated control mice (Fig. 6A). Quantitative immunoblots (Fig. 6B) revealed that CYP2B and CYP3A were induced in receptor (−/−) mice to only 63% of their induction in (+/+) mice (100%). Female mice contained an additional CYP2B protein (migrating between the two immunoreactive proteins in male microsomes) that is constitutively expressed and inducible by phenobarbital in both glucocorticoid receptor (+/+) and (−/−) female mice (Fig. 6). It is possible that this additional protein is CYP2B9 because compared with males, female mice express greater amounts of CYP2B9 (Nemoto and Sakurai, 1995; Jarukamjorn et al., 1999).
Discussion
Although recent reports identify PXR as a mediator of steroid and drug induction of CYP3A, several lines of evidence suggest that the glucocorticoid receptor may also be required for induction of this protein: 1) the weak activation of mouse or human PXR (Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998) or even no activation (Bertilsson et al., 1998) of human PXR by dexamethasone, 2) synergistic interaction between PCN and dexamethasone for induction of the endogenous rat CYP3A genes (Schuetz and Guzelian, 1984; Burger et al., 1992), 3) the glucocorticoid receptor and traditional GREs in the CYP3A gene were reported to mediate dexamethasone induction of the rat CYP3A1 promoter (Pereira et al., 1998), 4) the antiglucocorticoid (PXR ligand) RU486 blocks dexamethasone transcriptional activation of the CYP3A1 (Burger et al., 1992) and CYP3A4 (Ogg et al., 1999) promoters, and 5) rifampin is an efficacious inducer of mouse CYP3A (Wrighton et al., 1985b; Yanagimoto et al., 1997) but a weak activator of mouse PXR (Lehmann et al., 1998). These results suggested that there may be additional transcription factors (e.g., the glucocorticoid receptor) required for dexamethasone and rifampin induction of CYP3A. Using the glucocorticoid receptor (+/+) and (−/−) mice, we demonstrate that CYP3A induction by glucocorticoids, antiglucocorticoids, and rifampin proceeds via a mechanism that does not require the glucocorticoid receptor. Moreover, there appears to be no influence of the glucocorticoid receptor on basal expression of the CYP3As. In addition, the inability of rifampin to induce TAT or to displace dexamethasone from the mouse or human glucocorticoid receptor suggests that rifampin cannot be a ligand for this receptor.
We compared the induction of CYP3A with that of CYP2B in the glucocorticoid receptor (+/+) and (−/−) mice to determine whether the glucocorticoid receptor was a shared regulatory characteristic. In striking contrast to CYP3A, our results demonstrate that the glucocorticoid receptor is obligatory for both maximal constitutive expression of CYP2B and for either dexamethasone or PCN to induce CYP2B. This result seems paradoxical at first because PCN is an antiglucocorticoid and cannot activate the glucocorticoid receptor (Schuetz and Guzelian, 1984). Although questions regarding the precise molecular mechanisms of CYP2B induction by PCN and dexamethasone remain to be determined (i.e., is PXR mediating steroid induction of CYP2B?), based on our results, we propose a model in which dexamethasone and PCN induction of CYP2B requires both PXR and the glucocorticoid receptor. In support of this hypothesis, PXR is the only transcription factor identified that can mediate PCN signaling (Kliewer et al., 1998). In addition, we previously demonstrated that the mechanism by which dexamethasone induces CYP2B shares similar characteristics with nonclassic glucocorticoid induction of CYP3A (Kocarek et al., 1994), a mechanism now known to be mediated by PXR (Kliewer et al., 1998;Lehmann et al., 1998). Intriguingly, a xenochemical response module in the mouse Cyp2b10 and rat CYP2B1 and2B2 genes (Park et al., 1996; Stoltz et al., 1998) contains nuclear hormone response elements (including a DR4, a potential target sequence for PXR; Blumberg et al., 1998) and a putative GRE (Stoltz et al., 1998).
There are conflicting data in the literature regarding the contribution of glucocorticoids to phenobarbital induction of CYPs. Although most reports found that phenobarbital induction of rat or mouse CYP2B either required or was potentiated by glucocorticoids, high concentrations of dexamethasone can suppress phenobarbital induction of CYP2B in rats (Kocarek et al., 1994; Sidhu and Omicinski, 1995; Honkakoski and Negishi, 1998). Mice with targeted gene disruption of the glucocorticoid receptor offered a way to determine its contribution to phenobarbital induction in mice in vivo. Our results demonstrate that the glucocorticoid receptor is not essential for phenobarbital to induce CYP2B but does contribute to maximum phenobarbital induction of CYP2B in mouse liver. Surprisingly, a parallel effect of glucocorticoid receptor abrogation on the magnitude of phenobarbital induction of CYP3A was also found, suggesting the receptor may affect a common phenobarbital induction mechanism (Sueyoshi et al., 1999).
Our result that rifampin was not capable of binding with either the human or mouse glucocorticoid receptor is in stark contrast to an original report (Calleja et al., 1998) that rifampin is a nonsteroidal ligand for the human glucocorticoid receptor in HepG2 human hepatoblastoma cells. However, similar to our results, two other groups, using A549 human alveolar cells (Jaffuel et al., 1999) or mouse AtT20 pituitary corticotroph cells and COS-7 monkey kidney cells cotransfected with human glucocorticoid receptor (Ray et al., 1998), have failed to find that rifampin interacts with the glucocorticoid receptor. One possible explanation for the discrepancy between the original (Calleja et al., 1998) versus all subsequent reports is that in HepG2 cells, the authors may have been looking at PXR-mediated activation of the MMTV (mouse mammary tumor virus) promoter. Given that some HepG2 cells express a low level of PXR (E.G.S., unpublished observation) and that the MMTV enhancer has a potential nuclear receptor binding motif previously shown to bind LXR/RXR heterodimers (Willy et al., 1995) it is possible that rifampin activated PXR/RXR bound and activated MMTV in their experiments. Because AtT20, A549, and COS-7 cells presumably lack PXR, these cell lines would be incapable of rifampin activation of MMTV. In addition, although our data demonstrate that rifampin is not binding to the ligand binding pocket of the receptor in the classic sense, we cannot rule out rifampin activating GCR via another part of the receptor or even via another associated protein.
A recent report found that DMSO, the vehicle used in the rifampin treatments, can increase rat CYP3A by stabilizing the CYP3A protein in primary rat hepatocytes (Zangar and Novak, 1998). To confirm that rifampin, and not DMSO, was inducing CYP3A, we compared rifampin induction of CYP3A in ethanol or DMSO and found that CYP3A was equivalently induced when administered in either vehicle (not shown). Similarly, DMSO had no effect on either basal CYP2B or on the extent of CYP2B induction by phenobarbital (not shown), consistent with the lack of effect of DMSO on induction of CYP2B in primary mouse hepatocytes (Honkakoski and Negishi, 1998) and demonstrating that the lack of CYP2B induction by rifampin was not due to the DMSO vehicle. Thus, the effects of rifampin on these cytochromes are specific to rifampin and not a side effect of the DMSO.
Our results now reveal what may be an important species difference in rifampin induction of CYP2B. At the dose and duration administered, rifampin clearly did not induce mouse CYP2B, whereas we previously identified rifampin as an efficacious inducer of CYP2B6 in primary cultures of human hepatocytes (Strom et al., 1996). Because rifampin induced hepatic CYP3A, it clearly was biologically active in the hepatocyte. However, in the livers of these same mice, CYP2B appeared to be refractory to rifampin treatment and even appeared to be somewhat decreased. These results are similar to previous reports that even within the same species, rifampin can have both inductive and repressive effects on hepatic CYPs (Lange et al., 1984). Moreover, within a single CYP gene family (the CYP3As), there are noted species differences in rifampin activation of the rat (unresponsive) versus mouse and human (responsive) CYP3A proteins (Kocarek et al., 1995).
Could differences in genetic backgrounds (i.e., progeny are 129 × C57BL/6 heterozygotes) contribute to differences in CYP3A or CYP2B induction between the animals under study? The progeny would be expected to have random segregation of 129 or C57BL/6 P-450 allelic determinants. Unfortunately, 100% of isogenic glucocorticoid receptor (−/−) animals die at birth (Cole et al., 1995), necessitating the use of heterozygote progeny. Although there are well documented strain differences in the induction of some P-450s in mice (e.g., the CYP1A family), in a survey of 14 different strains of mice (including C57BL/6) we failed to identify any strain in which CYP3A was not induced by either dexamethasone or PCN (E.G.S., unpublished observation). Moreover, in this study, the induction of CYP3A proteins in male mice by dexamethasone, PCN, or rifampin was the same in all mice tested regardless of glucocorticoid receptor genotype and regardless of the admixture of genetic background. Finally, because CYP2B and CYP3A are induced in both parental strains (C57BL/6J and 129J) mice by dexamethasone (not shown), the complete loss of CYP2B induction by this agent in glucocorticoid receptor (−/−) mice can only be attributed to disruption of the glucocorticoid receptor locus.
Understanding the way in which drugs induce the CYPs (particularly whether they share identical regulatory mechanisms) is biologically relevant because it ultimately leads to better models for screening and predicting drug interactions that occur due to the induction of individual CYPs. Can we extrapolate these results to the regulation of human CYPs? We propose that analogous to mouse CYP3A, the glucocorticoid receptor will not be required for the induction of human CYP3A4 by glucocorticoids or rifampin because human PXR is activated by these agents and binds to a conserved hormone response element in the CYP3A4 gene (Lehmann et al., 1998). Moreover, we were unable to find an interaction of rifampin with human glucocorticoid receptor. However, if there are functional GREs in the human CYP3A genes, it is still possible that the glucocorticoid receptor could contribute to maximum glucocorticoid induction of human CYP3A. Importantly, rifampin and glucocorticoids are not selective inducers of human CYP3A but induce other isozymes of CYP, including CYP2B6 (Strom et al., 1996) and activities associated with CYP2C19 (Zhou et al., 1990) in human liver. It remains to be determined whether glucocorticoid induction of human CYP2B6 parallels our results with mouse CYP2B and absolutely requires the glucocorticoid receptor.
Because CYP3A and CYP2B proteins are up-regulated by many of the same drugs and xenobiotics, it was originally assumed that they were induced by a common mechanism, analogous to the paradigm of the aryl hydrocarbon receptor mechanism and pleiotropic induction of multiple detoxification proteins. However, we and others have demonstrated that induction of CYP3A and CYP2B differs in several respects. For example: 1) the time course of induction of these two cytochromes differs dramatically after treatment with PCN or phenobarbital (Hardwick et al., 1983), 2) protein synthesis inhibitors have opposite effects on the induction of these cytochromes by steroids and phenobarbital (Burger et al., 1990), 3) induction of these cytochromes can be differentiated by dose of inducer (Kocarek et al., 1990), and 4) phenobarbital induction of these two genes may proceed by different nuclear receptors (Honkakoski et al., 1998; Lehmann et al., 1998). We now add that glucocorticoid and antiglucocorticoid inductions of CYP2B and CYP3A have distinctly different requirements for the glucocorticoid receptor.
Footnotes
-
Send reprint requests to: Dr. Erin Schuetz, Department of Pharmaceutical Sciences, St. Jude Children's Research Hospital, 332 N. Lauderdale, Memphis, TN 38105. E-mail: erin.schuetz{at}stjude.org
-
This work was supported by National Institutes of Health Research Grants ES08658, ES05851, P30-CA21765, and DA04265 (L.M.B.); a block grant from the National Health and Medical Research Council of Australia; and the American Lebanese Syrian Associated Charities (ALSAC).
- Abbreviations used are::
- PXR
- pregnane X receptor
- DMEM
- Dulbecco's modified Eagle's medium
- TAT
- tyrosine aminotransferase
- GRE
- glucocorticoid response element
- PCN
- pregnenolone 16α-carbonitrile
- Received August 11, 1999.
- Accepted November 9, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}